
Identification of Novel miRNAs and Their Target Genes in the Response to Abscisic Acid in Arabidopsis

 ,
,
Abstract
:1. Introduction
2. Results
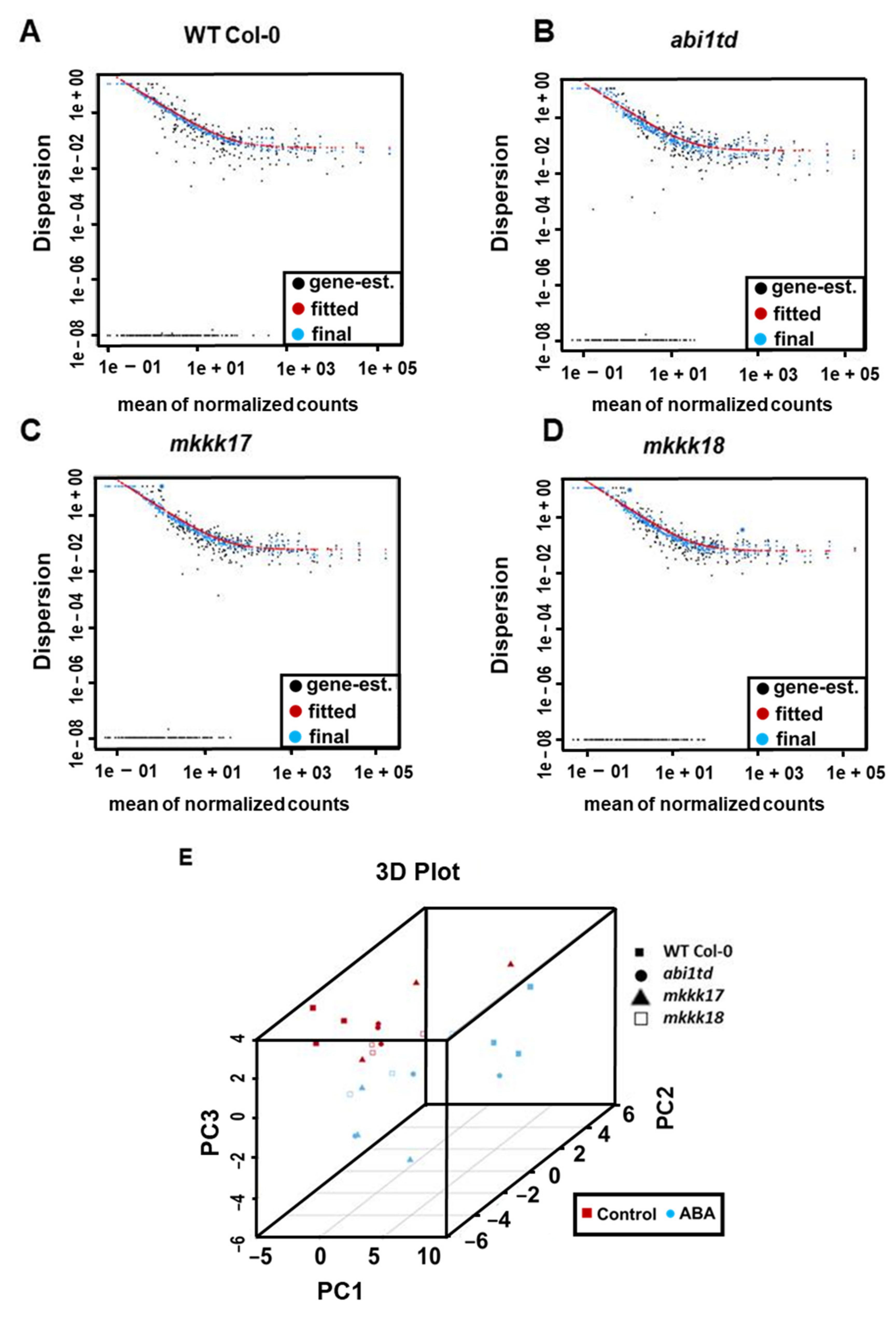
2.1. Small RNA Sequencing of Arabidopsis WT Col-0 and abi1td, mkkk17, and mkkk18 Mutants after ABA Treatment
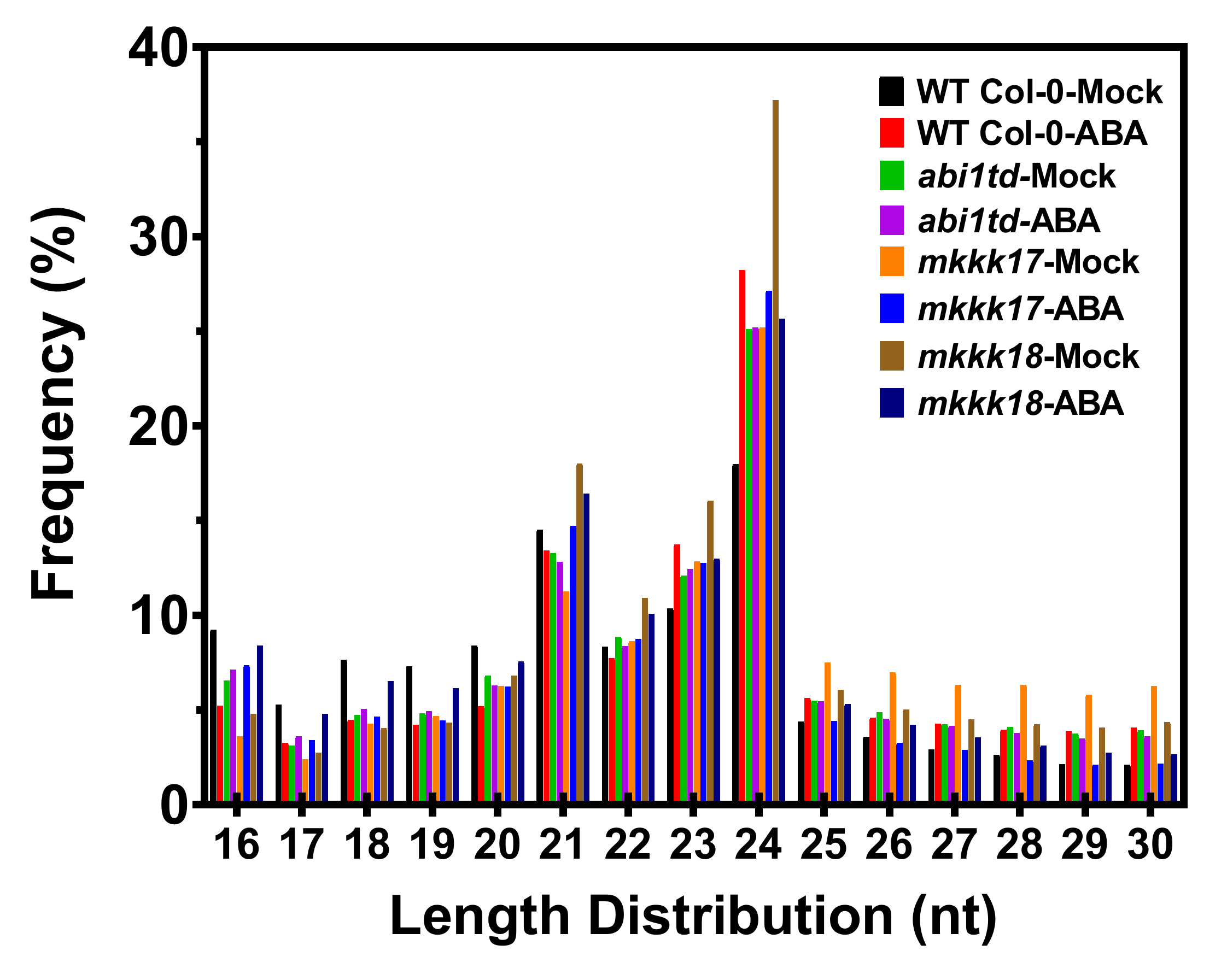
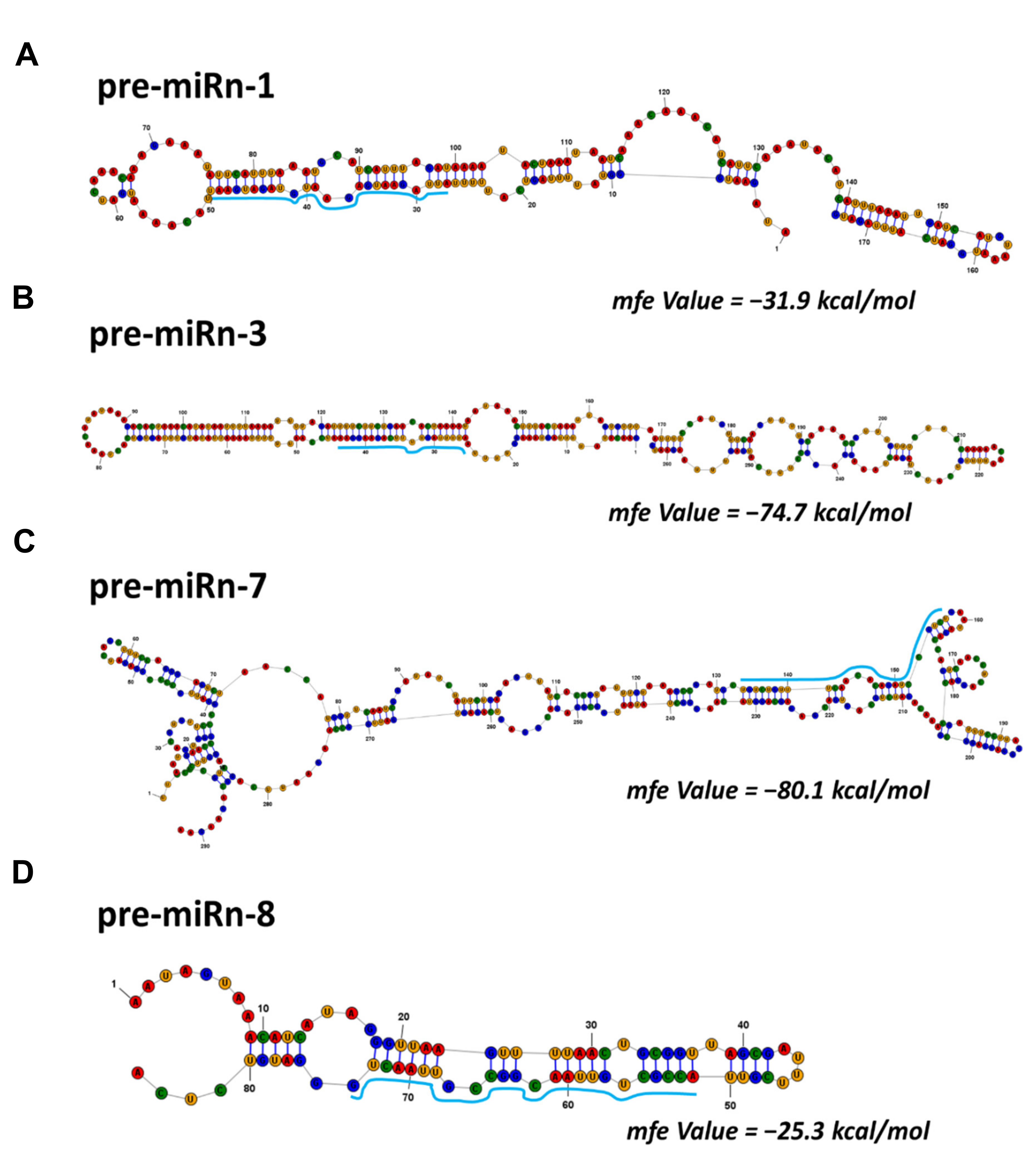
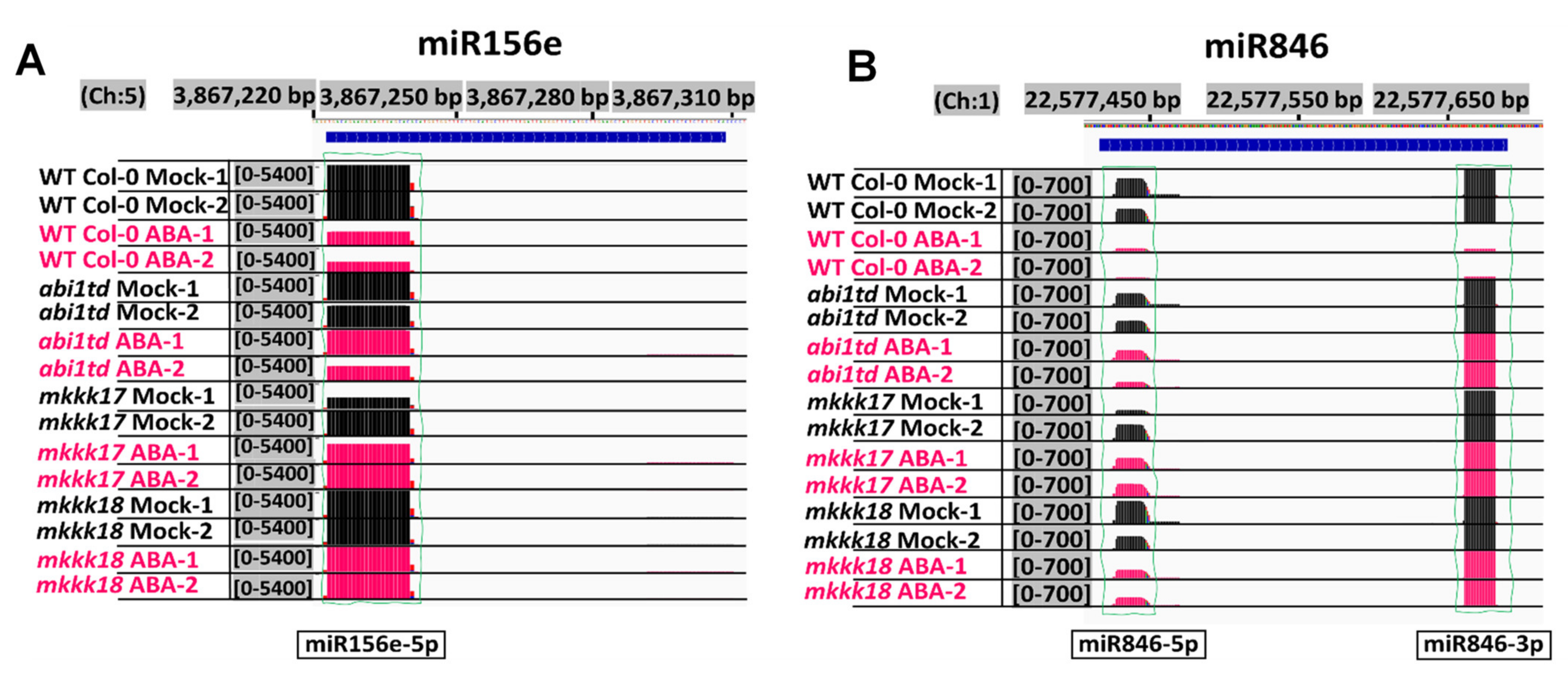
2.2. Identification of Known and Novel miRNAs
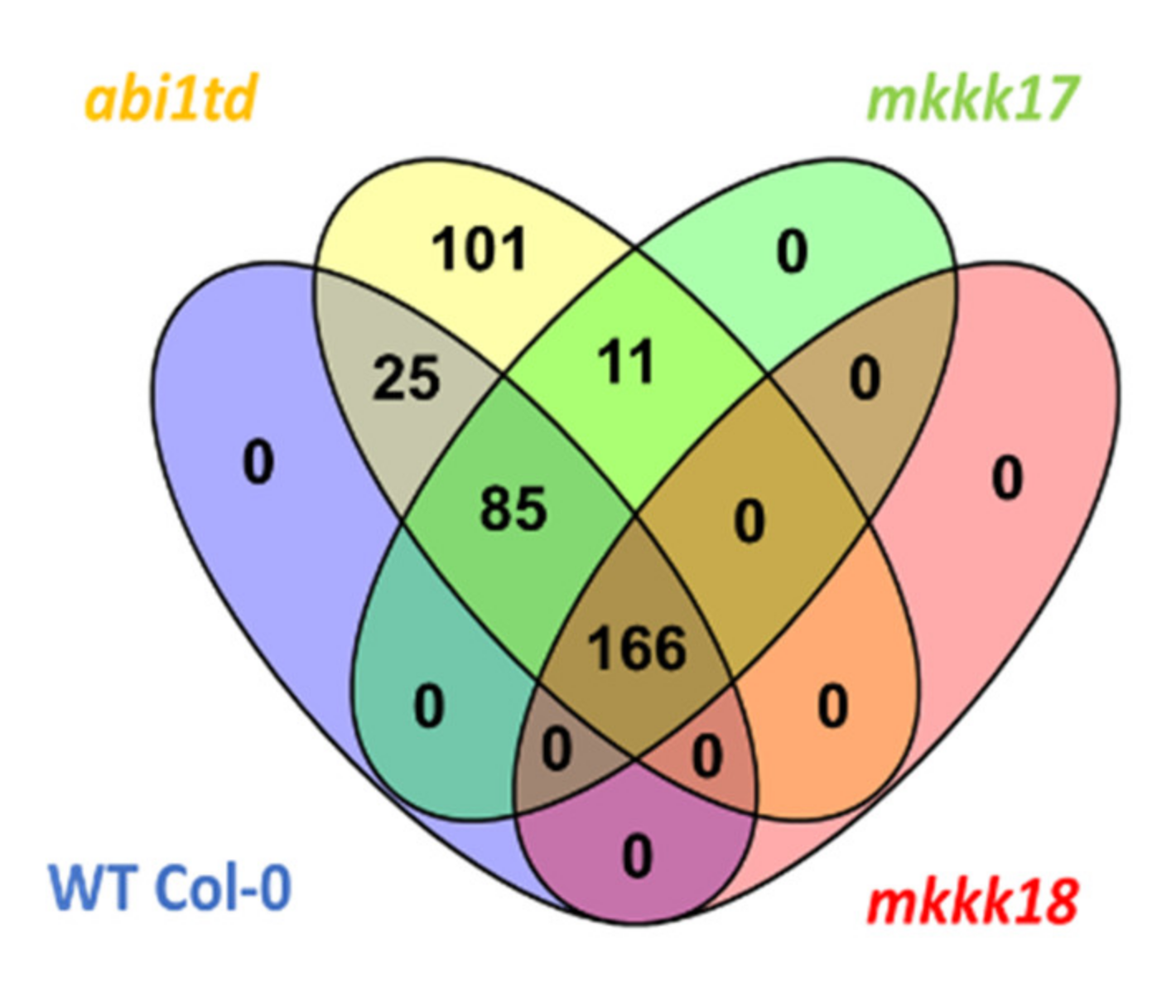
2.3. Differentially Expressed miRNA Screening
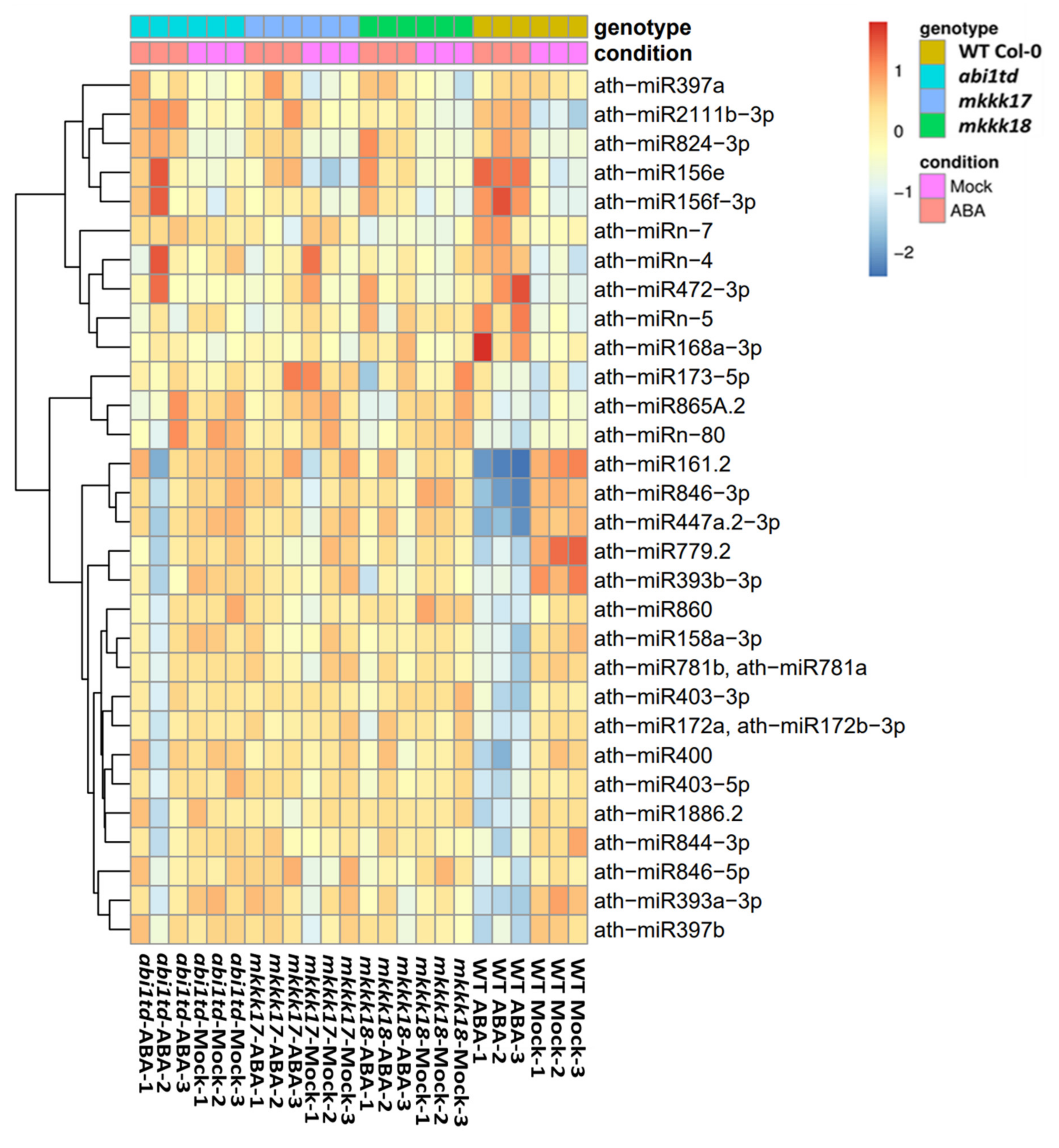
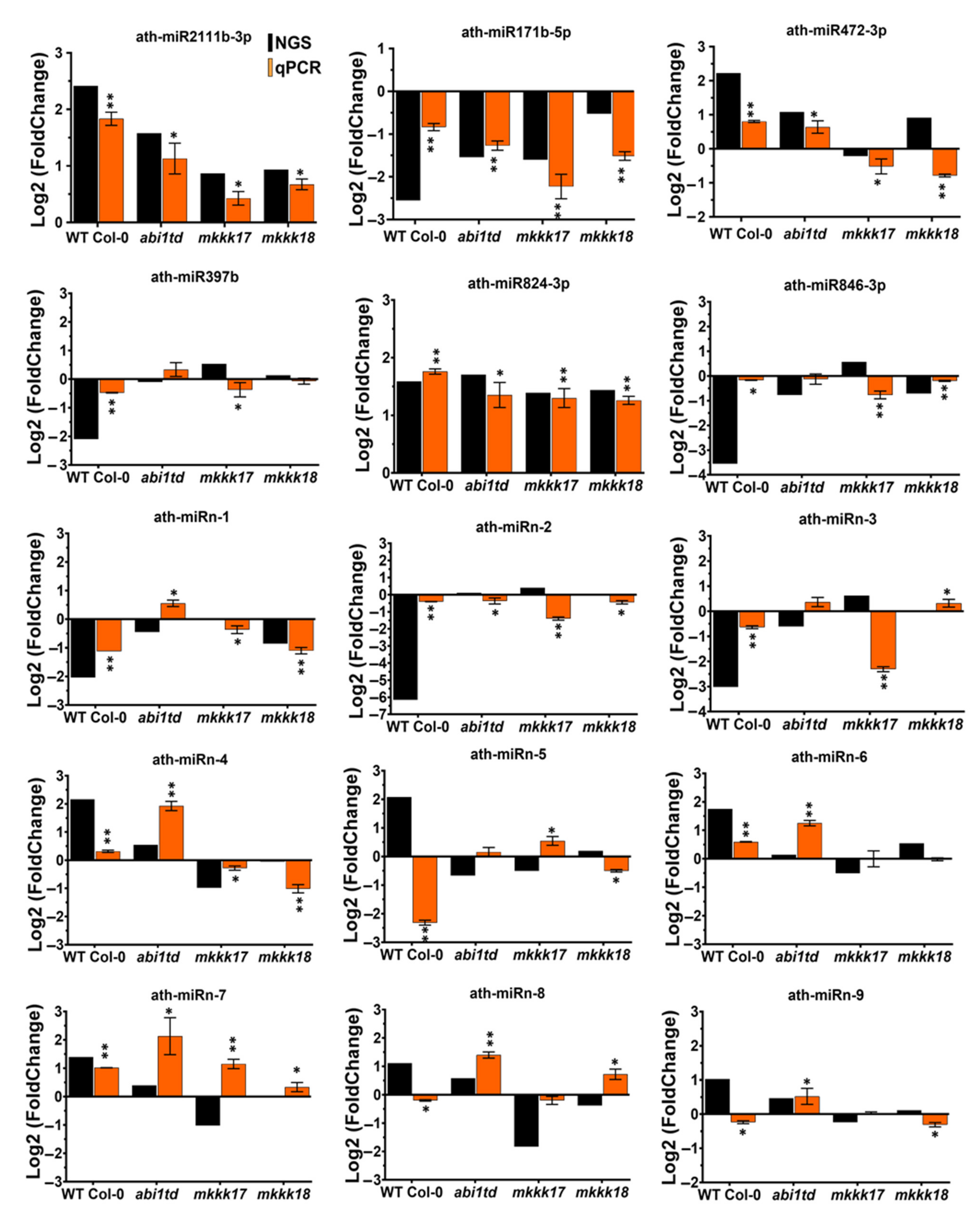
2.4. Expression Profiling by qRT-PCR of Known and Novel miRNAs
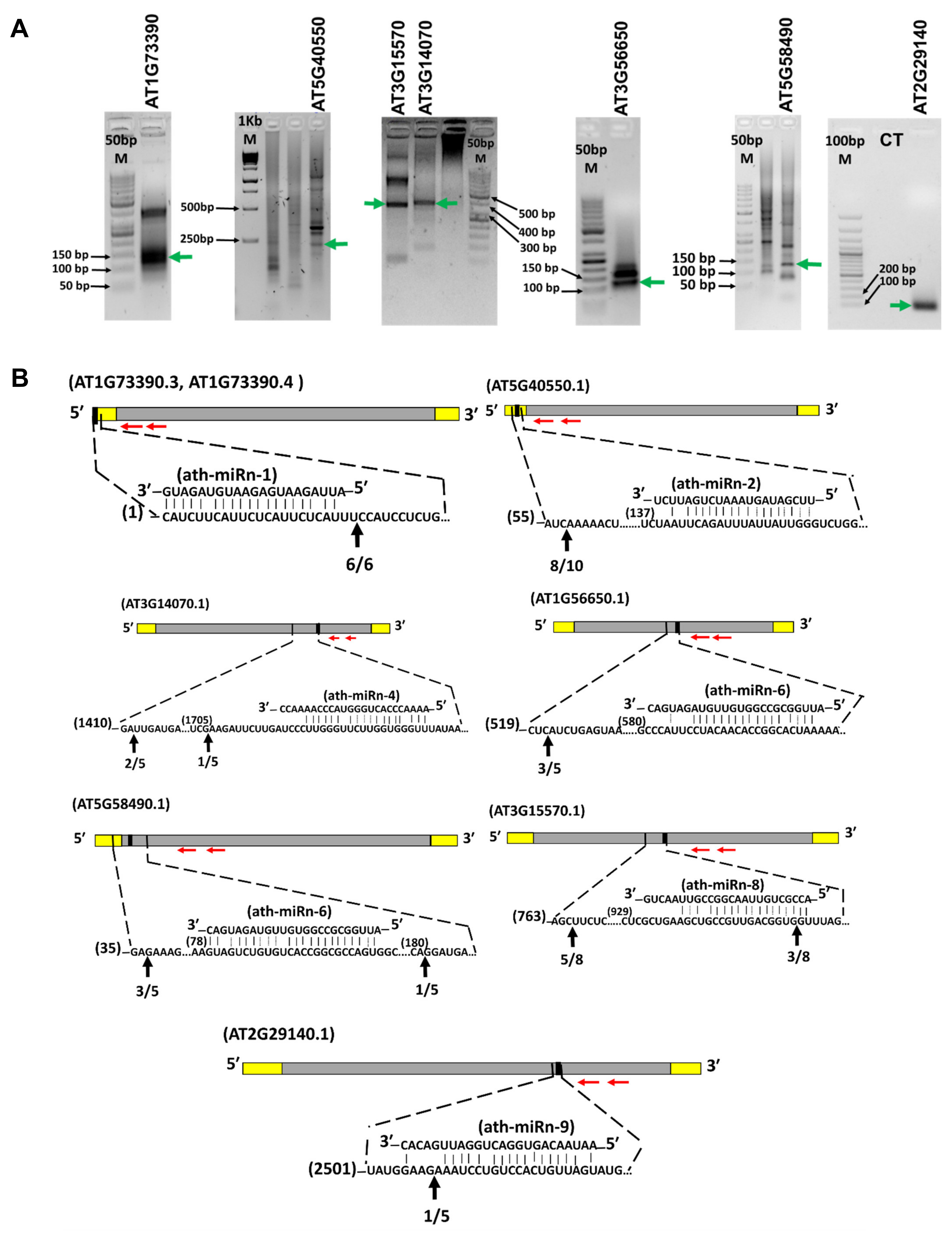
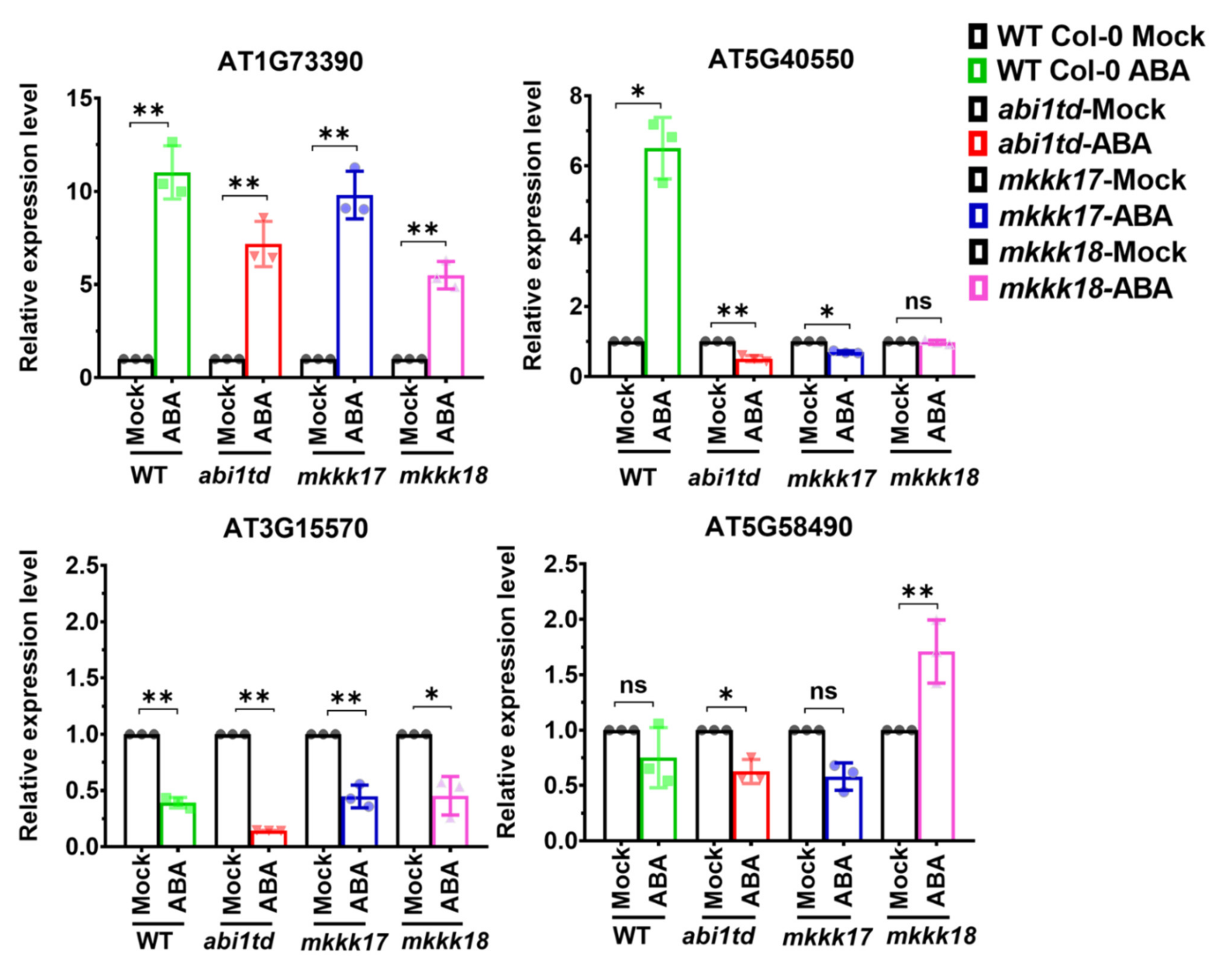
2.5. Target mRNA Prediction, 5′ RLM-RACE Validation, and Target Gene Expression by qRT-PCR
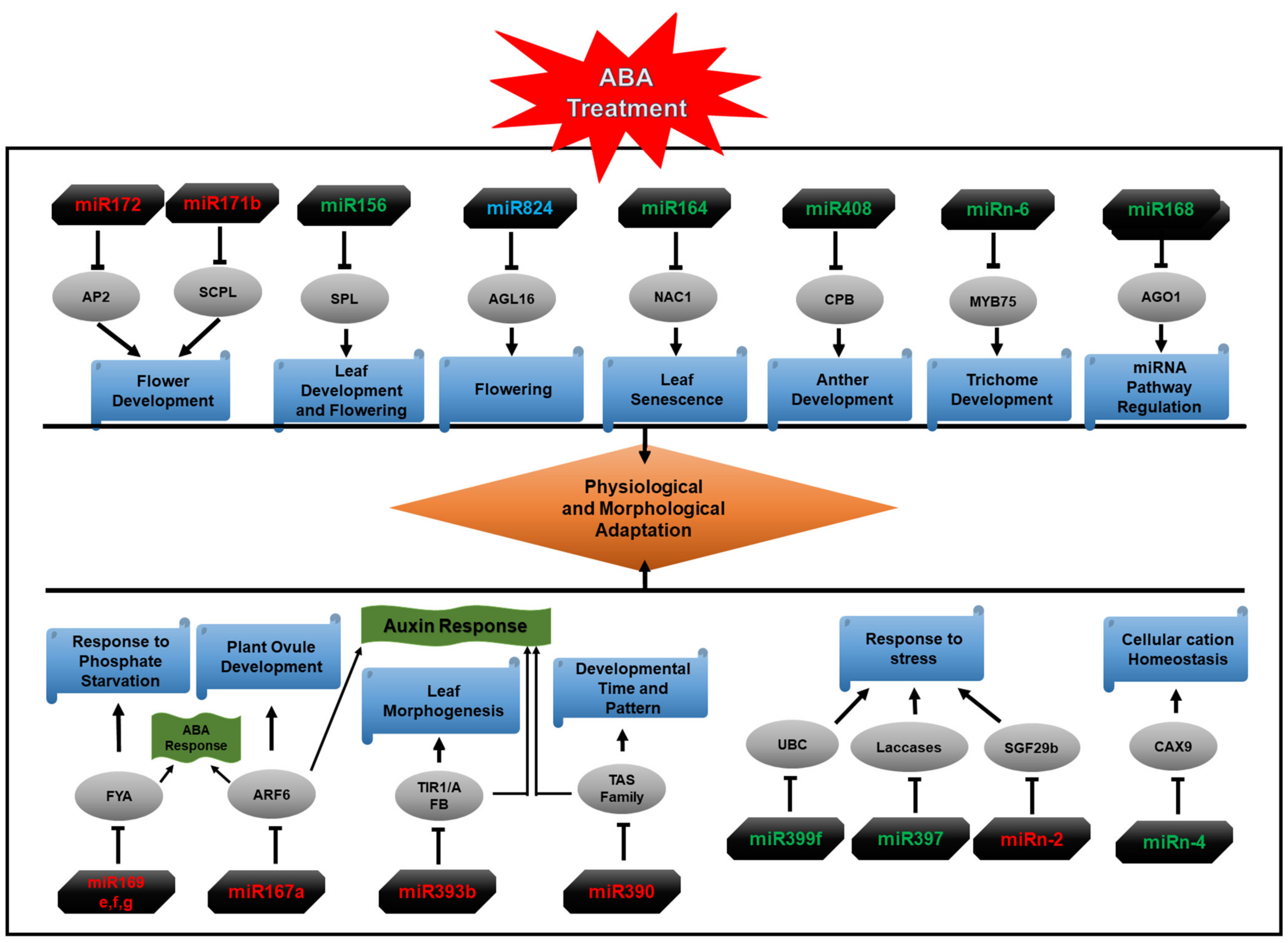
2.6. Functional Enrichment of miRNA Target Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sample Preparation for Sequencing
4.2. Sequencing and Processing of Reads
4.3. Differential Expression Analysis
4.4. Validation of miRNA Expression with Stem-Loop qRT-PCR
4.5. Target Validation by 5′ RLM-RACE-PCR and miRNA Target Gene Expression by qRT-PCR
4.6. Target Gene Identification, Gene Ontology, and KEGG Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finkelstein, R.R.; Gampala, S.S.L.; Rock, C.D. Abscisic acid signaling in seeds and seedlings. Plant Cell 2002, 14, 15–46. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.M. Abscisic acid: Hidden architect of root system structure. Plants 2015, 4, 548–572. [Google Scholar] [CrossRef] [PubMed]
- Danquah, A.; De Zélicourt, A.; Boudsocq, M.; Neubauer, J.; Frei Dit Frey, N.; Leonhardt, N.; Pateyron, S.; Gwinner, F.; Tamby, J.P.; Ortiz-Masia, D.; et al. Identification and characterization of an ABA-activated MAP kinase cascade in Arabidopsis thaliana. Plant J. 2015, 82, 232–244. [Google Scholar] [CrossRef]
- Mitula, F.; Tajdel, M.; Cieśla, A.; Kasprowicz-Maluśki, A.; Kulik, A.; Babula-Skowrońska, D.; Michalak, M.; Dobrowolska, G.; Sadowski, J.; Ludwików, A.; et al. Arabidopsis ABA-Activated Kinase MAPKKK18 is Regulated by Protein Phosphatase 2C ABI1 and the Ubiquitin-Proteasome Pathway. Plant Cell Physiol. 2015, 56, 2351–2367. [Google Scholar] [CrossRef]
- Menges, M.; Dóczi, R.; Ökrész, L.; Morandini, P.; Mizzi, L.; Soloviev, M.; Murray, J.A.H.; Bögre, L. Comprehensive gene expression atlas for the Arabidopsis MAP kinase signalling pathways. New Phytol. 2008, 179, 643–662. [Google Scholar] [CrossRef]
- Umezawa, T.; Nakashima, K.; Miyakawa, T.; Kuromori, T.; Tanokura, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular basis of the core regulatory network in ABA responses: Sensing, signaling and transport. Plant Cell Physiol. 2010, 51, 1821–1839. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Rock, C.D. MIR846 and MIR842 comprise a cistronic MIRNA pair that is regulated by abscisic acid by alternative splicing in roots of Arabidopsis. Plant Mol. Biol. 2013, 81, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Xie, Z.; Allen, E.; Fahlgren, N.; Calamar, A.; Givan, S.A.; Carrington, J.C. Expression of Arabidopsis MIRNA genes. Plant Physiol. 2005, 138, 2145–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F.; Gasciolli, V.; Crété, P.; Vaucheret, H. The Nuclear dsRNA Binding Protein HYL1 Is Required for MicroRNA Accumulation and Plant Development, but Not Posttranscriptional Transgene Silencing. Curr. Biol. 2004, 14, 346–351. [Google Scholar] [CrossRef]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Yang, Z.; Li, J.; Minakhina, S.; Yang, M.; Padgett, R.W.; Steward, R.; Chen, X. Methylation as a crucial step in plant microRNA biogenesis. Science 2005, 307, 932–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mee, Y.P.; Wu, G.; Gonzalez-Sulser, A.; Vaucheret, H.; Poethig, R.S. Nuclear processing and export of microRNAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 3691–3696. [Google Scholar] [CrossRef] [Green Version]
- Vaucheret, H.; Vazquez, F.; Crété, P.; Bartel, D.P. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004, 18, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Baumberger, N.; Baulcombe, D.C. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 11928–11933. [Google Scholar] [CrossRef] [Green Version]
- Voinnet, O. Origin, Biogenesis, and Activity of Plant MicroRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, N.; Wang, H.; Kasahara, H.; Liu, J.; MacPherson, C.; Machida, Y.; Kamiya, Y.; Hannah, M.A.; Chuaa, N.H. IAA-Ala Resistant3, an evolutionarily conserved target of miR167, mediates Arabidopsis root architecture changes during high osmotic stress. Plant Cell 2012, 24, 3590–3602. [Google Scholar] [CrossRef] [Green Version]
- Reyes, J.L.; Chua, N.H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef]
- Yoon, E.K.; Yang, J.H.; Lim, J.; Kim, S.H.S.K.; Kim, S.H.S.K.; Lee, W.S. Auxin regulation of the microRNA390-dependent transacting small interfering RNA pathway in Arabidopsis lateral root development. Nucleic Acids Res. 2009, 38, 1382–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acquadro, A.; Barchi, L.; Portis, E.; Nourdine, M.; Carli, C.; Monge, S.; Valentino, D.; Lanteri, S. Whole genome resequencing of four Italian sweet pepper landraces provides insights on sequence variation in genes of agronomic value. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, A.; Zhang, X.; Hou, Y.; Shangguan, M.; Gajjeraman, P.; Li, Y.; Wei, C. Genome-wide identification of conserved and novel microRNAs in one bud and two tender leaves of tea plant (Camellia sinensis) by small RNA sequencing, microarray-based hybridization and genome survey scaffold sequences. BMC Plant Biol. 2017, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Ali, A.; Saifi, M.; Saxena, P.; Ahlawat, S.; Abdin, M.Z. Identification and the potential involvement of miRNAs in the regulation of artemisinin biosynthesis in A. annua. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Li, R.; Chen, D.; Wang, T.; Wan, Y.; Li, R.; Fang, R.; Wang, Y.; Hu, F.; Zhou, H.; Li, L.; et al. High throughput deep degradome sequencing reveals microRNAs and their targets in response to drought stress in mulberry (Morus alba). PLoS ONE 2017, 12, 1–30. [Google Scholar] [CrossRef]
- Wang, A.; Hu, J.; Gao, C.; Chen, G.; Wang, B.; Lin, C.; Song, L.; Ding, Y.; Zhou, G. Genome-wide analysis of long non-coding RNAs unveils the regulatory roles in the heat tolerance of Chinese cabbage (Brassica rapa ssp. chinensis). Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Su, S.; Jin, L.; Peng, R.; Sun, D.; Ji, H.; Yu, Y.; Xu, J. Identification of microRNAs and their targets in inflorescences of an Ogura-type cytoplasmic male-sterile line and its maintainer fertile line of turnip (Brassica rapa ssp. rapifera) via high-throughput sequencing and degradome analysis. PLoS ONE 2020, 15, e0236829. [Google Scholar] [CrossRef]
- Xu, X.; Chen, X.; Chen, Y.; Zhang, Q.; Su, L.; Chen, X.; Chen, Y.; Zhang, Z.; Lin, Y.; Lai, Z. Genome-wide identification of miRNAs and their targets during early somatic embryogenesis in Dimocarpus longan Lour. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Wang, Q.; Wei, L.; Liang, Y.; Dai, J.; Xia, G.; Liu, S. Small RNA and degradome sequencing used to elucidate the basis of tolerance to salinity and alkalinity in wheat. BMC Plant Biol. 2018, 18, 1–17. [Google Scholar] [CrossRef]
- Duan, H.; Lu, X.; Lian, C.; An, Y.; Xia, X.; Yin, W. Genome-wide analysis of microRNA responses to the phytohormone abscisic acid in Populus euphratica. Front. Plant Sci. 2016, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Stefano, P.; Ivana, M.; Arianna, G.; Letizia, Z.; Angelo, G.; Rosella, C.; Roberta, B.; Vittorio, C.; Antonella, C.; Maurizio, M.; et al. Identification of microRNAs and relative target genes in Moringa oleifera leaf and callus. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Schleiff, E.; Simm, S. miRNAs involved in transcriptome remodeling during pollen development and heat stress response in Solanum lycopersicum. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in Arabidopsis thaliana by miRr156 and its target SPL3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-Regulated SPL Transcription Factors Define an Endogenous Flowering Pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, A.M.; Nodine, M.D.; Gaj, M.D. MiR160 and miR166/165 contribute to the LEC2-mediated auxin response involved in the somatic embryogenesis induction in Arabidopsis. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.S.; Li, J.; Stahle, M.I.; Dubroué, A.; Gubler, F.; Millar, A.A. Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc. Natl. Acad. Sci. USA 2007, 104, 16371–16376. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.; Rock, C.D. Jacalin lectin At5g28520 is regulated by ABA and miR846. Plant Signal. Behav. 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Sreenivasulu, N.; Sopory, S.K.; Kavi Kishor, P.B. Deciphering the regulatory mechanisms of abiotic stress tolerance in plants by genomic approaches. Gene 2007, 388, 1–13. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic Acid: Emergence of a Core Signaling Network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunkar, R.; Zhu, J.K. Novel and stress regulated microRNAs and other small RNAs from Arabidopsis w inside box sign. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Ma, Z.; Liu, Z. Non-Coding RNA Transcription and RNA-Directed DNA Methylation in Arabidopsis. Mol. Plant 2014, 7, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Lewsey, M.G.; Hardcastle, T.J.; Melnyk, C.W.; Molnar, A.; Valli, A.; Urich, M.A.; Nery, J.R.; Baulcombe, D.C.; Ecker, J.R. Mobile small RNAs regulate genome-wide DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, E801–E810. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhu, J. RNA-directed DNA methylation. Curr. Opin. Plant Biol. 2011, 14, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronemus, M.; Vaughn, M.W.; Martienssen, R.A. MicroRNA-Targeted and Small Interfering RNA-Mediated mRNA Degradation Is Regulated by Argonaute, Dicer, and RNA-Dependent RNA Polymerase in Arabidopsis. Plant Cell 2006, 18, 1559–1574. [Google Scholar] [CrossRef] [Green Version]
- Paola, D.; De Paola, D.; Cattonaro, F.; Pignone, D.; Sonnante, G. The miRNAome of globe artichoke: Conserved and novel micro RNAs and target analysis The miRNAome of globe artichoke: Conserved and novel micro RNAs and target analysis. BMC Genom. 2012, 13, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepansky, A. Regulation of lysine catabolism in Arabidopsis through concertedly regulated synthesis of the two distinct gene products of the composite AtLKR/SDH locus. J. Exp. Bot. 2005, 56, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Akter, N.; Sobahan, M.A.; Uraji, M.; Ye, W.; Hossain, M.A.; Mori, I.C.; Nakamura, Y.; Murata, Y. Effects of Depletion of Glutathione on Abscisic Acid- and Methyl Jasmonate-Induced Stomatal Closure in Arabidopsis thaliana. Biosci. Biotechnol. Biochem. 2012, 76, 2032–2037. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Lv, D.; Wang, P.; Wang, X.; Chen, J.; Miao, C.; Song, C. An Arabidopsis Glutathione Peroxidase Functions as Both a Redox Transducer and a Scavenger in Abscisic Acid and Drought Stress Responses. Plant Cell 2006, 18, 2749–2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wang, J.; Li, S.; Kakan, X.; Zhou, Y.; Miao, Y.; Wang, F.; Qin, H.; Huang, R. Ascorbic acid integrates the antagonistic modulation of ethylene and abscisic acid in the accumulation of reactive oxygen species. Plant Physiol. 2019, 179, 1861–1875. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Verslues, P.E. Mechanisms independent of abscisic acid (ABA) or proline feedback have a predominant role in transcriptional regulation of proline metabolism during low water potential and stress recovery. Plant Cell Environ. 2010, 33, 1838–1851. [Google Scholar] [CrossRef]
- Rapala-Kozik, M.; Wolak, N.; Kujda, M.; Banas, A.K. The upregulation of thiamine (vitamin B1) biosynthesis in Arabidopsis thaliana seedlings under salt and osmotic stress conditions is mediated by abscisic acid at the early stages of this stress response. BMC Plant Biol. 2012, 12, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gong, H.; Li, D.; Zhou, R.; Zhao, F.; Zhang, X.; You, J. Integrated small RNA and Degradome sequencing provide insights into salt tolerance in sesame (Sesamum indicum L.). BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dai, L.; Ai, J.; Wang, Y.; Ren, F. Identification and functional prediction of cold-related long non-coding RNA (lncRNA) in grapevine. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Park, S.Y.; Choi, J.H.; Oh, D.H.; Johnson, J.C.; Dassanayake, M.; Jeong, D.H.; Oh, M.H. Genome-wide analysis of brassinosteroid responsive small RNAs in Arabidopsis thaliana. Genes Genom. 2020, 42, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, G.; Khandelwal, A.; Kapoor, M. High-throughput sequencing and differential expression analysis of miRNAs in response to Brassinosteroid treatment in Arabidopsis thaliana. Funct. Integr. Genom. 2019, 19, 597–615. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, K.; Han, Z.; Wang, P.; Fang, Y. Genome-wide identification of Arabidopsis long noncoding RNAs in response to the blue light. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balyan, S.; Kumar, M.; Mutum, R.D.; Raghuvanshi, U.; Agarwal, P.; Mathur, S.; Raghuvanshi, S. Identification of miRNA-mediated drought responsive multi-tiered regulatory network in drought tolerant rice, Nagina. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef]
- Jeyaraj, A.; Wang, X.; Wang, S.; Liu, S.; Zhang, R.; Wu, A.; Wei, C. Identification of Regulatory Networks of MicroRNAs and Their Targets in Response to Colletotrichum gloeosporioides in Tea Plant (Camellia sinensis L.). Front. Plant Sci. 2019, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, D.; Spriggs, A.; Yang, J.; Pogson, B.J.; Dennis, E.S.; Wilson, I.W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot. 2010, 61, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Jatan, R.; Chauhan, P.S.; Lata, C. High-throughput sequencing and expression analysis suggest the involvement of Pseudomonas putida RA-responsive micrornas in growth and development of Arabidopsis. Int. J. Mol. Sci. 2020, 21, 5468. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zuo, Z.; Qiu, J.L. Identification and Characterization of ABA-Responsive MicroRNAs in Rice. J. Genet. Genom. 2015, 42, 393–402. [Google Scholar] [CrossRef]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of Plant MicroRNA Targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Carianopol, C.S.; Chan, A.L.; Dong, S.; Provart, N.J.; Lumba, S.; Gazzarrini, S. An abscisic acid-responsive protein interaction network for sucrose non-fermenting related kinase1 in abiotic stress response. Commun. Biol. 2020, 3, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kreynes, A.E.; Yong, Z.; Liu, X.M.; Wong, D.C.J.; Castellarin, S.D.; Ellis, B.E. Biological impacts of phosphomimic AtMYB75. Planta 2020, 251, 1–20. [Google Scholar] [CrossRef]
- Liu, Y.; Li, M.; Li, T.; Chen, Y.; Zhang, L.; Zhao, G.; Zhuang, J.; Zhao, W.; Gao, L.; Xia, T. Airborne fungus-induced biosynthesis of anthocyanins in Arabidopsis thaliana via jasmonic acid and salicylic acid signaling. Plant Sci. 2020, 300, 110635. [Google Scholar] [CrossRef]
- Bach-Pages, M.; Homma, F.; Kourelis, J.; Kaschani, F.; Mohammed, S.; Kaiser, M.; van der Hoorn, R.A.L.; Castello, A.; Preston, G.M. Discovering the RNA-binding proteome of plant leaves with an improved RNA interactome capture method. Biomolecules 2020, 10, 661. [Google Scholar] [CrossRef] [PubMed]
- Joshna, C.R.; Saha, P.; Atugala, D.; Chua, G.; Muench, D.G. Plant PUF RNA-binding proteins: A wealth of diversity for post-transcriptional gene regulation. Plant Sci. 2020, 297, 110505. [Google Scholar] [CrossRef]
- Palatnik, J.F.; Allen, E.; Wu, X.; Schommer, C.; Schwab, R.; Carrington, J.C.; Weigel, D. Control of leaf morphogenesis by microRNAs. Nature 2003, 425, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Wang, Q.; Sun, R.; Zhang, B. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J. Exp. Bot. 2015, 66, 789–804. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Wang, Y.; Wang, L.; Yang, L.; Wang, R.; Li, X. miR172b Controls the Transition to Autotrophic Development Inhibited by ABA in Arabidopsis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, D.; Chun, H.J.; Kang, S.; Shin, G.; Park, S.J.; Hong, H.; Kim, C.; Kim, D.H.; Lee, S.Y.; Kim, M.C.; et al. A role for Arabidopsis miR399f in salt, drought, and ABA signaling. Mol. Cells 2016, 39, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, B.D.; Musialak-Lange, M.; Nuc, P.; May, P.; Buhtz, A.; Kehr, J.; Walther, D.; Scheible, W.R. Identification of nutrient-responsive Arabidopsis and rapeseed microRNAs by comprehensive real-time polymerase chain reaction profiling and small RNA sequencing. Plant Physiol. 2009, 150, 1541–1555. [Google Scholar] [CrossRef] [Green Version]
- Hou, G.; Du, C.; Gao, H.; Liu, S.; Sun, W.; Lu, H.; Kang, J.; Xie, Y.; Ma, D.; Wang, C. Identification of microRNAs in developing wheat grain that are potentially involved in regulating grain characteristics and the response to nitrogen levels. BMC Plant Biol. 2020, 20, 1–21. [Google Scholar] [CrossRef]
- Li, W.X.; Oono, Y.; Zhu, J.; He, X.J.; Wu, J.M.; Iida, K.; Lu, X.Y.; Cui, X.; Jin, H.; Zhu, J.K. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 2008, 20, 2238–2251. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Ding, H.; Zhu, J.-K.; Zhang, F.; Li, W.-X. Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol. 2011, 190, 906–915. [Google Scholar] [CrossRef] [Green Version]
- Lundmark, M.; Kørner, C.J.; Nielsen, T.H. Global analysis of microRNA in Arabidopsis in response to phosphate starvation as studied by locked nucleic acid-based microarrays. Physiol. Plant. 2010, 140, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Hu, X.; Cai, W.; Huang, W.; Zhou, X.; Luo, Q.; Yang, H.; Wang, J.; Huang, J. Arabidopsis miR171-Targeted Scarecrow-Like Proteins Bind to GT cis-Elements and Mediate Gibberellin-Regulated Chlorophyll Biosynthesis under Light Conditions. PLoS Genet. 2014, 10, 20–21. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Schmidhalter, U. Drought and salinity: A comparison of their effects on mineral nutrition of plants. J. Plant Nutr. Soil Sci. 2005, 168, 541–549. [Google Scholar] [CrossRef]
- Hsieh, L.C.; Lin, S.I.; Shih, A.C.C.; Chen, J.W.; Lin, W.Y.; Tseng, C.Y.; Li, W.H.; Chiou, T.J. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 2009, 151, 2120–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, P.S.; Borza, T.; Critchley, A.T.; Hiltz, D.; Norrie, J.; Prithiviraj, B. Ascophyllum nodosum extract mitigates salinity stress in Arabidopsis thaliana by modulating the expression of miRNA involved in stress tolerance and nutrient acquisition. PLoS ONE 2018, 13, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Barciszewska-Pacak, M.; Milanowska, K.; Knop, K.; Bielewicz, D.; Nuc, P.; Plewka, P.; Pacak, A.M.; Vazquez, F.; Karlowski, W.; Jarmolowski, A.; et al. Arabidopsis microRNA expression regulation in a wide range of abiotic stress responses. Front. Plant Sci. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Pilon, M. MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J. Biol. Chem. 2008, 283, 15932–15945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Shao, C.; Ma, X.; Wang, H.; Chen, M. Expression-Based Functional Investigation of the Organ-Specific MicroRNAs in Arabidopsis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Zhao, D.; Xia, X.; Wei, M.; Sun, J.; Meng, J.; Tao, J. Overexpression of herbaceous peony miR156e-3p improves anthocyanin accumulation in transgenic Arabidopsis thaliana lateral branches. 3 Biotech 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yant, L.; Mathieu, J.; Dinh, T.T.; Ott, F.; Lanz, C.; Wollmann, H.; Chen, X.; Schmid, M. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 2010, 22, 2156–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.L.; Wu, C.C.; Huang, H.C.; Chen, H.J.; Hsieh, H.L.; Juan, H.F. Identification of microRNA 395a in 24-epibrassinolide-regulated root growth of Arabidopsis thaliana using microRNA arrays. Int. J. Mol. Sci. 2013, 14, 14270–14286. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.F.; Tian, Q.; Reed, J.W. Arabidopis microRNA 167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 2006, 133, 4211–4218. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.H.; Tian, X.; Li, Y.J.; Wu, C.A.; Zheng, C.C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Ludwików, A.; Kierzek, D.; Gallois, P.; Zeef, L.; Sadowski, J. Gene expression profiling of ozone-treated Arabidopsis abi1td insertional mutant: Protein phosphatase 2C ABI1 modulates biosynthesis ratio of ABA and ethylene. Planta 2009, 230, 1003–1017. [Google Scholar] [CrossRef]
- Shi, J.; Dong, M.; Li, L.; Liu, L.; Luz-Madrigal, A.; Tsonis, P.A.; Del Rio-Tsonis, K.; Liang, C. MirPRo-a novel standalone program for differential expression and variation analysis of miRNAs. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.R.; Yeoh, J.M.; Coruh, C.; Axtell, M.J. Improved Placement of Multi-mapping Small RNAs. G3 Genes Genomes Genet. 2016, 6, 2103–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, 155–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.; Chow, C.; Wu, N.; Hou, P.; Chang, W. EXPath: A database of comparative expression analysis inferring metabolic pathways for plants. BMC Genom. 2015, 16, S6. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype and Condition | WT Col-0 Mock | WT Col-0 ABA | abi1td Mock | abi1td ABA | mkkk17 Mock | mkkk17 ABA | mkkk18 Mock | mkkk18 ABA | |
|---|---|---|---|---|---|---|---|---|---|
| Unannotated | Processed Reads | 9,484,333 (±305,722) | 7,782,658 (±3,032,385) | 10,327,250 (±1,263,563) | 7,488,293 (±305,722) | 12,597,403 (±306,139) | 7,375,998 (±801,769) | 10,332,359 (±593,100) | 8,095,522 (±2,410,235) |
| Aligned Reads | 9,377,237 (±291,693) (98.87%) | 7,592,660 (±2,990,770) (97.44%) | 10,160,442 (±1,251,305) (98.38%) | 7,356,650 (±933,688) (98.27%) | 12,361,791 (±306,825) (98.13%) | 7,256,364 (±783,546) (98.19%) | 9,977,310 (±531,749) (96.58%) | 7,837,873 (±2,319,507) (96.5%) | |
| alignment_not_unique | 6,052,568 (±97,495) | 4,860,241 (±2,226,793) | 6,670,315 (±1,163,741) | 4,657,135 (±640,197) | 8,888,020 (±692,945) | 4,130,142 (±523,786) | 6,089,703 (±284,811) | 4,864,610 (±1,607,750) | |
| ambiguous | 84,483 (±28,631) | 157,990 (±91,477) | 205,679 (±51,593) | 168,844 (±107,752) | 242,764 (±65,097) | 140,124 (±30,453) | 252,149 (±25,429) | 124,278 (±90,401) | |
| no_feature | 1,095,681 (±320,538) | 1,176,768 (±373,427) | 1,386,570 (±112,773) | 1,013,711 (±335,090) | 1,439,914 (±210,521) | 1,326,686 (±171,840) | 1,781,633 (±301,685) | 1,153,640 (±495,774) | |
| not_aligned | 1,234,288 (±309,658) | 589,360 (±116,084) | 808,774 (±151,073) | 651,428 (±363,168) | 634,204 (±356,362) | 731,196 (±258,725) | 759,329 (±144,704) | 1,010,286 (±153,079) | |
| Annotated | lncRNA | 15,888 (±4458) | 17,414 (±4273) | 19,856 (±2187) | 15,312 (±4441) | 19,546 (±3844) | 18,929 (±3166) | 24,911 (±4465) | 16,666 (±6589) |
| miRNA | 360,701 (±94,021) | 143,893 (±30,443) | 327,809 (±70,037) | 189,858 (±67,813) | 246,958 (±126,301) | 311,619 (±64,928) | 391,059 (±79,742) | 248,964 (±57,159) | |
| ncRNA | 63,272 (±20,395) | 16,918 (±7013) | 53,668 (±7655) | 31,245 (±17,723) | 45,512 (±24,622) | 54,202 (±6738) | 71,275 (±14,488) | 38,867 (±12,868) | |
| nontranslating_CDS | 403 (±92) | 431 (±163) | 497 (±62) | 395 (±122) | 617 (±59) | 400 (±26) | 604 (±96) | 401 (±196) | |
| protein_coding | 282,368 (±32,142) | 316,490 (±142,682) | 341,844 (±30,871) | 242,310 (±44,467) | 484,528 (±36,738) | 257,798 (±31,391) | 354,631 (±58,945) | 271,948 (±109,900) | |
| rRNA | 103,274 (±8762) | 131,380 (±33,382) | 118,960 (±11,677) | 106,636 (±22,877) | 156,854 (±22,778) | 102,371 (±21,861) | 110,539 (±2074) | 103,332 (±24,713) | |
| snRNA | 2309 (±540) | 2771 (±1457) | 2922 (±272) | 2168 (±517) | 3696 (±762) | 2619 (±180) | 7647 (±6067) | 2469 (±917) | |
| snoRNA | 34,739 (±11,459) | 37,995 (±19,310) | 59,513 (±12,832) | 54,093 (±24,850) | 52,302 (±12,099) | 27,799 (±4818) | 56,471 (±17,001) | 25,981 (±20,829) | |
| tRNA | 154,356 (±24,306) | 331,003 (±226,926) | 330,838 (±67,629) | 355,153 (±211,607) | 382,484 (±123,782) | 272,109 (±91,642) | 432,406 (±108,854) | 234,075 (±163,704) |
| Number of Reads † | Fold Change (FC) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| miRNA ID | WT Col-0 Mock | WT Col-0 ABA | abi1td Mock | abi1td ABA | mkkk17 Mock | mkkk17 ABA | mkkk18 Mock | mkkk18 ABA | WT Col-0 | abi1td | mkkk17 | mkkk18 |
| miR166e-3p | 20,943 (±5369) | 24,743 (±7424) | 16,015 (±4897) | 16,298 (±511) | 11,265 (±3877) | 23,781 (±4607) | 20,115 (±4559) | 25,951 (±13,351) | 0.56 * | 0.26 | −0.12 | 0.59 |
| miR165b | 5687 (±1575) | 6133 (±1725) | 4164 (±1477) | 3676 (±159) | 2601 (±942) | 6084 (±1578) | 5118 (±1218) | 6266 (±4403) | 0.43 | 0.07 | 0.02 | 0.51 |
| miR159a | 6732 (±1309) | 4025 (±978) | 4846 (±1625) | 4444 (±1783) | 2473 (±1437) | 7751 (±2094) | 5157 (±1625) | 5706 (±1375) | −0.41 | 0.01 | 0.56 | 0.39 |
| miR158a-3p | 7803 (±2455) | 1474 (±384) | 6887 (±2683) | 4128 (±2559) | 4000 (±2460) | 7378 (±2977) | 6757 (±1703) | 4582 (±712) | −1.99 ** | −0.66 | −0.18 | −0.33 |
| miR159b-3p | 2886 (±584) | 1468 (±320) | 2072 (±856) | 1357 (±463) | 986 (±630) | 2807 (±713) | 2193 (±750) | 1930 (±344) | −0.64 ** | −0.45 | 0.46 | 0.06 |
| miR398b-3p | 1551 (±125) | 1327 (±367) | 786 (±368) | 818 (±109) | 473 (±208) | 1202 (±455) | 906 (±205) | 1459 (±608) | 0.09 | 0.29 | 0.16 | 0.91 |
| miR167a-3p | 1458 (±500) | 1025 (±406) | 1736 (±523) | 1058 (±123) | 1475 (±398) | 1331 (±499) | 3001 (±318) | 1097 (±21) | −0.15 | −0.48 | −1.32 ** | −1.24 ** |
| miR160c-5p | 448 (±125) | 558.23 (±97) | 351 (±138) | 334 (±44) | 215 (±91) | 592 (±58) | 463 (±72) | 467.57 (±181) | 0.69 ** | 0.16 | 0.29 | 0.22 |
| miR827 | 1381 (±228) | 597 (±139) | 791 (±204) | 897 (±250) | 643 (±418) | 1917 (±436) | 1617 (±560) | 1044 (±406) | −0.88 ** | 0.32 | 0.54 | −0.38 |
| miR403-3p | 1120 (±317) | 293 (±89) | 1133 (±206) | 926 (±588) | 760 (±337) | 1721 (±381) | 1822 (±256) | 1162 (±394) | −1.48 ** | −0.27 | 0.03 | −0.44 |
| miR396a-5p | 700 (±149) | 317 (±34) | 480 (±134) | 462 (±260) | 345 (±223) | 861 (±115) | 762 (±96) | 570 (±95) | −0.75 ** | 0 | 0.28 | −0.2 |
| miR161.1 | 455 (±117) | 396 (±99) | 332 (±79) | 356 (±52) | 263 (±112) | 532 (±170) | 494 (±110) | 492 (±157) | 0.15 | 0.29 | −0.15 | 0.22 |
| miR167a-5p | 401 (±103) | 331 (±67) | 382 (±105) | 356 (±99) | 246 (±128) | 535 (±51) | 487 (±81) | 431 (±93) | 0.09 | 0.04 | 0 | 0.05 |
| miR156f-5p | 363 (±60) | 248 (±23) | 291 (±128) | 249 (±154) | 152 (±73) | 417 (±132) | 367 (±65) | 365 (±39) | −0.19 | −0.13 | 0.23 | 0.18 |
| miR408-3p | 261 (±15) | 427 (±202) | 207 (±202) | 288 (±66) | 142 (±70) | 467 (±82) | 214 (±39) | 404 (±39) | 0.98 ** | 0.75 * | 0.56 * | 1.13 ** |
| miRNA ID | Target Gene ID | Expectation | Inhibition | Target Description |
|---|---|---|---|---|
| ath-miRn-1 | AT1G73390.3 | 2.5 | Cleavage | Endosomal targeting BRO1-like domain-containing protein |
| ath-miRn-1 | AT1G73390.4 | 2.5 | Cleavage | Endosomal targeting BRO1-like domain-containing protein |
| ath-miRn-2 | AT5G40550.1 | 3 | Cleavage | SGF29 tudor-like domain |
| ath-miRn-4 | AT3G14070.1 | 3.5 | Cleavage | CAX9, CCX3, ATCCX3, cation exchanger 9 |
| ath-miRn-6 | AT1G56650.1 | 3 | Cleavage | PAP1, MYB75, SIAA1, ATMYB75, production of anthocyanin pigment 1 |
| ath-miRn-6 | AT5G58490.1 | 3 | Cleavage | NAD(P)-binding Rossmann-fold superfamily protein |
| ath-miRn-8 | AT3G15570.1 | 3 | Cleavage | Phototropic-responsive NPH3 family protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehdi, S.M.M.; Krishnamoorthy, S.; Szczesniak, M.W.; Ludwików, A. Identification of Novel miRNAs and Their Target Genes in the Response to Abscisic Acid in Arabidopsis. Int. J. Mol. Sci. 2021, 22, 7153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137153
Mehdi SMM, Krishnamoorthy S, Szczesniak MW, Ludwików A. Identification of Novel miRNAs and Their Target Genes in the Response to Abscisic Acid in Arabidopsis. International Journal of Molecular Sciences. 2021; 22(13):7153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137153
Chicago/Turabian StyleMehdi, Syed Muhammad Muntazir, Sivakumar Krishnamoorthy, Michal Wojciech Szczesniak, and Agnieszka Ludwików. 2021. "Identification of Novel miRNAs and Their Target Genes in the Response to Abscisic Acid in Arabidopsis" International Journal of Molecular Sciences 22, no. 13: 7153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137153