
Bisphenol A Modulates Autophagy and Exacerbates Chronic Kidney Damage in Mice

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
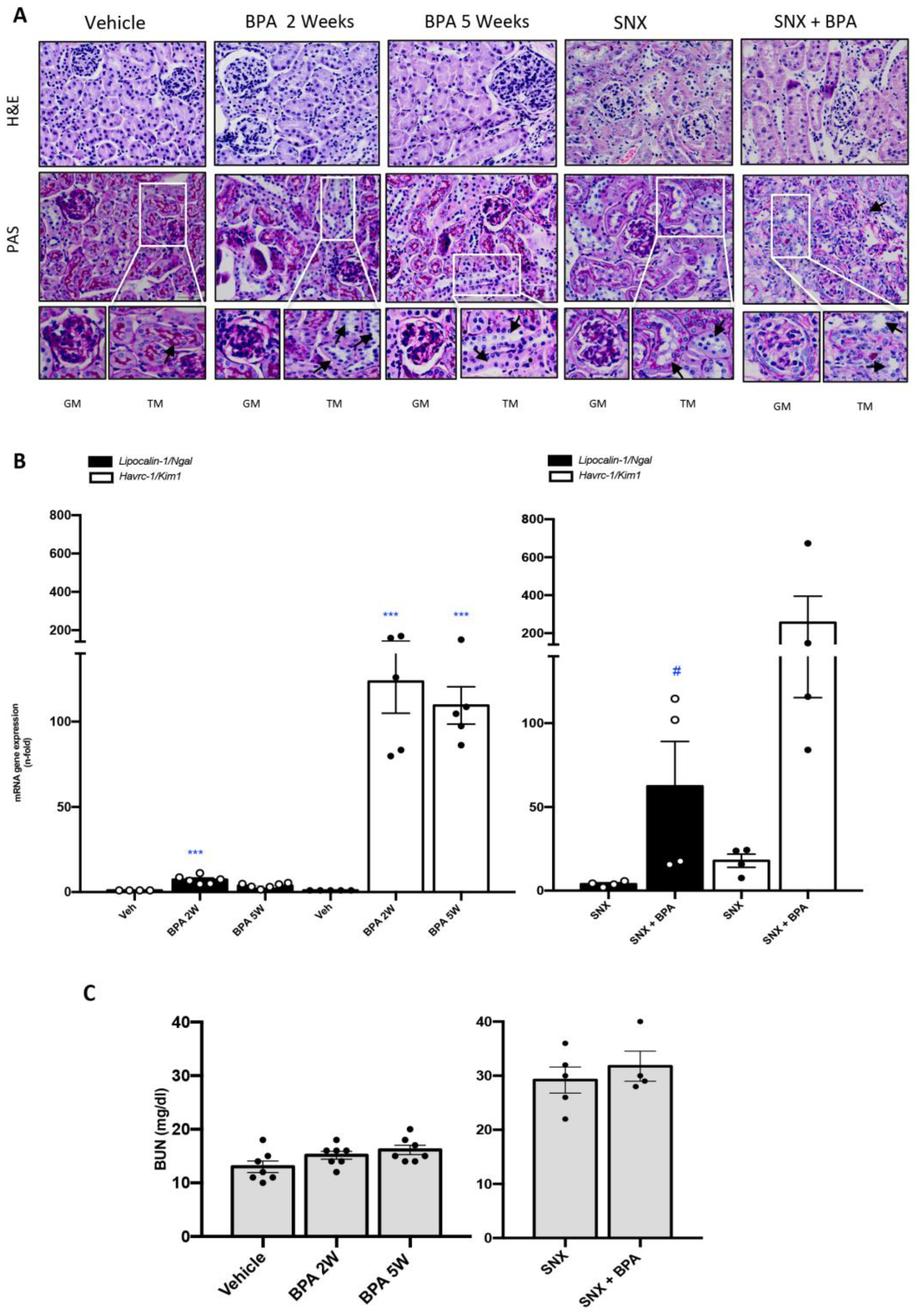
2.1. Chronic Systemic Administration of BPA Induces Tubular Damage in the Kidney of Healthy Mice and Exacerbates Kidney Damage in Experimental CKD
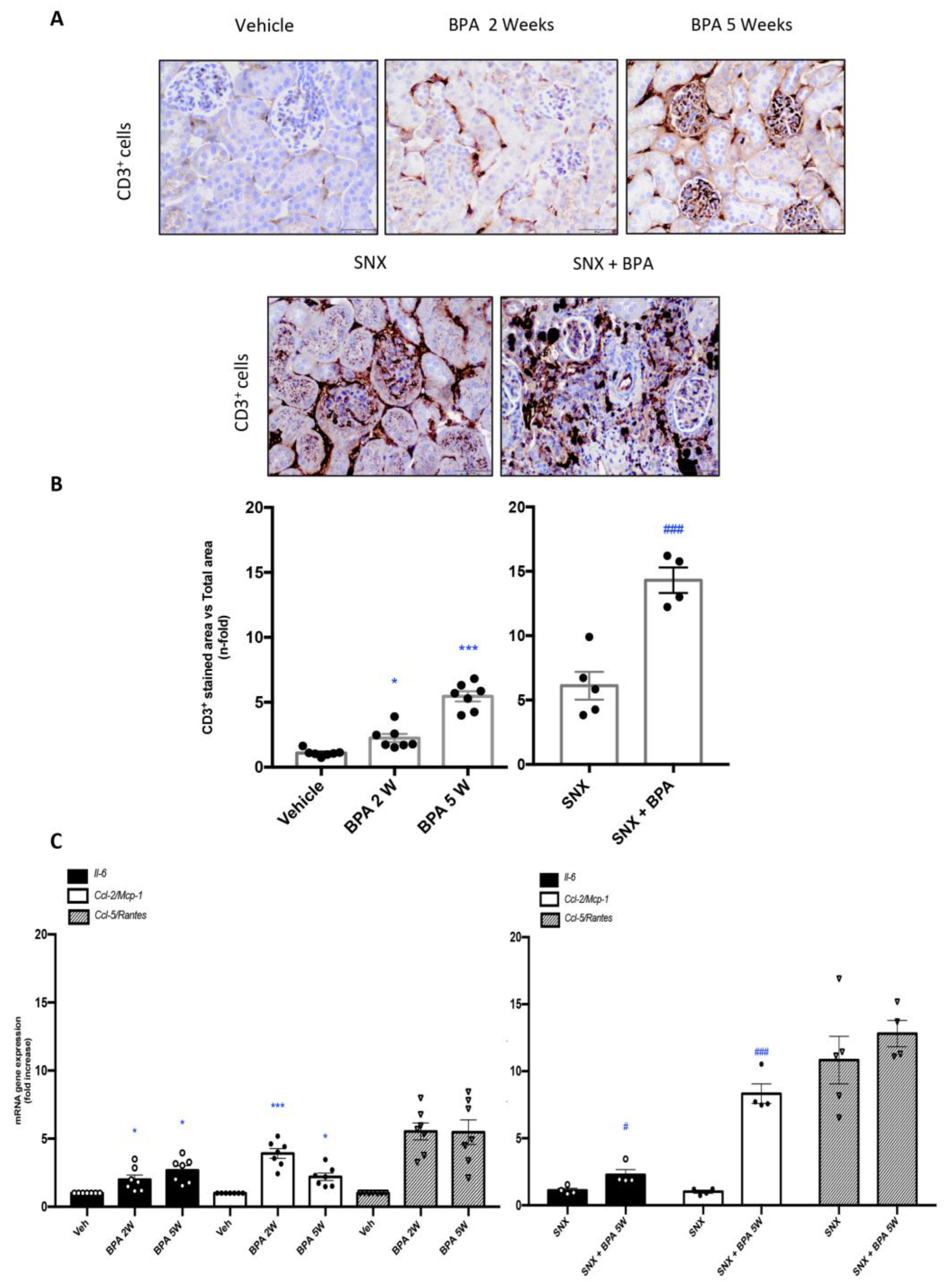
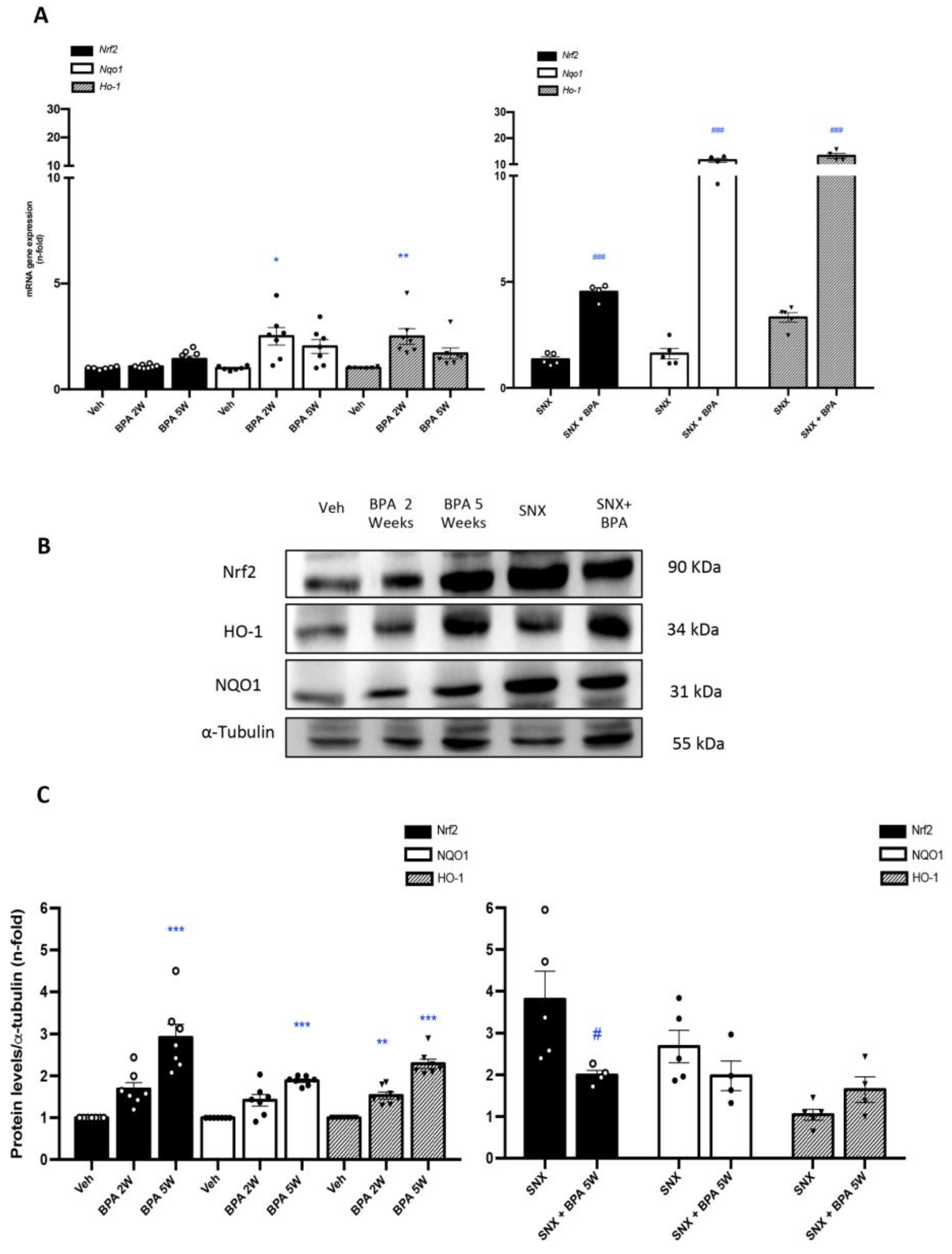
2.2. Chronic Administration of BPA Causes Inflammatory Cell Infiltration in Healthy and Injured Kidneys by Upregulation of Proinflammatory Factors and Redox-Mediated Processes
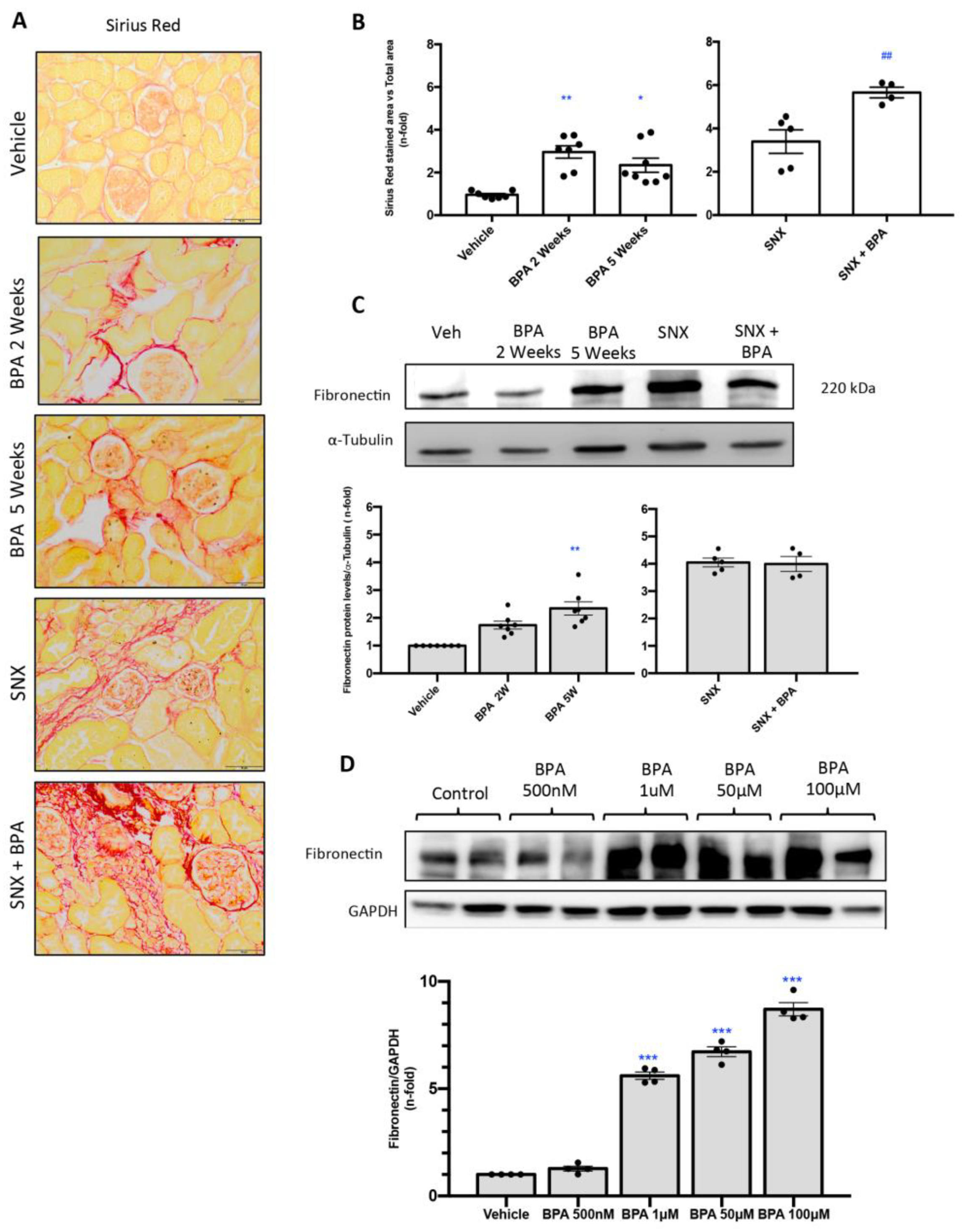
2.3. Chronic Administration of BPA to Mice Induces Renal Fibrosis in Healthy Mice
2.4. Stimulation with BPA Induces the Expression of Profibrotic Marker Fibronectin in Human Renal Proximal Tubuloepithelial Cells (HK-2)
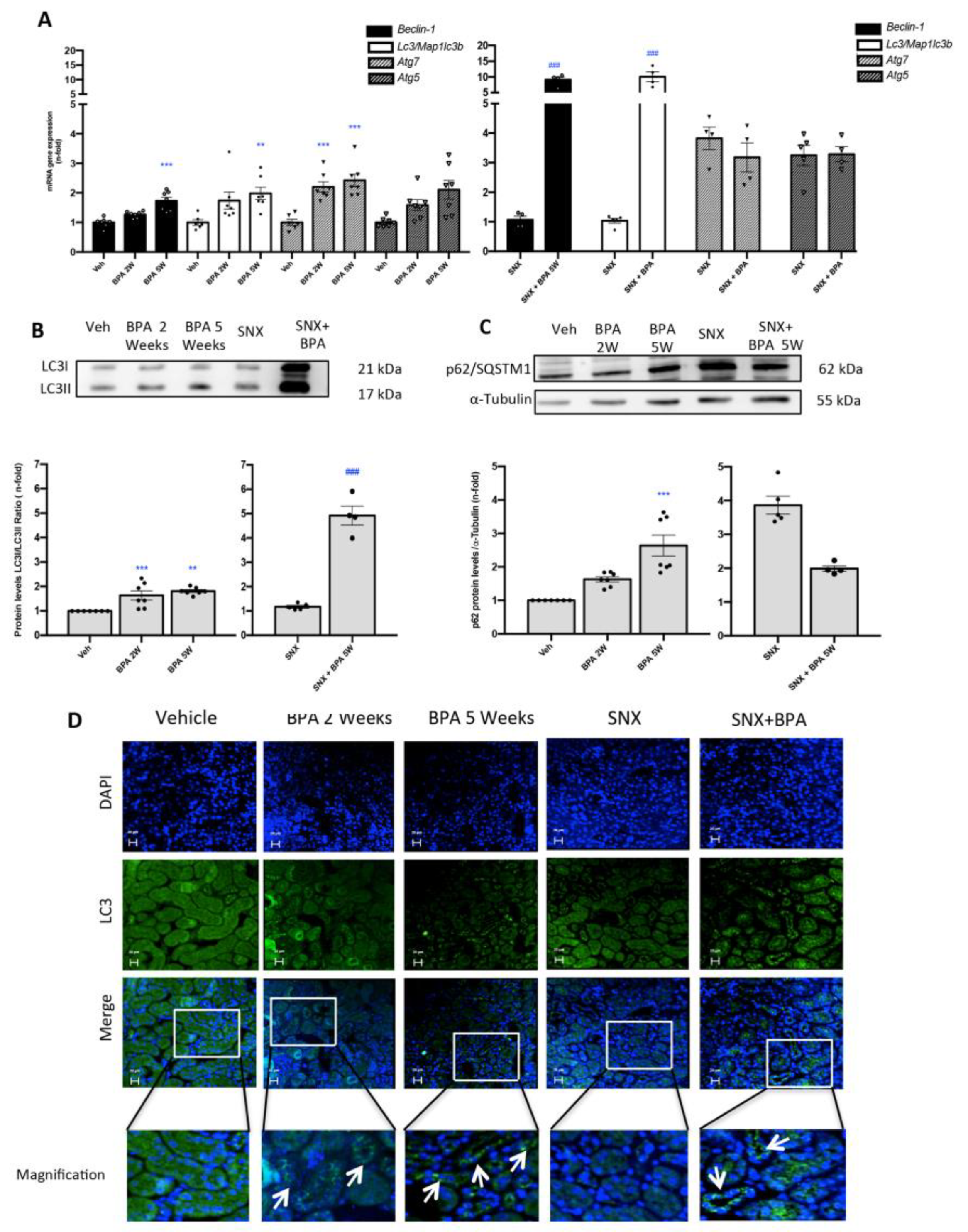
2.5. Chronic Administration of BPA to Mice Induces the Blockade of Autophagosome Maturation in the Kidney
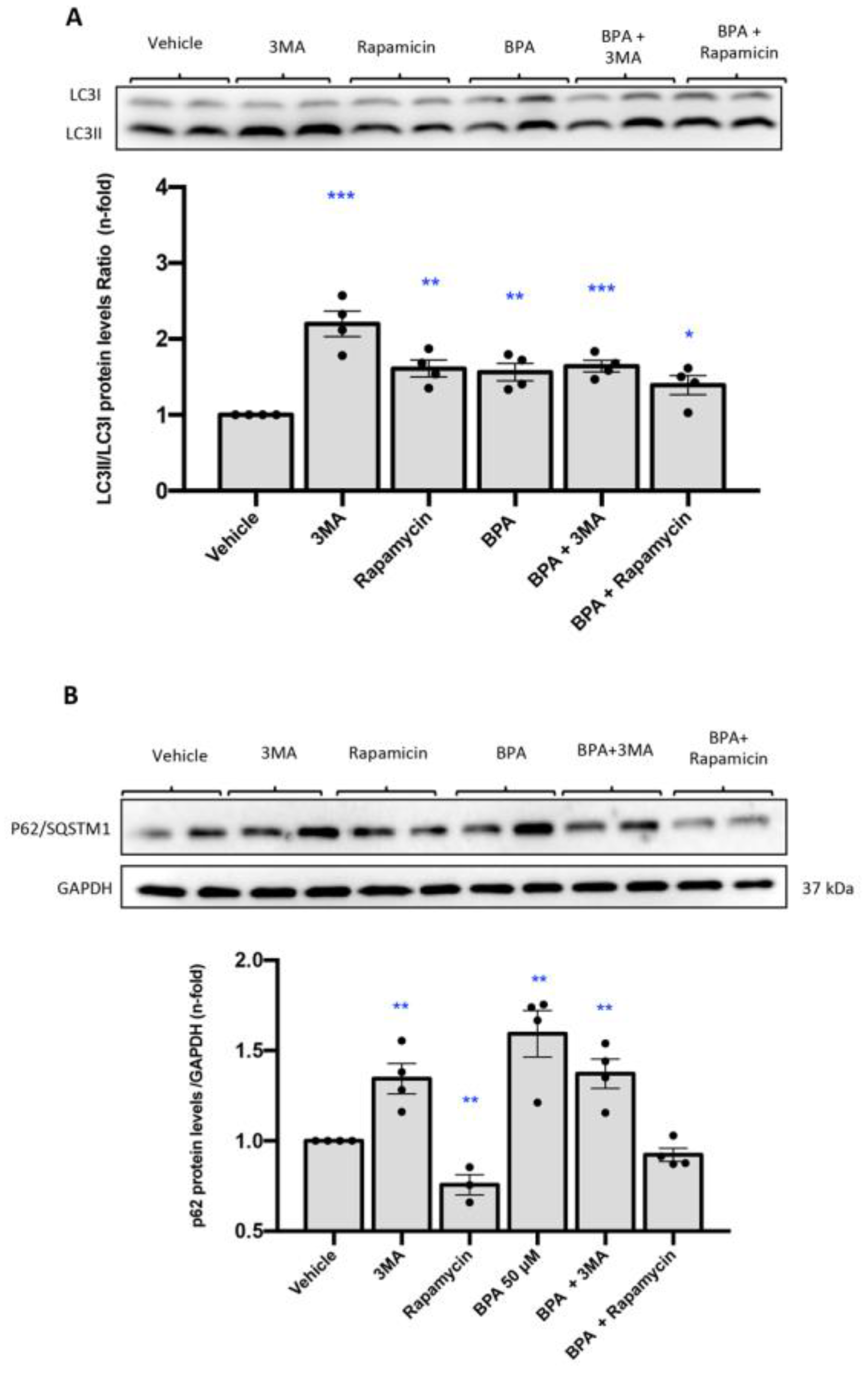
2.6. Stimulation with BPA Induces the Blockade of Maturation of Autophagosome in Human Renal Proximal Tubuloepithelial Cells (HK-2)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Cultured Studies
4.3. In Vivo Studies
- (A).
- Harmful effect of BPA in healthy mice: (i) vehicle; (ii) BPA at 2 weeks; (iii) BPA at 5 weeks;
- (B).
- Harmful effect of BPA in established CKD: (iv) SNX vehicle; (v) SNX with BPA at 5 weeks.
4.4. Gene Expression Studies
4.5. Protein Studies
4.6. Renal Histology and Immunohistochemistry
4.7. Immunofluorescence Staining of LC3B
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- González-Parra, E.; Herrero, J.A.; Elewa, U.; Bosch, R.J.; Arduán, A.O.; Egido, J. Bisphenol A in chronic kidney disease. Int. J. Nephrol. 2013, 2013, 437857. [Google Scholar] [CrossRef]
- Rubin, B.S. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol. 2011, 127, 27–34. [Google Scholar] [CrossRef]
- Kang, J.H.; Kondo, F.; Katayama, Y. Human exposure to bisphenol A. Toxicology 2006, 226, 79–89. [Google Scholar] [CrossRef]
- Thomson, B.M.; Grounds, P.R. Bisphenol A in canned foods in New Zealand: An exposure assessment. Food Addit. Contam. 2005, 22, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.K.; Chuang, J.C.; Morgan, M.K.; Lordo, R.A.; Sheldon, L.S. An observational study of the potential exposures of preschool children to pentachlorophenol, bisphenol-A, and nonylphenol at home and daycare. Environ. Res. 2007, 103, 9–20. [Google Scholar] [CrossRef]
- Testai, E.; Hartemann, P.; Rodríguez-Farre, E.; Rastogi, S.C.; Bustos, J.; Gundert-Remy, U.; Hensten, A.; Kopperud, H.M.; Olea, N.; Piersma, A.; et al. The safety of the use of bisphenol A in medical devices. Regul. Toxicol. Pharmacol. 2016, 79, 106–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekant, W.; Völkel, W. Human exposure to bisphenol A by biomonitoring: Methods, results and assessment of environmental exposures. Toxicol. Appl. Pharmacol. 2008, 228, 114–134. [Google Scholar] [CrossRef]
- Dallio, M.; Diano, N.; Masarone, M.; Gravina, A.G.; Patanè, V.; Romeo, M.; Di Sarno, R.; Errico, S.; Nicolucci, C.; Abenavoli, L.; et al. Chemical effect of bisphenol A on non-alcoholic fatty liver disease. Int. J. Environ. Res. Public Health 2019, 16, 3134. [Google Scholar] [CrossRef] [Green Version]
- Wade, M.; Delawder, V.; Reneau, P.; dos Santos, J.M. The effect of BPA exposure on insulin resistance and type 2 diabetes—The impact of muscle contraction. Med. Hypotheses 2020, 140. [Google Scholar] [CrossRef] [PubMed]
- Vom Saal, F.S.; Nagel, S.C.; Coe, B.L.; Angle, B.M.; Taylor, J.A. The estrogenic endocrine disrupting chemical bisphenol A (BPA) and obesity. Mol. Cell. Endocrinol. 2012, 354, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.J.; Lee, S.Y.; Kim, K.Y.; Hong, Y.P. Acute testis toxicity of bisphenol A diglycidyl ether in Sprague-Dawley rats. J. Prev. Med. Public Health 2010, 43, 131–137. [Google Scholar] [CrossRef]
- Li, M.; Bi, Y.; Qi, L.; Wang, T.; Xu, M.; Huang, Y.; Xu, Y.; Chen, Y.; Lu, J.; Wang, W.; et al. Exposure to bisphenol A is associated with low-grade albuminuria in Chinese adults. Kidney Int. 2012, 81, 1131–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trasande, L.; Attina, T.M.; Trachtman, H. Bisphenol A exposure is associated with low-grade urinary albumin excretion in children of the United States. Kidney Int. 2013, 83, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Mas, S.; Bosch-Panadero, E.; Abaigar, P.; Camarero, V.; Mahillo, I.; Civantos, E.; Sanchez-Ospina, D.; Ruiz-Priego, A.; Egido, J.; Ortiz, A.; et al. Influence of dialysis membrane composition on plasma bisphenol A levels during online hemodiafiltration. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Bosch-Panadero, E.; Mas, S.; Sanchez-Ospina, D.; Camarero, V.; Pérez-Gómez, M.V.; Saez-Calero, I.; Abaigar, P.; Ortiz, A.; Egido, J.; González-Parra, E. The choice of hemodialysis membrane affects bisphenol A levels in blood. J. Am. Soc. Nephrol. 2016, 27, 1566–1574. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Panadero, E.; Mas, S.; Civantos, E.; Abaigar, P.; Camarero, V.; Ruiz-Priego, A.; Ortiz, A.; Egido, J.; González-Parra, E. Bisphenol A is an exogenous toxin that promotes mitochondrial injury and death in tubular cells. Environ. Toxicol. 2018, 33, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Yoon, J.H. Exposure to environmental pollutants and a marker of early kidney injury in the general population: Results of a nationally representative cross-sectional study based on the Korean National Environmental Health Survey (KoNEHS) 2012–2014. Sci. Total Environ. 2019, 681, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; De Smet, R. Pathophysiologic effects of uremic retention solutes. J. Am. Soc. Nephrol. 1999, 10, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.P.; Souza, A.; Farrell, L.; Pollak, M.R.; Lewis, G.D.; Steele, D.J.R.; Thadhani, R.; Clish, C.B.; Greka, A.; Gerszten, R.E. Metabolite profiling identifies markers of uremia. J. Am. Soc. Nephrol. 2010, 21, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijers, B.K.I.; Evenepoel, P. The gut-kidney axis: Indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol. Dial. Transplant. 2011, 26, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Gassman, N.R. Induction of oxidative stress by bisphenol A and its pleiotropic effects. Environ. Mol. Mutagen. 2017, 58, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, N.; Paul-Bellon, R.; Fénichel, P. A commentary on “Involvement of activating ERK1/2 trough G protein coupled receptor 30 and estrogen receptor α/β in low doses of bisphenol A promoting growth of Sertoli TM4 cells”. Toxicol. Lett. 2015, 237, 165–166. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Shibata, M.; Lu, T.; Furuya, T.; Degterev, A.; Mizushima, N.; Yoshimori, T.; MacDonald, M.; Yankner, B.; Yuan, J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006, 281, 14474–14485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.E. Autophagy in kidney disease. Annu. Rev. Physiol. 2020, 82, 297–322. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.W.; Yuan, Y.; Chen, J.H.; Lin, W.Q. Kidney disease models: Tools to identify mechanisms and potential therapeutic targets. Zool. Res. 2018, 39, 72–86. [Google Scholar] [PubMed]
- Rossmann, P.; Říha, I.; Matoušovic, K.; Bohdanecká, M.; Bukovský, A. Experimental ablation nephropathy: Fine structure, morphometry, cell membrane epitopes, glomerular polyanion and effect of subsequent transplantation. Pathol. Res. Pract. 1990, 186, 491–506. [Google Scholar] [CrossRef]
- Amin, R.P.; Vickers, A.E.; Sistare, F.; Thompson, K.L.; Roman, R.J.; Lawton, M.; Kramer, J.; Hamadeh, H.K.; Collins, J.; Grissom, S.; et al. Identification of putative gene-based markers of renal toxicity. Environ. Health Perspect. 2004, 112, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Vaidya, V.S.; Liu, J.; Waalkes, M.P.; Edwards, J.R.; Lamar, P.C.; Bernard, A.M.; Dumont, X.; Bonventre, J.V. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007, 72, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, U.O.; Abboud, H.E. Chemokines and renal disease. Am. J. Kidney Dis. 1995, 26, 982–994. [Google Scholar] [CrossRef]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The P62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar]
- Hengstler, J.G.; Foth, H.; Gebel, T.; Kramer, P.J.; Lilienblum, W.; Schweinfurth, H.; Völkel, W.; Wollin, K.M.; Gundert-Remy, U. Critical evaluation of key evidence on the human health hazards of exposure to bisphenol A. Crit. Rev. Toxicol. 2011, 41, 263–291. [Google Scholar] [CrossRef] [Green Version]
- Rochester, J.R.; Bolden, A.L. Bisphenol S and F: A systematic review and comparison of the hormonal activity of bisphenol A substitutes. Environ. Health Perspect. 2015, 123, 643–650. [Google Scholar] [CrossRef]
- Rubin, B.S.; Schaeberle, C.M.; Soto, A.M. The case for BPA as an obesogen: Contributors to the controversy. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Meli, R.; Monnolo, A.; Annunziata, C.; Pirozzi, C.; Ferrante, M.C. Oxidative stress and BPA toxicity: An antioxidant approach for male and female reproductive dysfunction. Antioxidants 2020, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Kuklenyik, Z.; Reidy, J.A.; Caudill, S.P.; Ekong, J.; Needham, L.L. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ. Health Perspect. 2005, 113, 391–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, J.M.; Kalkbrenner, A.E.; Calafat, A.M.; Yolton, K.; Ye, X.; Dietrich, K.N.; Lanphear, B.P. Impact of early-life bisphenol A exposure on behavior and executive function in children. Pediatrics 2011, 128, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerge, D.R.; Twaddle, N.C.; Woodling, K.A.; Fisher, J.W. Pharmacokinetics of bisphenol A in neonatal and adult rhesus monkeys. Toxicol. Appl. Pharmacol. 2010, 248, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.H.; Wu, Y.; Liu, M.; Attina, T.M.; Naidu, M.; Karthikraj, R.; Kannan, K.; Warady, B.A.; Furth, S.; Vento, S.; et al. Serially assessed bisphenol A and phthalate exposure and association with kidney function in children with chronic kidney disease in the US and Canada: A longitudinal cohort study. PLoS Med. 2020, 17. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, T.; Shi, Y.; Zhuang, F.; Lu, J.; Zhu, Q.; Ding, F. Bisphenol A analogs in patients with chronic kidney disease and dialysis therapy. Ecotoxicol. Environ. Saf. 2019, 185. [Google Scholar] [CrossRef]
- Sangai, N.P.; Verma, R.J.; Trivedi, M.H. Testing the efficacy of quercetin in mitigating bisphenol A toxicity in liver and kidney of mice. Toxicol. Ind. Health 2014, 30, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Çiğ, B.; Yildizhan, K. Resveratrol diminishes bisphenol A-induced oxidative stress through TRPM2 channel in the mouse kidney cortical collecting duct cells. J. Recept. Signal Transduct. 2020, 40, 570–583. [Google Scholar] [CrossRef]
- Exacerbating Lupus Nephritis Following BPA Exposure Is Associated with Abnormal Autophagy in MRL/Lpr Mice. Available online: https://pubmed.ncbi.nlm.nih.gov/32194912/ (accessed on 30 May 2021).
- Balci, A.; Ozkemahli, G.; Erkekoglu, P.; Zeybek, N.D.; Yersal, N.; Kocer-Gumusel, B. Histopathologic, apoptotic and autophagic, effects of prenatal bisphenol A and/or di(2-ethylhexyl) phthalate exposure on prepubertal rat testis. Environ. Sci. Pollut. Res. 2020, 27, 20104–20116. [Google Scholar] [CrossRef]
- Alboghobeish, S.; Mahdavinia, M.; Zeidooni, L.; Samimi, A.; Oroojan, A.A.; Alizadeh, S.; Dehghani, M.A.; Ahangarpour, A.; Khorsandi, L. Efficiency of naringin against reproductive toxicity and testicular damages induced by bisphenol A in rats. Iran. J. Basic Med. Sci. 2019, 22, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, I.; Kum, Ş.; Tuğlu, M.İ. Histological investigations on thymus of male rats prenatally exposed to bisphenol A. Chemosphere 2018, 206, 1–8. [Google Scholar] [CrossRef]
- Eweda, S.; Newairy, A.; Abdou, H.; Gaber, A. Bisphenol A-induced oxidative damage in the hepatic and cardiac tissues of rats: The modulatory role of sesame lignans. Exp. Ther. Med. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, P.; Fernandez, T.; García-Arévalo, M.; Alonso-Magdalena, P.; Nadal, A.; Perillan, C.; Arguelles, J. Effects of bisphenol A treatment during pregnancy on kidney development in mice: A stereological and histopathological study. J. Dev. Orig. Health Dis. 2018, 9, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Alderson, H.V.; Ritchie, J.P.; Pagano, S.; Middleton, R.J.; Pruijm, M.; Vuilleumier, N.; Kalra, P.A. The associations of blood kidney injury molecule-1 and neutrophil gelatinase-associated lipocalin with progression from CKD to ESRD. Clin. J. Am. Soc. Nephrol. 2016, 11, 2141–2149. [Google Scholar] [CrossRef]
- Garlo, K.G.; White, W.B.; Bakris, G.L.; Zannad, F.; Wilson, C.A.; Kupfer, S.; Vaduganathan, M.; Morrow, D.A.; Cannon, C.P.; Charytan, D.M. Kidney biomarkers and decline in EGFR in patients with type 2 diabetes. Clin. J. Am. Soc. Nephrol. 2018, 13, 398–405. [Google Scholar] [CrossRef] [Green Version]
- Jha, J.C.; Banal, C.; Chow, B.S.M.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [Green Version]
- Ozbek, E. Induction of oxidative stress in kidney. Int. J. Nephrol. 2012, 2012, 465897. [Google Scholar] [CrossRef] [Green Version]
- Avci, B.; Bahadir, A.; Tuncel, O.K.; Bilgici, B. Influence of α-tocopherol and α-lipoic acid on bisphenol-A-induced oxidative damage in liver and ovarian tissue of rats. Toxicol. Ind. Health 2016, 32, 1381–1390. [Google Scholar] [CrossRef]
- Kobayashi, K.; Liu, Y.; Ichikawa, H.; Takemura, S.; Minamiyama, Y. Effects of bisphenol A on oxidative stress in the rat brain. Antioxidants 2020, 9, 240. [Google Scholar] [CrossRef] [Green Version]
- Bábíčková, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Leaf, I.A.; Duffield, J.S. What can target kidney fibrosis? Nephrol. Dial. Transplant. 2017, 32, I89–I97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-α: Central factors in the altered cytokine network of uremia—The good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.J.; Ferguson, K.K.; Anzalota Del Toro, L.V.; Alshawabkeh, A.N.; Cordero, J.F.; Meeker, J.D. Associations between urinary phenol and paraben concentrations and markers of oxidative stress and inflammation among pregnant women in Puerto Rico. Int. J. Hyg. Environ. Health 2015, 218, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, F.; Jiang, L.; Liu, X.; Geng, C.; Wang, W.; Zhong, L.; Yang, G.; Chen, M. Bisphenol A induces oxidative stress-associated DNA damage in INS-1 cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 769, 29–33. [Google Scholar] [CrossRef]
- Aboul Ezz, H.S.; Khadrawy, Y.A.; Mourad, I.M. The effect of bisphenol A on some oxidative stress parameters and acetylcholinesterase activity in the heart of male albino rats. Cytotechnology 2015, 67, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Saura, M.; Marquez, S.; Reventun, P.; Olea-Herrero, N.; Arenas, M.I.; Moreno-Gómez-Toledano, R.; Gómez-Parrizas, M.; Muñóz-Moreno, C.; González-Santander, M.; Zaragoza, C.; et al. Oral administration of bisphenol A induces high blood pressure through angiotensin II/CaMKII-dependent uncoupling of ENOS. FASEB J. 2014, 28, 4719–4728. [Google Scholar] [CrossRef]
- Akram, R.; Iqbal, R.; Hussain, R.; Jabeen, F.; Ali, M. Evaluation of oxidative stress, antioxidant enzymes and genotoxic potential of bisphenol A in fresh water bighead carp (Aristichthys nobils) fish at low concentrations. Environ. Pollut. 2021, 268. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Kong, Y.; Ommati, M.M.; Tang, Z.; Li, H.; Li, L.; Zhao, C.; Shi, Z.; Wang, J. Bisphenol A-induced apoptosis, oxidative stress and DNA damage in cultured rhesus monkey embryo renal epithelial Marc-145 cells. Chemosphere 2019, 234, 682–689. [Google Scholar] [CrossRef]
- Yazdani, M.; Andresen, A.M.S.; Gjøen, T. Short-term effect of bisphenol-A on oxidative stress responses in Atlantic salmon kidney cell line: A transcriptional study. Toxicol. Mech. Methods 2016, 26, 295–300. [Google Scholar] [CrossRef]
- Kobroob, A.; Peerapanyasut, W.; Chattipakorn, N.; Wongmekiat, O. Damaging effects of bisphenol A on the kidney and the protection by melatonin: Emerging evidences from in vivo and in vitro studies. Oxidative Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Babu, S.; Uppu, S.; Claville, M.O.; Uppu, R.M. Prooxidant actions of bisphenol A (BPA) phenoxyl radicals: Implications to BPA-related oxidative stress and toxicity. Toxicol. Mech. Methods 2013, 23, 273–280. [Google Scholar] [CrossRef]
- Yang, Y.J.; Hong, Y.C.; Oh, S.Y.; Park, M.S.; Kim, H.; Leem, J.H.; Ha, E.H. Bisphenol A exposure is associated with oxidative stress and inflammation in postmenopausal women. Environ. Res. 2009, 109, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lim, K.T. Expression of TNF-α and IL-6 in HMC-1 cells treated with bisphenol A is attenuated by plant-originating glycoprotein (75 Κda) by blocking p38 MAPK. Naunyn Schmiedeberg’s Arch. Pharmacol. 2010, 382, 51–61. [Google Scholar] [CrossRef]
- Alekhya Sita, G.J.; Gowthami, M.; Srikanth, G.; Krishna, M.M.; Rama Sireesha, K.; Sajjarao, M.; Nagarjuna, K.; Nagarjuna, M.; Chinnaboina, G.K.; Mishra, A.; et al. Protective role of luteolin against bisphenol A-induced renal toxicity through suppressing oxidative stress, inflammation, and upregulating Nrf2/ARE/HO-1 pathway. IUBMB Life 2019, 71, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Isaka, Y.; Yoshimori, T. Autophagy and kidney inflammation. Autophagy 2017, 13, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Tanaka, M. P62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J. Cell Sci. 2011, 124, 2692–2701. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Ichimura, Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010, 584, 1374–1378. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Tao, Q.; Yu, L.; Li, L.; Bai, T.; Song, X.; Hu, H.; Li, Y.; Tan, X. Activation of autophagy contributes to the renoprotective effect of postconditioning on acute kidney injury and renal fibrosis. Biochem. Biophys. Res. Commun. 2018, 504, 641–646. [Google Scholar] [CrossRef]
- Leventhal, J.S.; Wyatt, C.M.; Ross, M.J. Recycling to discover something new: The role of autophagy in kidney disease. Kidney Int. 2017, 91, 4–6. [Google Scholar] [CrossRef]
- Chen, W.T.; Hung, K.C.; Wen, M.S.; Hsu, P.Y.; Chen, T.H.; Wang, H.D.; Fang, J.T.; Shie, S.S.; Wang, C.Y. Impaired leukocytes autophagy in chronic kidney disease patients. Cardiorenal Med. 2013, 3, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.-A.; Wu, V.C.-C.; Wang, C.-Y. Autophagy in chronic kidney diseases. Cells 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Cai, G.-Y.; Duan, Z.-Y.; Liu, S.-W.; Wu, J.; Lv, Y.; Hou, K.; Li, Z.-X.; Zhang, X.-G.; Chen, X.-M. Urinary sediment miRNAs reflect tubulointerstitial damage and therapeutic response in IgA nephropathy. BMC Nephrol. 2017, 18, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Ye, M.; Zhao, L.; Zou, W.; Shen, W.; Zhang, H.; Gong, J.; He, Q. The novel involvement of podocyte autophagic activity in the pathogenesis of lupus nephritis. Histol. Histopathol. 2018, 33, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Law, H.K.W. The role of autophagy in lupus nephritis. Int. J. Mol. Sci. 2015, 16, 25154–25167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Kuwana, H.; Shimamura, Y.; Ogata, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T.; Morita, T.; Sasaki, S.; et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin. Exp. Nephrol. 2010, 14, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravichandran, K.; Edelstein, C.L. Polycystic kidney disease: A case of suppressed autophagy? Semin. Nephrol. 2014, 34, 27–33. [Google Scholar] [CrossRef]
- Distefano, G.; Boca, M.; Rowe, I.; Wodarczyk, C.; Ma, L.; Piontek, K.B.; Germino, G.G.; Pandolfi, P.P.; Boletta, A. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell. Biol. 2009, 29, 2359–2371. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, O.; Tharaux, P.L.; Huber, T.B. Autophagy in kidney disease and aging: Lessons from rodent models. Kidney Int. 2016, 90, 950–964. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Nam, S.A.; Song, H.C.; Ko, J.S.; Park, S.H.; Kim, H.L.; Choi, E.J.; Kim, Y.S.; Kim, J.; Kim, Y.K. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology 2012, 17, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Kim, S., II; Lee, S.-Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, C.; Wang, C.; Duan, P.; Huang, W.; Chen, W.; Tang, S.; Yang, K. Bisphenol A induces autophagy and apoptosis concurrently involving the Akt/mTOR pathway in testes of pubertal SD rats. Environ. Toxicol. 2017, 32, 1977–1989. [Google Scholar] [CrossRef]
- Morris, A. Endocrine disruptors: Does BPA disrupt autophagy in the liver? Nat. Rev. Endocrinol. 2017, 13, 250. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Gomez, I.G.; Ren, S.; Hudkins, K.; Roach, A.; Alpers, C.E.; Shankland, S.J.; D’Agati, V.D.; Duffield, J.S. Deficient autophagy results in mitochondrial dysfunction and FSGS. J. Am. Soc. Nephrol. 2015, 26, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Ricart, M.; Torramade-Moix, S.; Pascual, G.; Palomo, M.; Moreno-Castaño, A.B.; Martinez-Sanchez, J.; Vera, M.; Cases, A.; Escolar, G. Endothelial damage, inflammation and immunity in chronic kidney disease. Toxins 2020, 12, 361. [Google Scholar] [CrossRef]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in renal diseases: New and old players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Kendziorski, J.A.; Belcher, S.M. Strain-specific induction of endometrial periglandular fibrosis in mice exposed during adulthood to the endocrine disrupting chemical bisphenol A. Reprod. Toxicol. 2015, 58, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elswefy, S.E.S.; Abdallah, F.R.; Atteia, H.H.; Wahba, A.S.; Hasan, R.A. Inflammation, oxidative stress and apoptosis cascade implications in bisphenol A-induced liver fibrosis in male rats. Int. J. Exp. Pathol. 2016, 97, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Bruno, K.A.; Mathews, J.E.; Yang, A.L.; Frisancho, J.A.; Scott, A.J.; Greyner, H.D.; Molina, F.A.; Greenaway, M.S.; Cooper, G.M.; Bucek, A.; et al. BPA Alters estrogen receptor expression in the heart after viral infection activating cardiac mast cells and T cells leading to perimyocarditis and fibrosis. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Leelahavanichkul, A.; Yan, Q.; Hu, X.; Eisner, C.; Huang, Y.; Chen, R.; Mizel, D.; Zhou, H.; Wright, E.C.; Kopp, J.B.; et al. AnIt is OKgiotensin ii overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010, 78, 1136–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Kimura, N.; Totsukawa, K. Effect of compound exposure to bisphenol A and nonylphenol on the development of fertility of fetal mice. J. Mamm. Ova Res. 2007, 24, 35–41. [Google Scholar] [CrossRef]
- Samova, S.; Doctor, H.; Verma, R. In vivo analysis of bisphenol A induced dose-dependent adverse effects in cauda epididymis of mice. Interdiscip. Toxicol. 2018, 11, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Neha, P.; Sangai, J.R.V. Quercetin alleviates bisphenol A-induced changes in nucleic acid and protein contents in mice. Acta Pol. Pharm. Drug Res. 2011, 68, 867–873. [Google Scholar]
- Yoshino, H.; Ichihara, T.; Kawabe, M.; Imai, N.; Hagiwara, A.; Asamoto, M.; Shirai, T. Lack of significant alteration in the prostate or testis of F344 rat offspring after transplacental and lactational exposure to bisphenol A. J. Toxicol. Sci. 2002, 27, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Ichihara, T.; Yoshino, H.; Imai, N.; Tsutsumi, T.; Kawabe, M.; Tamano, S.; Inaguma, S.; Suzuki, S.; Shirai, T. Lack of carcinogenic risk in the prostate with transplacental and lactational exposure to bisphenol A in rats. J. Toxicol. Sci. 2003, 28, 165–171. [Google Scholar] [CrossRef]
- Cstee Comments on: Risk Assessment Report on: Bisphenol A Human Health. Available online: https://ec.europa.eu/health/ph_risk/committees/sct/documents/out156_en.pdf (accessed on 30 May 2021).
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Valdivielso, J.M.; Sanz, A.B.; Bosch-Panadero, E.; Rodrigues-Díez, R.R.; Egido, J.; Ortiz, A.; González-Parra, E.; Ruiz-Ortega, M. TRAF3 modulation: Novel Mechanism for the anti-inflammatory effects of the vitamin D receptor agonist paricalcitol in renal disease. J. Am. Soc. Nephrol. 2020, 31, 2026–2042. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priego, A.R.; Parra, E.G.; Mas, S.; Morgado-Pascual, J.L.; Ruiz-Ortega, M.; Rayego-Mateos, S. Bisphenol A Modulates Autophagy and Exacerbates Chronic Kidney Damage in Mice. Int. J. Mol. Sci. 2021, 22, 7189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137189
Priego AR, Parra EG, Mas S, Morgado-Pascual JL, Ruiz-Ortega M, Rayego-Mateos S. Bisphenol A Modulates Autophagy and Exacerbates Chronic Kidney Damage in Mice. International Journal of Molecular Sciences. 2021; 22(13):7189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137189
Chicago/Turabian StylePriego, Alberto Ruiz, Emilio González Parra, Sebastián Mas, José Luis Morgado-Pascual, Marta Ruiz-Ortega, and Sandra Rayego-Mateos. 2021. "Bisphenol A Modulates Autophagy and Exacerbates Chronic Kidney Damage in Mice" International Journal of Molecular Sciences 22, no. 13: 7189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137189