
A Systematic Review of Parkinson’s Disease Pharmacogenomics: Is There Time for Translation into the Clinics?


Abstract
:1. Introduction
2. Results
2.1. Drug Specific Pharmacogenomic Properties
2.1.1. Pharmacogenomics of the Therapeutic Response to L-dopa
2.1.2. Pharmacogenomics of the Side-Effects to L-dopa
2.1.3. Dopamine Receptor Agonists
2.1.4. COMT Inhibitors
2.1.5. MAO Inhibitors
2.2. Genotype Specific Treatment and Pharmacogenomic Properties
2.2.1. LRRK2
2.2.2. SNCA
2.2.3. GBA
2.2.4. PRKN/PINK1/DJ1
3. Discussion
4. Materials and Methods
5. Conclusions
- Most evidence from the analyzed studies is found via secondary endpoints, which limits their power, with small sample size also being a diminishing factor.
- Conflicting reports between varied populations could be a consequence of low sample sizes and unaccounted interactions, which ultimately leads to low confidence in the data currently available.
- The most promising avenues for clinical implementation of pharmacogenetics lie in the current findings of impulse control disorders and hyperhomocysteinemia, where the available data are more consistent.
- Most of the studies focus on L-dopa and DA, and greater focus should also be given to other PD treatment options such as MAO-B and COMT inhibitors.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Vuletic, V.; Racki, V.; Chudy, D.; Bogdanovic, N. Deep Brain Stimulation in Non-motor Symptoms of Neurodegenerative diseases. In Neuromodulation Guiding the Advance of Research and Therapy [Working Title]; IntechOpen: London, UK, 2019. [Google Scholar]
- Heinzel, S.; Berg, D.; Gasser, T.; Chen, H.; Yao, C.; Postuma, R.B. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Fereshtehnejad, S.-M.; Zeighami, Y.; Dagher, A.; Postuma, R.B. Clinical criteria for subtyping Parkinson’s disease: Biomarkers and longitudinal progression. Brain 2017, 140, 1959–1976. [Google Scholar] [CrossRef]
- Ciccacci, C.; Borgiani, P. Pharmacogenomics in Parkinson’s disease: Which perspective for developing a personalized medicine? Neural Regen. Res. 2019, 14, 75–76. [Google Scholar] [CrossRef]
- Olanow, C.W.; Obeso, J.A.; Stocchi, F. Continuous dopamine-receptor treatment of Parkinson’s disease: Scientific rationale and clinical implications. Lancet Neurol. 2006, 5, 677–687. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Advances in understanding genomic markers and pharmacogenetics of Parkinsons disease. Expert Opin. Drug Metab. Toxicol. 2016, 12, 433–448. [Google Scholar] [CrossRef]
- Džoljić, E.; Novaković, I.; Krajinovic, M.; Grbatinić, I.; Kostić, V. Pharmacogenetics of drug response in Parkinson’s disease. Int. J. Neurosci. 2015, 125, 635–644. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Corvol, J.C.; Poewe, W. Pharmacogenetics of Parkinson’s Disease in Clinical Practice. Mov. Disord. Clin. Pract. 2017, 4, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, K.; Milani, L. Translating pharmacogenomics into clinical decisions: Do not let the perfect be the enemy of the good. Hum. Genomics 2019, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson Disease—Clinical Annotations. Available online: https://www.pharmgkb.org/disease/PA445254/clinicalAnnotation (accessed on 30 April 2020).
- Salat, D.; Tolosa, E. Levodopa in the treatment of Parkinson’s disease: Current status and new developments. J. Parkinsons Dis. 2013, 3, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craddock, N.; Owen, M.J.; O’Donovan, M.C. The catechol-O-methyl transferase (COMT) gene as a candidate for psychiatric phenotypes: Evidence and lessons. Mol. Psychiatry 2006, 11, 446–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, D.J.; Newman, T.K.; Savitz, J.; Ramesar, R. Warriors versus worriers: The role of COMT gene variants. CNS Spectr. 2006, 11, 745–748. [Google Scholar] [CrossRef]
- Bialecka, M.; Kurzawski, M.; Klodowska-Duda, G.; Opala, G.; Tan, E.K.; Drozdzik, M. The association of functional catechol-O-methyltransferase haplotypes with risk of Parkinsons disease, levodopa treatment response, and complications. Pharmacogenet. Genomics 2008, 18, 815–821. [Google Scholar] [CrossRef]
- Cheshire, P.; Bertram, K.; Ling, H.; O’Sullivan, S.S.; Halliday, G.; McLean, C.; Bras, J.; Foltynie, T.; Storey, E.; Williams, D.R. Influence of single nucleotide polymorphisms in COMT, MAO-A and BDNF genes on dyskinesias and levodopa use in Parkinson’s disease. Neurodegener. Dis. 2013, 13, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Białecka, M.; Droździk, M.; Kłodowska-Duda, G.; Honczarenko, K.; Gawrońska-Szklarz, B.; Opala, G.; Stankiewicz, J. The effect of monoamine oxidase B (MAOB) and catechol-O-methyltransferase (COMT) polymorphisms on levodopa therapy in patients with sporadic Parkinson’s disease. Acta Neurol. Scand. 2004, 110, 260–266. [Google Scholar] [CrossRef]
- Contin, M.; Martinelli, P.; Mochi, M.; Riva, R.; Albani, F.; Baruzzi, A. Genetic polymorphism of catechol-O-methyltransferase and levodopa pharmacokinetic-pharmacodynamic pattern in patient with parkinson’s disease. Mov. Disord. 2005, 20, 734–739. [Google Scholar] [CrossRef]
- Sampaio, T.F.; dos Santos, E.U.D.; de Lima, G.D.C.; dos Anjos, R.S.G.; da Silva, R.C.; Asano, A.G.C.; Asano, N.M.J.; Crovella, S.; de Souza, P.R.E. MAO-B and COMT Genetic Variations Associated With Levodopa Treatment Response in Patients With Parkinson’s Disease. J. Clin. Pharmacol. 2018, 58, 920–926. [Google Scholar] [CrossRef]
- Lee, M.S.; Lyoo, C.H.; Ulmanen, I.; Syvänen, A.C.; O Rinne, J. Genotypes of catechol-O-methyltransferase and response to levodopa treatment in patients with Parkinson’s disease. Neurosci. Lett. 2001, 298, 131–134. [Google Scholar] [CrossRef]
- Becker, M.L.; Visser, L.E.; Van Schaik, R.H.N.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H.C. OCT1 polymorphism is associated with response and survival time in anti-Parkinsonian drug users. Neurogenetics 2011, 12, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Altmann, V.; Schumacher-Schuh, A.F.; Rieck, M.; Callegari-Jacques, S.M.; Rieder, C.R.M.; Hutz, M.H. Influence of genetic, biological and pharmacological factors on levodopa dose in Parkinson’s disease. Pharmacogenomics 2016, 17, 481–488. [Google Scholar] [CrossRef]
- De Lau, L.M.L.; Verbaan, D.; Marinus, J.; Heutink, P.; Van Hilten, J.J. Catechol-O-methyltransferase Val158Met and the risk of dyskinesias in Parkinson’s disease. Mov. Disord. 2012, 27, 132–135. [Google Scholar] [CrossRef]
- Watanabe, M.; Harada, S.; Nakamura, T.; Ohkoshi, N.; Yoshizawa, K.; Hayashi, A.; Shoji, S. Association between catechol-O-methyltransferase gene polymorphisms and wearing-off and dyskinesia in Parkinson’s disease. Neuropsychobiology 2003, 48, 190–193. [Google Scholar] [CrossRef]
- Martín-Flores, N.; Fernández-Santiago, R.; Antonelli, F.; Cerquera, C.; Moreno, V.; Martí, M.J.; Ezquerra, M.; Malagelada, C. MTOR Pathway-Based Discovery of Genetic Susceptibility to L-DOPA-Induced Dyskinesia in Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2092–2100. [Google Scholar] [CrossRef]
- Foltynie, T.; Cheeran, B.; Williams-Gray, C.H.; Edwards, M.J.; Schneider, S.A.; Weinberger, D.; Rothwell, J.C.; Barker, R.A.; Bhatia, K.P. BDNF val66met influences time to onset of levodopa induced dyskinesia in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, E.U.D.; Sampaio, T.F.; Tenório dos Santos, A.D.; Bezerra Leite, F.C.; da Silva, R.C.; Crovella, S.; Asano, A.G.C.; Asano, N.M.J.; de Souza, P.R.E. The influence of SLC6A3 and DRD2 polymorphisms on levodopa-therapy in patients with sporadic Parkinson’s disease. J. Pharm. Pharmacol. 2019, 71, 206–212. [Google Scholar] [CrossRef]
- Rieck, M.; Schumacher-Schuh, A.F.; Altmann, V.; Francisconi, C.L.; Fagundes, P.T.; Monte, T.L.; Callegari-Jacques, S.M.; Rieder, C.R.; Hutz, M.H. DRD2 haplotype is associated with dyskinesia induced by levodopa therapy in Parkinson’s disease patients. Pharmacogenomics 2012, 13, 1701–1710. [Google Scholar] [CrossRef]
- Strong, J.A.; Dalvi, A.; Revilla, F.J.; Sahay, A.; Samaha, F.J.; Welge, J.A.; Gong, J.; Gartner, M.; Yue, X.; Yu, L. Genotype and smoking history affect risk of levodopa-induced dyskinesias in Parkinson’s disease. Mov. Disord. 2006, 21, 654–659. [Google Scholar] [CrossRef]
- Zappia, M.; Annesi, G.; Nicoletti, G.; Arabia, G.; Annesi, F.; Messina, D.; Pugliese, P.; Spadafora, P.; Tarantino, P.; Carrideo, S.; et al. Sex differences in clinical and genetic determinants of levodopa peak-dose dyskinesias in Parkinson disease: An exploratory study. Arch. Neurol. 2005, 62, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Oliveri, R.L.; Annesi, G.; Zappia, M.; Civitelli, D.; Montesanti, R.; Branca, D.; Nicoletti, G.; Spadafora, P.; Pasqua, A.A.; Cittadella, R.; et al. Dopamine D2 receptor gene polymorphism and the risk of levodopa-induced dyskinesias in PD. Neurology 1999, 53, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cho, J.; Lee, E.K.; Park, S.S.; Jeon, B.S. Differential genetic susceptibility in diphasic and peak-dose dyskinesias in Parkinson’s disease. Mov. Disord. 2011, 26, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.; Hofer, A.; Grapengiesser, A.; Gasser, T.; Kupsch, A.; Roots, I.; Brockmöller, J. L-Dopa-induced adverse effects in PD and dopamine transporter gene polymorphism. Neurology 2003, 60, 1750–1755. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Z.L.; Chen, B. Association study of dopamine D2, D3 receptor gene polymorphisms with motor fluctuations in PD. Neurology 2001, 56, 1757–1759. [Google Scholar] [CrossRef]
- Paus, S.; Gadow, F.; Knapp, M.; Klein, C.; Klockgether, T.; Wüllner, U. Motor complications in patients form the German Competence Network on Parkinson’s disease and the DRD3 Ser9Gly polymorphism. Mov. Disord. 2009, 24, 1080–1084. [Google Scholar] [CrossRef]
- Schumacher-Schuh, A.F.; Altmann, V.; Rieck, M.; Tovo-Rodrigues, L.; Monte, T.L.; Callegari-Jacques, S.M.; Medeiros, M.S.; Rieder, C.R.M.; Hutz, M.H. Association of common genetic variants of HOMER1 gene with levodopa adverse effects in Parkinson’s disease patients. Pharm. J. 2014, 14, 289–294. [Google Scholar] [CrossRef]
- Purcaro, C.; Vanacore, N.; Moret, F.; Di Battista, M.E.; Rubino, A.; Pierandrei, S.; Lucarelli, M.; Meco, G.; Fattapposta, F.; Pascale, E. DAT gene polymorphisms (rs28363170, rs393795) and levodopa-induced dyskinesias in Parkinson’s disease. Neurosci. Lett. 2019, 690, 83–88. [Google Scholar] [CrossRef]
- De Bonis, M.L.; Tessitore, A.; Pellecchia, M.T.; Longo, K.; Salvatore, A.; Russo, A.; Ingrosso, D.; Zappia, V.; Barone, P.; Galletti, P.; et al. Impaired transmethylation potential in Parkinson’s disease patients treated with l-Dopa. Neurosci. Lett. 2010, 468, 287–291. [Google Scholar] [CrossRef]
- Gorgone, G.; Currò, M.; Ferlazzo, N.; Parisi, G.; Parnetti, L.; Belcastro, V.; Tambasco, N.; Rossi, A.; Pisani, F.; Calabresi, P.; et al. Coenzyme Q10, hyperhomocysteinemia and MTHFR C677T polymorphism in levodopa-treated Parkinson’s disease patients. NeuroMolecular Med. 2012, 14, 84–90. [Google Scholar] [CrossRef]
- Yuan, R.Y.; Sheu, J.J.; Yu, J.M.; Hu, C.J.; Tseng, I.J.; Ho, C.S.; Yeh, C.Y.; Hung, Y.L.; Chiang, T.R. Methylenetetrahydrofolate reductase polymorphisms and plasma homocysteine in levodopa-treated and non-treated Parkinson’s disease patients. J. Neurol. Sci. 2009, 287, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Frauscher, B.; Högl, B.; Maret, S.; Wolf, E.; Brandauer, E.; Wenning, G.K.; Kronenberg, M.F.; Kronenberg, F.; Tafti, M.; Poewe, W. Association of daytime sleepiness with COMT polymorphism in patients with parkinson disease: A pilot study. Sleep 2004, 27, 733–736. [Google Scholar] [CrossRef] [Green Version]
- Rissling, I.; Frauscher, B.; Kronenberg, F.; Tafti, M.; Stiasny-Kolster, K.; Robyr, A.-C.; Körner, Y.; Oertel, W.H.; Poewe, W.; Högl, B.; et al. Daytime sleepiness and the COMT val158met polymorphism in patients with Parkinson disease. Sleep 2006, 29, 108–111. [Google Scholar] [PubMed] [Green Version]
- Rissling, I.; Körner, Y.; Geller, F.; Stiasny-Kolster, K.; Oertel, W.H.; Möller, J.C. Preprohypocretin polymorphisms in Parkinson disease patients reporting “sleep attacks”. Sleep 2005, 28, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Rissling, I.; Geller, F.; Bandmann, O.; Stiasny-Kolster, K.; Körner, Y.; Meindorfner, C.; Krüger, H.P.; Oertel, W.H.; Möller, J.C. Dopamine receptor gene polymorphisms in Parkinson’s disease patients reporting “sleep attacks”. Mov. Disord. 2004, 19, 1279–1284. [Google Scholar] [CrossRef]
- Rieck, M.; Schumacher-Schuh, A.F.; Altmann, V.; Callegari-Jacques, S.M.; Rieder, C.R.M.; Hutz, M.H. Association between DRD2 and DRD3 gene polymorphisms and gastrointestinal symptoms induced by levodopa therapy in Parkinson’s disease. Pharm. J. 2018, 18, 196–200. [Google Scholar] [CrossRef]
- Redenšek, S.; Flisar, D.; Kojovic, M.; Kramberger, M.G.; Georgiev, D.; Pirtošek, Z.; Trošt, M.; Dolžan, V. Dopaminergic pathway genes influence adverse events related to dopaminergic treatment in Parkinson’s disease. Front. Pharmacol. 2019, 9. [Google Scholar] [CrossRef]
- Nombela, C.; Rowe, J.B.; Winder-Rhodes, S.E.; Hampshire, A.; Owen, A.M.; Breen, D.P.; Duncan, G.W.; Khoo, T.K.; Yarnall, A.J.; Firbank, M.J.; et al. Genetic impact on cognition and brain function in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain 2014, 137, 2743–2758. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Hampshire, A.; Barker, R.A.; Owen, A.M. Attentional control in Parkinson’s disease is dependent on COMT val 158 met genotype. Brain 2008, 131, 397–408. [Google Scholar] [CrossRef]
- Goetz, C.G.; Burke, P.F.; Leurgans, S.; Berry-Kravis, E.; Blasucci, L.M.; Raman, R.; Zhou, L. Genetic variation analysis in Parkinson disease patients with and without hallucinations: Case-control study. Arch. Neurol. 2001, 58, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makoff, A.J.; Graham, J.M.; Arranz, M.J.; Forsyth, J.; Li, T.; Aitchison, K.J.; Shaikh, S.; Grünewald, R.A. Association study of dopamine receptor gene polymorphisms with drug-induced hallucinations in patients with idiopathic Parkinson’s disease. Pharmacogenetics 2000, 10, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Si, Y.M.; Liu, Z.L.; Yu, L. Cholecystokinin, cholecystokinin-A receptor and cholecystokinin-B receptor gene polymorphisms in Parkinson’s disease. Pharmacogenetics 2003, 13, 365–369. [Google Scholar] [CrossRef]
- De Luca, V.; Annesi, G.; De Marco, E.V.; De Bartolomeis, A.; Nicoletti, G.; Pugliese, P.; Muscettola, G.; Barone, P.; Quattrone, A. HOMER1 promoter analysis in Parkinson’s disease: Association study with psychotic symptoms. Neuropsychobiology 2009, 59, 239–245. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, J.; Guo, Y.; Dong, G.; Zou, W.; Chen, Z. Association between BDNF G196A (Val66Met) polymorphism and cognitive impairment in patients with Parkinson’s disease: A meta-analysis. Braz. J. Med. Biol. Res. 2019, 52, e8443. [Google Scholar] [CrossRef] [Green Version]
- Gatto, E.M.; Aldinio, V. Impulse Control Disorders in Parkinson’s Disease. A Brief and Comprehensive Review. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Cormier-Dequaire, F.; Bekadar, S.; Anheim, M.; Lebbah, S.; Pelissolo, A.; Krack, P.; Lacomblez, L.; Lhommée, E.; Castrioto, A.; Azulay, J.P.; et al. Suggestive association between OPRM1 and impulse control disorders in Parkinson’s disease. Mov. Disord. 2018, 33, 1878–1886. [Google Scholar] [CrossRef]
- Zainal Abidin, S.; Tan, E.L.; Chan, S.-C.; Jaafar, A.; Lee, A.X.; Abd Hamid, M.H.N.; Abdul Murad, N.A.; Pakarul Razy, N.F.; Azmin, S.; Ahmad Annuar, A.; et al. DRD and GRIN2B polymorphisms and their association with the development of impulse control behaviour among Malaysian Parkinson’s disease patients. BMC Neurol. 2015, 15, 59. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Lee, E.K.; Park, S.S.; Lim, J.Y.; Kim, H.J.; Kim, J.S.; Jeon, B.S. Association of DRD3 and GRIN2B with impulse control and related behaviors in Parkinson’s disease. Mov. Disord. 2009, 24, 1803–1810. [Google Scholar] [CrossRef]
- Castro-Martínez, X.H.; García-Ruiz, P.J.; Martínez-García, C.; Martínez-Castrillo, J.C.; Vela, L.; Mata, M.; Martínez-Torres, I.; Feliz-Feliz, C.; Palau, F.; Hoenicka, J. Behavioral addictions in early-onset Parkinson disease are associated with DRD3 variants. Park. Relat. Disord. 2018, 49, 100–103. [Google Scholar] [CrossRef]
- Arbouw, M.E.L.; Movig, K.L.L.; Egberts, T.C.G.; Poels, P.J.E.; Van Vugt, J.P.P.; Wessels, J.A.M.; Van Der Straaten, R.J.H.M.; Neef, C.; Guchelaar, H.J. Clinical and pharmacogenetic determinants for the discontinuation of non-ergoline dopamine agonists in Parkinson’s disease. Eur. J. Clin. Pharmacol. 2009, 65, 1245–1251. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Z.; Tang, B.S.; Yan, X.X.; Liu, J.; Ouyang, D.S.; Nie, L.N.; Fan, L.; Li, Z.; Ji, W.; Hu, D.L.; et al. Association of the DRD2 and DRD3 polymorphisms with response to pramipexole in Parkinson’s disease patients. Eur. J. Clin. Pharmacol. 2009, 65, 679–683. [Google Scholar] [CrossRef]
- Xu, S.; Liu, J.; Yang, X.; Qian, Y.; Xiao, Q. Association of the DRD2 CAn-STR and DRD3 Ser9Gly polymorphisms with Parkinson’s disease and response to dopamine agonists. J. Neurol. Sci. 2017, 372, 433–438. [Google Scholar] [CrossRef]
- Zhi, Y.; Yuan, Y.; Si, Q.; Wang, M.; Shen, Y.; Wang, L.; Zhang, H.; Zhang, K. The Association between DRD3 Ser9Gly Polymorphism and Depression Severity in Parkinson’s Disease. Parkinsons. Dis. 2019, 2019, 1642087. [Google Scholar] [CrossRef] [PubMed]
- Paus, S.; Grünewald, A.; Klein, C.; Knapp, M.; Zimprich, A.; Janetzky, B.; Möller, J.C.; Klockgether, T.; Wüllner, U. The DRD2 TaqIA polymorphism and demand of dopaminergic medication in Parkinson’s disease. Mov. Disord. 2008, 23, 599–602. [Google Scholar] [CrossRef]
- McDonell, K.E.; van Wouwe, N.C.; Harrison, M.B.; Wylie, S.A.; Claassen, D.O. Taq1A polymorphism and medication effects on inhibitory action control in Parkinson disease. Brain Behav. 2018, 8, 1008. [Google Scholar] [CrossRef]
- Erga, A.H.; Dalen, I.; Ushakova, A.; Chung, J.; Tzoulis, C.; Tysnes, O.B.; Alves, G.; Pedersen, K.F.; Maple-Grødem, J. Dopaminergic and opioid pathways associated with impulse control disorders in Parkinson’s disease. Front. Neurol. 2018, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvol, J.C.; Bonnet, C.; Charbonnier-Beaupel, F.; Bonnet, A.M.; Fiévet, M.H.; Bellanger, A.; Roze, E.; Meliksetyan, G.; Ben Djebara, M.; Hartmann, A.; et al. The COMT Val158Met polymorphism affects the response to entacapone in Parkinson’s disease: A randomized crossover clinical trial. Ann. Neurol. 2011, 69, 111–118. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, H.S.; Cho, E.K.; Lim, J.H.; Rinne, J.O. COMT genotype and effectiveness of entacapone in patients with fluctuating Parkinson’s disease. Neurology 2002, 58, 564–567. [Google Scholar] [CrossRef]
- Chong, D.J.; Suchowersky, O.; Szumlanski, C.; Weinshilboum, R.M.; Brant, R.; Campbell, N.R.C. The relationship between COMT genotype and the clinical effectiveness of tolcapone, a COMT inhibitor, in patients with Parkinson’s disease. Clin. Neuropharmacol. 2000, 23, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Trenkwalder, C.; Kuoppamäki, M.; Vahteristo, M.; Müller, T.; Ellmén, J. Increased dose of carbidopa with levodopa and entacapone improves “off” time in a randomized trial. Neurology 2019, 92, E1487–E1496. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Ramírez, J.; Gamazon, E.R.; Mirkov, S.; Chen, P.; Wu, K.; Sun, C.; Cox, N.J.; Cook, E.; Das, S.; et al. Genetic factors affecting gene transcription and catalytic activity of UDP-glucuronosyltransferases in human liver. Hum. Mol. Genet. 2014, 23, 5558–5569. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, H.; Nakajima, M.; Katoh, M.; Hara, Y.; Tachibana, O.; Yamashita, J.; McLeod, H.L.; Yokoi, T. A novel polymorphism in the promoter region of human UGT1A9 gene (UGT1A9*22) and its effects on the transcriptional activity. Pharmacogenetics 2004, 14, 329–332. [Google Scholar] [CrossRef]
- Ferrari, M.; Martignoni, E.; Blandini, F.; Riboldazzi, G.; Bono, G.; Marino, F.; Cosentino, M. Association of UDP-glucuronosyltransferase 1A9 polymorphisms with adverse reactions to catechol-O-methyltransferase inhibitors in Parkinson’s disease patients. Eur. J. Clin. Pharmacol. 2012, 68, 1493–1499. [Google Scholar] [CrossRef]
- Masellis, M.; Collinson, S.; Freeman, N.; Tampakeras, M.; Levy, J.; Tchelet, A.; Eyal, E.; Berkovich, E.; Eliaz, R.E.; Abler, V.; et al. Dopamine D2 receptor gene variants and response to rasagiline in early Parkinson’s disease: A pharmacogenetic study. Brain 2016, 139, 2050–2062. [Google Scholar] [CrossRef] [Green Version]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoccolella, S.; Lamberti, P.; Armenise, E.; De Mari, M.; Lamberti, S.V.; Mastronardi, R.; Fraddosio, A.; Iliceto, G.; Livrea, P. Plasma homocysteine levels in Parkinson’s disease: Role of antiparkinsonian medications. Park. Relat. Disord. 2005, 11, 131–133. [Google Scholar] [CrossRef]
- Kraemmer, J.; Smith, K.; Weintraub, D.; Guillemot, V.; Nalls, M.A.; Cormier-Dequaire, F.; Moszer, I.; Brice, A.; Singleton, A.B.; Corvol, J.C. Clinical-genetic model predicts incident impulse control disorders in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1106–1111. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S. Impulse Control Disorders in Parkinson’s Disease: Pathophysiology, Effect of Genetic Polymorphism and Future Research Directions. Austin J. Clin. Neurol. 2017, 4, 1100. [Google Scholar] [CrossRef]
- Agúndez, J.A.G.; García-Martín, E.; Alonso-Navarro, H.; Jiménez-Jiménez, F.J. Anti-Parkinson’s disease drugs and pharmacogenetic considerations. Expert Opin. Drug Metab. Toxicol. 2013, 9, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Bonifácio, M.J.; Palma, P.N.; Almeida, L.; Soares-da-Silva, P. Catechol-O-methyltransferase and Its Inhibitors in Parkinson’s Disease. CNS Drug Rev. 2007, 13, 352–379. [Google Scholar] [CrossRef]
- Borges, N. Tolcapone in Parkinson’s disease: Liver toxicity and clinical efficacy. Expert Opin. Drug Saf. 2005, 4, 69–73. [Google Scholar] [CrossRef]
- Martignoni, E.; Cosentino, M.; Ferrari, M.; Porta, G.; Mattarucchi, E.; Marino, F.; Lecchini, S.; Nappi, G. Two patients with COMT inhibitor-induced hepatic dysfunction and UGT1A9 genetic polymorphism. Neurology 2005, 65, 1820–1822. [Google Scholar] [CrossRef]
- Kaplowitz, N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov. 2005, 4, 489–499. [Google Scholar] [CrossRef]
- Bonifácio, M.J.; Torrão, L.; Loureiro, A.I.; Palma, P.N.; Wright, L.C.; Soares-Da-Silva, P. Pharmacological profile of opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br. J. Pharmacol. 2015, 172, 1739–1752. [Google Scholar] [CrossRef] [Green Version]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Guay, D.R.P. Rasagiline (TVP-1012): A new selective monoamine oxidase inhibitor for Parkinson’s disease. Am. J. Geriatr. Pharmacother. 2006, 4, 330–346. [Google Scholar] [CrossRef]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [Green Version]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Nalls, M.A.; McLean, C.Y.; Rick, J.; Eberly, S.; Hutten, S.J.; Gwinn, K.; Sutherland, M.; Martinez, M.; Heutink, P.; Williams, N.M.; et al. Diagnosis of Parkinson’s disease on the basis of clinical and genetic classification: A population-based modelling study. Lancet Neurol. 2015, 14, 1002–1009. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.; Zhang, Y.; Pan, H.; Xu, Q.; Guo, J.; Tang, B.; Sun, Q. Clinical heterogeneity among LRRK2 variants in Parkinson’s disease: A Meta-analysis. Front. Aging Neurosci. 2018, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- Yahalom, G.; Kaplan, N.; Vituri, A.; Cohen, O.S.; Inzelberg, R.; Kozlova, E.; Korczyn, A.D.; Rosset, S.; Friedman, E.; Hassin-Baer, S. Dyskinesias in patients with Parkinson’s disease: Effect of the leucine-rich repeat kinase 2 (LRRK2) G2019S mutation. Park. Relat. Disord. 2012, 18, 1039–1041. [Google Scholar] [CrossRef]
- Cacabelos, R.; Carrera, I.; Alejo, R.; Fernández-Novoa, L.; Cacabelos, P.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Tellado, I.; Cacabelos, N.; et al. Pharmacogenetics of Atremorine-Induced Neuroprotection and Dopamine Response in Parkinson’s Disease. Planta Med. 2019, 85, 1351–1362. [Google Scholar] [CrossRef]
- Nishioka, K.; Ross, O.A.; Ishii, K.; Kachergus, J.M.; Ishiwata, K.; Kitagawa, M.; Kono, S.; Obi, T.; Mizoguchi, K.; Inoue, Y.; et al. Expanding the clinical phenotype of SNCA duplication carriers. Mov. Disord. 2009, 24, 1811–1819. [Google Scholar] [CrossRef]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti--Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.C.; Honoré, A.; et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 202–210. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Q.Y.; Zhao, Y.W.; Shu, L.; Guo, J.F.; Xu, Q.; Yan, X.X.; Tang, B.S. Effect of GBA mutations on phenotype of Parkinson’s disease: A study on Chinese population and a meta-analysis. Parkinsons Dis. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.L.; Graham, E.; Critchley, P.; Schrag, A.E.; Wood, N.W.; Lees, A.J.; Bhatia, K.P.; Quinn, N. Parkin disease: A phenotypic study of a large case series. Brain 2003, 126, 1279–1292. [Google Scholar] [CrossRef]
- Benamer, H.T.S.; De Silva, R. LRRK2 G2019S in the North African population: A review. Eur. Neurol. 2010, 63, 321–325. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Pu, J. Leucine-Rich Repeat Kinase 2 in Parkinson’s Disease: Updated from Pathogenesis to Potential Therapeutic Target. Eur. Neurol. 2018, 79, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z. LRRK2 in Parkinson’s disease: In vivo models and approaches for understanding pathogenic roles. FEBS J. 2009, 276, 6445–6454. [Google Scholar] [CrossRef] [Green Version]
- Alkanli, N.; Ay, A. The Relationship between Alpha-Synuclein (SNCA) Gene Polymorphisms and Development Risk of Parkinson’s Disease. In Synucleins—Biochemistry and Role in Diseases; IntechOpen: London, UK, 2020. [Google Scholar]
- Wang, Z.; Gao, G.; Duan, C.; Yang, H. Progress of immunotherapy of anti-α-synuclein in Parkinson’s disease. Biomed. Pharmacother. 2019, 115, 108843. [Google Scholar] [CrossRef]
- Lin, J.Y.; Xie, C.L.; Zhang, S.F.; Yuan, W.; Liu, Z.G. Current experimental studies of gene therapy in Parkinson’s disease. Front. Aging Neurosci. 2017, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Avenali, M.; Blandini, F.; Cerri, S. Glucocerebrosidase Defects as a Major Risk Factor for Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 97. [Google Scholar] [CrossRef] [Green Version]
- Sassone, J.; Valtorta, F.; Ciammola, A. Early Dyskinesias in Parkinson’s Disease Patients With Parkin Mutation: A Primary Corticostriatal Synaptopathy? Front. Neurosci. 2019, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Kuhl McGuire, M. Next in Line for Parkinson’s Therapies: PRKN and PINK1 Parkinson’s Disease. Available online: https://www.michaeljfox.org/news/next-line-parkinsons-therapies-prkn-and-pink1 (accessed on 20 May 2020).
- Rahman, A.A.; Morrison, B.E. Contributions of VPS35 Mutations to Parkinson’s Disease. Neuroscience 2019, 401, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372. [Google Scholar] [CrossRef]


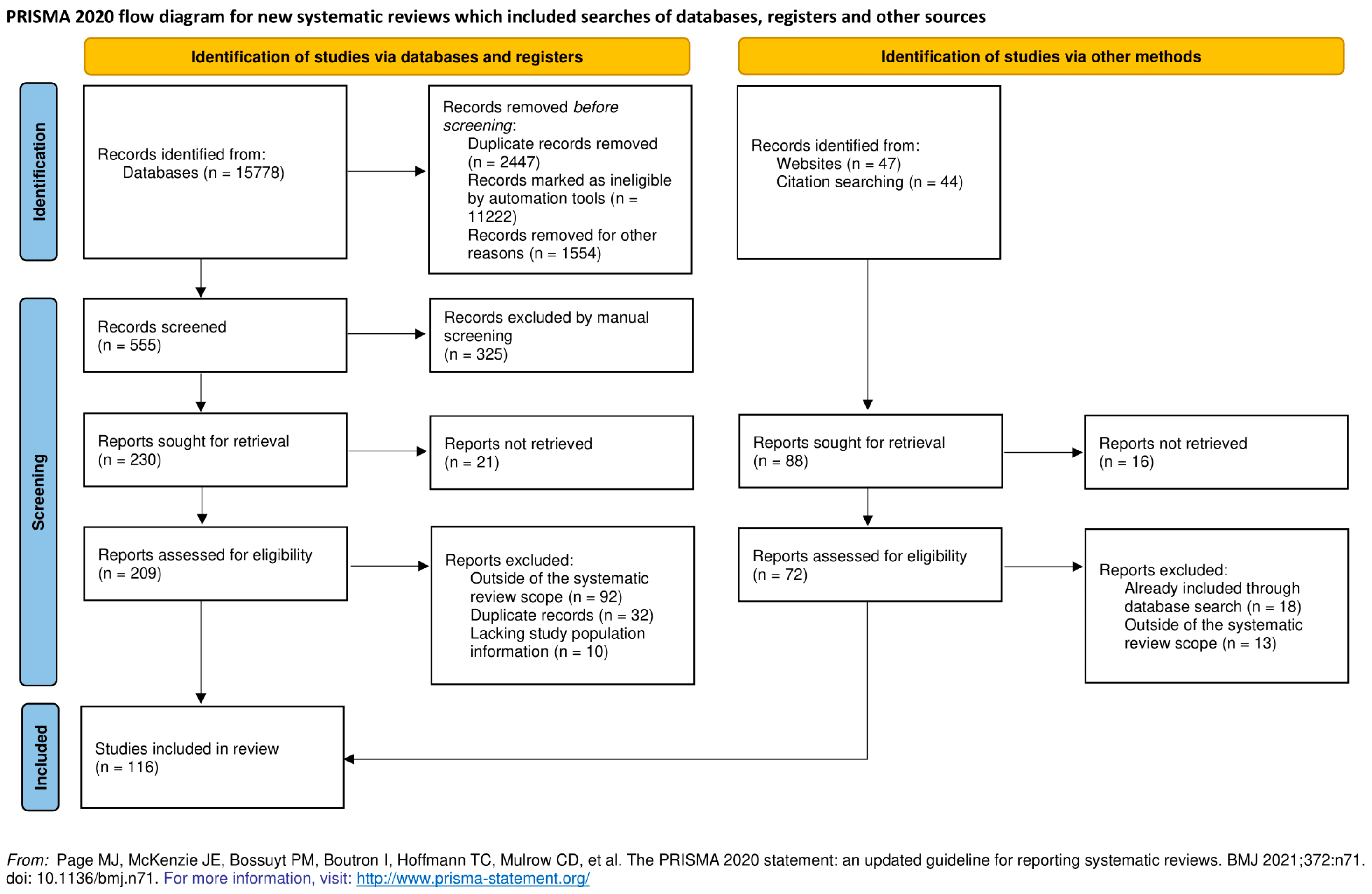{kind=link}
| References | Study Design | Population (Ethnicity) | Main Finding |
|---|---|---|---|
| Bialecka M et al. [19] | Genetic screening for COMT SNP-s and the association of COMT haplotypes with the dose and complications of levodopa therapy in PD patients | 679 study participants (322 PD and 357 controls)—participants genotyped for four SNPs in the COMT gene | The frequency of G_C_G_G (high activity) haplotype carriers was higher in late onset PD patients (p = 0.04) compared with controls |
| Cheshire et al. [20] | Influence of SNP in COMT, MAO-A and BDNF on LID | 285 Parkinson disease patients | Individual SNPS in BDNF, COMT and MAO-A required higher doses of levodopa |
| Bialecka M et al. [21] | Prospective study over 5 years | 95 patients with sporadic PD divided into 2 groups (group 1 treated with levodopa < 500 mg/daily; group 2 > 500 mg/daily) | Higher frequency of COMT (L/L) homozygotes in the group treated with lower doses of levodopa |
| Contin M et al. [22] | Prospective study—serial measurement of plasma levodopa, finger-tapping and dyskinesia ratings | 104 patients with PD | No clinically relevant levodopa response associated with the COMT polymorphism |
| Sampaio et al. [23] | Genetic screening for MAO-B and COMT SNP followed by a multivariate analysis (sex, duration of disease, levodopa treatment duration) | 162 Brazillian PD patients treated with levodopa split into 2 groups according to levodopa dose | Patients carrying MAO-B(rs1799836) A and AA genotypes and COMT (Rs4680) LL genotype suffered more frequently from LID; male population with MAO-B treated with higher doses of levodopa(p = 0.04) |
| Lee et al. [24] | Genetic screening for COMT SNP-s in PD, multiple system atrophy and controls; association with response to levodopa | 73 Korean patients with PD, 29 with MSA and 49 controls | No significant association between the rs4680 polypmorphism and the response to L-dopa |
| Becker et al. [25] | Genetic screening for OCT1 SNP and the correlation with L-dopa dosage | 7983 Caucasians aged 55 years and older who had a first prescription for levodopa between July 1st 1991 and January 1st 2008 | Higher L-dopa doses were needed for patients with SLC22A1 gene rs622342A>C |
| Altmann et al. [26] | Multivariate analysis of genetic polymorphisms in relation to L-dopa dosage | 224 Parkinson’s disease patients | Lower required doses of L-dopa in patients with SV2C rs30196 polymorphism and in SLC6A3 polymorphism |
| de Lau LM et al. [27] | Prospective study | 219 patients with PD without dyskinesias at baseline | The A-allele of the COMT Val158Met polymorphism was related to an increased risk of developing dyskinesias during follow-up |
| Watanabe et al. [28] | Genetic screening of COMT Val-108-Met polymorphism | 121 Japanese patients with Parkinson’s disease (PD) and 100 controls. | Patients with homozygosity for the low-activity allele showed a tendency to exhibit the “wearing-off” phenomenon compared with controls |
| Martin-Flores et al. [29] | Genetic screening for mTOR genetic pathway mutations | 401 patients with PD of European origin from the northeastern part of the Iberian peninsula | Patients with SNP’s in the mTOR signalling cascade could have increased severity and onset of L-dopa induced dyskinesias. SNP rs1043098 and rs1043098 in the EIF4EBP2 gene, RICTOR rs2043112, and PRKCA rs4790904 had increased LID onset. Patients with SNP in HRAS rs12628, PRKN rs1801582 and also with a four-loci epistatic combination involving RPS6KB1 rs1292034, HRAS rs12628, RPS6KA2 rs6456121 and FCHSD1 rs456998 had increased LID severity |
| Foltynie et al. [30] | Genetic screening for BDNF genotypes | 315 patients from the UK, unknown origin | Patients with the functional met allele BDNF polymorphism is associated with a significantly higher risk of developing dyskinesias |
| dos Santos et al. [31] | Targeted genotyping for DRD2/ANKK1 (rs1800497) and SLC6A3/DAT1 (rs28363170) phenotypes | 195 idiopathic Brazilian PD patients | Association between the occurrence of dyskinesia with an increased greater disease severity, higher L-DOPA dose and use of dopamine agonist |
| Rieck et al. [32] | Targeted genotyping for the variants in the DRD2/ANKK1 gene region (-141CIns/Del, rs2283265, rs1076560, C957T, TaqIA and rs2734849) | 199 Brazilian PD patients | Carriers of the TTCTA haplotype show an increased risk for the presence of dyskinesia (p = 0.007; 1.538 [95% CI: 1.126–2.101]) |
| Strong et al. [33] | Targeted genotyping of the mu opioid receptor gene and the DRD2 gene | 92 USA PD patients of unknown origin that had levodopa induced dyskinesias | G-allele of the A118G polymorphism of the mu opioid receptor is associated with an increased risk of earlier dyskinesia onset. Early dyskinesia was linked to the DRD2 14 and 14/15 alleles |
| Zappia et al. [34] | Genotyping analysis of the intronic CA dinucleotide short tandem repeat (CAn-STR) polymorphisms in the DRD2 gene | 215 PD patients from southern Italy | Genetic factors related to the DRD2 CAn-STR polymorphism were not independent predictors for PDD in the total population, but they had a strong protective effect on the appearance of PDD when the multivariate analysis was performed in men. In women, a genetic protective effect on PDD was not evident |
| Oliveri et al. [35] | Genotyping analysis of polymorphisms in the DRD1 and DRD2 genes | 136 sporadic PD patients and 224 population control subjects | DRD1 polymorphisms were not associated with the risk of developing PD or peak-dose dyskinesias. The 15 allele DRD2 gene polymorphism was more frequent in parkinsonian subjects than in control subjects. Frequency of both the 13 allele and the 14 allele DRD2 gene plymorphism was higher in non-dyskinetic than in the dyskinetic PD subjects |
| Lee et al. [36] | Targeted genotyping for six genetic variants (DRD2 Taq1A (=g.32806C>T, rs1800497), DRD3 p.S9G (rs6280), GRIN2B c.2664C>T (rs1806201), c.366C>G (rs7301328), c.-200T>G (rs1019385) and 5-HTTLPR) | 503 PD patients and 559 healthy controls of Korean origin | DRD3 p.S9G variant is exclusively associated with diphasic dyskinesia, with the AA genotype likely shortening the durations of the dyskinesias; Peak dose dyskinesias were not associated with any of the six analysed genetic variants |
| Kaiser et al. [37] | Retrospective noninterventional study focusing on DRD2, DRD3 and DRD4 gene polymorphisms. | 183 idiopathic German PD patients | The polymorphisms of DRD2, DRD3, and DRD4 were not associated with the risk to develop adverse effects of L -dopa. Patients with psychosis or dyskinesia carried the nine copy allele 40-bp VNTR of the DAT more frequently than nonafflicted patients |
| Wang et al. [38] | Genotype association study of DRD2 TaqIA, DRD3 BalI and MspI polymorphisms | 140 idiopathic USA PD patients | Findings suggest that DRD2 TaqIA polymorphism may be associated with an increased risk for developing motor fluctuations in PD |
| Paus et al. [39] | Database analysis; Association study of DRD3 Ser9Gly genotype | 690 German PD patients | Stepwise regression analysis revealed no effect of DRD3 Ser9Gly on chorea, dystonia or motor fluctuations in PD. Increased risk of sleep attacks in DRD4 48-bp VNTR polymorphism |
| Schumacher-Schuh et. [40] | Genotyping of the HOMER1 gene for rs4704559, rs10942891 and rs4704560 polymorphisms | 205 Brazilian PD patients | The rs4704559 G allele was associated with a lower prevalence of dyskinesia and visual hallucinations |
| Purcaro et al. [41] | Targeted genotyping of DAT gene polymorphisms (rs28363170, rs393795) | 181 Italian PD patients | DAT gene 10R/10R (rs28363170) and A carrier (rs393795) of the DAT gene reduces the risk of LID occurrence during long-term therapy with l-DOPA (OR = 0.31; 95% CI, 0.09–0.88) |
| De Bonis ML et al. [42] | Genotyping analysis of A SNP C667T (rs1801133) | 44 PD patients treated with L-Dopa (20 with concomitant dopamineagonists, group A) and 12 patients L-Dopa untreated | L-Dopa administration in hyperhomocysteinemic PD patients can lower intracellular concentration of (AdoMet) in erythrocytes (RBC) with hyperhomocysteinaemia causing a significant increase in S-Adenosylhomocysteine (AdoHcy) level; may lead to drug resistance through COMT upregulation |
| Gorgone et al. [43] | Genotype screening for the methylenetetrahydrofolate reductase (MTHFR) gene polymorphism | 60 Italian PD patients and 82 healthy subjects | Patients with a TT677 mutated genotype had higher homocysteine and Coenzyme Q10 levels and needed a lower L-dopa daily dose |
| Yual et al. [44] | Targeted genotyping of the MTHFR gene | 48 L-dopa treated patients, 28 non-treated PD patients and 110 control of Taiwanese origin | Genetic C677T and A1298C polymorphisms in 5,10-methylenetetrahydrofolate reductase (MTHFR) and levodopa therapy in Parkinson’s disease (PD) may increase homocysteine (Hcy) level |
| Frusher et al. [45] | Genotype screening of D4 receptor of COMT(alleles LL,LH,HH) and the correlation with daytime sleepiness | 46 PD patients | Higher ESS (Epworth Sleepiness Scale) in LH/LL alleles |
| Rissling et al. [46] | Genotype screening of COMT rs4680 polymorphism and the correlation with daytime sleepiness | 240 patients with PD (70 with the met-met (LL), 116 with the met-val (LH), and 54 with the val-val (HH) genotype | No clinical relevance of COMT in daytime sleepiness in PD |
| Rissling et al. [47] | Genotype screening of preprohypocretin polymorphisms and the correlation with sudden onset of sleep | 132 PD patients and 132 PD patients without sudden onset of sleep | A significant association between the (-909T/C) preprohypocretin polymorphism and sudden onset of sleep in Parkinson disease |
| Rissling et al. [48] | Genotype screening of D2,D3,D4 polymorphisms; association with sudden onset of sleep | 137 PD patients with SOS(sudden onset of sleep) and 137 PD patients without SOS | A significant association between the dopamine D2 receptor gene polymorphism Taq IA and SOS in PD |
| Rieck et al. [49] | Genotype association study of DRD2 and DRD3 gene polymorphisms and gastrointestinal symptoms induced by levodopa therapy | 217 Brazilian PD patients | DRD2 Ins/Ins and DRD3 Ser/Ser genotypes were independent and predictors of gastrointestinal symptoms associated with levodopa therapy |
| Redenšek et al. [50] | Retrospective cohort study | 31 unrelated PD patient | carriers of at least one DRD3 rs6280 C allele and CC homozygotes had higher odds for this adverse event |
| Nombela et al. [51] | Prospective cohort study focused on cognitive decline and COMT Val158Met (rs4680), MAPT (rs9468) H1 vs. H2 haplotype and APOE-e2, 3, 4 polymorphisms | 168 UK PD patients and 85 matched controls | All three analysed genotypes had significant association with cognitive decline, with associations relating to L-dopa therapy in the COMT gene |
| Williams-Gray et al. [52] | Genotype associative study on cognitive decline in association to COMT val(158)met genotype and dopaminergic medication | 29 medicated patients with early PD | Significant underactivation across the frontoparietal attentional network |
| Goetz et al. [53] | Case control study | 44 patients with PD and chronic hallucinations and 44 patients with PD without | Carriers of DRD3 rs6280 C allele may have higher odds of developing visual hallucinations |
| Makoff et al. [54] | Genotype association study of DRD2 and DRD3 polymorphims and drug-induced hallucinations | 155 white Caucasian PD patients from the UK | No association was found with the whole group of hallucinating patients and their controls. However, an association was found with late-onset hallucinations and the C allele of the TaqIA polymorphism, 10.5 kb 39 to DRD2 |
| Wang et al. [55] | Case control study | 160 Chinese patients with Parkinson’s disease and 160 controls | Visual hallucinations in PD are associated with cholecystokinin -45C>T polymorphism; also in the presence of the cholecystokinin-A receptor TC/CC genotype |
| De Luca et al. [56] | Genotyping association study of HOMER1 gene and development of psychotic symptoms in PD | 131 sporadic PD patients from southern Italy | allele A of the rs4704559 marker linked to increased susceptibility to psychotic symptoms in PD p = 0.004 |
| Wang et al. [57] | Meta-analysis focused on the association between BDNF G196A (Val66Met) polymorphism and cognitive impairment in PD patients | Six studies involving 532 PD patients and 802 controls | G196A (Val66Met) polymorphism is significantly associated with cognitive impairment in PD, especially in Caucasian populations |
| Gatto et al. [58] | Genotyping multivariate study of SNP-s and Impulse control disorder(ICD) | 276 patients with PD | OPRK1 polymorphism rs702764 significantly predicted incident ICD behaviour; polymorphisms of the DRD1, DRD2, DRD3, HTR2A and GRIN2B genes also associated with ICD in patients with PD |
| Cormier-Dequaire et al. [59] | Multicenter case-control genotype association study | 172 French Caucasian patients and 132 controls | No variant was significantly associated with impulse control disorders or related behaviors after correction for multiple testing, although the 2 top variants were close to significant (OPRM1 rs179991, p = 0.0013; Bonferroni adjusted p = 0.065; DAT1 40-base pair variable number tandem repeat; p = 0.0021; Bonferroni adjusted p = 0.105) |
| Zainal Abidin et al. [60] | Multivariate association study of SNPs and increased risk of ICD development | 52 Malaysian PD patients with 39 without ICB | DRD1 (rs4532 and rs4867798), DRD2/ANKK1 rs1800497] and glutamate (GRIN2B rs7301328) receptor genes confer increased risk of ICD development among PD patients |
| Lee et al. [61] | Genotype association study genotypes DRD3 p.S9G, DRD2 Taq1A, GRIN2B c.366C>G, c.2664C>T and c.-200T>G, and the promoter region of the serotonin transporter gene (5-HTTLPR) with the incidence of ICD’s | 404 Korean PD patients and 559 healthy controls | Variants of DRD3 p.S9G and GRIN2B c.366C>G may be associated with the appearance of ICD in PD |
| Castro-Martinez et al. [62] | Genotype association study of rs6280 DRD3 single nucleotide variation (SNV) (Ser9Gly) and incidence of ICD’s | 199 Hispanic PD patients | Behavioral addictions in PD are associated with an early onset of the disease, the rs6280 DRD3 SNV and the type of dopamine agonist |
| Arbouw et al. [63] | Genotype association study of pharmacogenetic determinants for the discontinuation of non-ergoline dopamine agonists | 90 Dutch PD patients | This study identified apomorphine use and levodopa dosages between 500 and 1000 mg as non-genetic and the 15× DRD2 CA repeat allele as genetic determinants for the discontinuation of non-ergoline DA treatment in patients with PD |
| Liu et al. [64] | Genotype association study focusing on the response to pramipexole in PD patients | 30 Chinese PD patients | DRD3 Ser9Gly polymorphisms are significantly associated with the therapeutic efficacy of pramipexole in Chinese patients with PD |
| Xu et al. [65] | Genotype association study of DRD2 CA n-STR and DRD3 Ser9Gly polymorphisms with Parkinson’s disease and response to dopamine agonists | 168 PD patients of Chinese origin and 182 controls | Genotype in DRD3 Ser9Gly was the main factor determining different doses of DAs and PD patients carrying Gly/Gly genotype require higher doses of pramipexole for effective treatment |
| Zhi et al. [66] | Genotype association study of DRD3 Ser9Gly polymorphism and depression severity in Parkinson’s disease | 61 PD patients of Chinese origin and 47 controls | D3 gene Ser9Gly polymorphism might be associated with the severity of depression characterized by anhedonia in PD patients |
| Paus et al. [67] | Genotype association study of the DRD2 TaqIA polymorphism and demand of dopaminergic medication in Parkinson’s disease | 607 PD German patients of varied origin | DRD2 TaqIA polymorphism alone has no pivotal role for interindividual variability of dopaminergic requirement in PD |
| McDonell et al. [68] | Genotype association study of DRD2 and DRD3 gene polymorphisms and the incidence of ICD | 28 USA PD patients | Patients with the rs1800497 Taq1A (A1) polymorphism (A1/A1 or A1/A2: 11 subjects) showed improved proficiency to suppress impulsive actions when on DAAg; conversely, patients with the A2/A2 allele (14 patients) became less proficient at suppressing incorrect response information on DA; Polymorphisms in rs6277 and rs6280 were not associated with a differential medication response |
| Erga et al. [69] | Whole-exome sequencing study of 17 genes connected to ICD | 119 Norwegian PD patients | Eleven SNPs were associated with ICDs, and the four SNPs with the most robust performance significantly increased ICD predictability (AUC = 0.81, 95% CI 0.73–0.90) compared to clinical data alone (DA use and age; AUC = 0.65, 95% CI 0.59–0.78); The strongest predictive factors were rs5326 in DRD1, which was associated with increased odds of ICDs, and rs702764 in OPRK1, which was associated with decreased odds of ICDs |
| Corvol et al. [70] | Randomized crossover clinical trial focused on the effect of COMT Val158Met polymorphism to the entacapone response in PD patients | 58 French PD patients | The COMTHH genotype in PD patients enhances the effect of entacapone on the pharmacodynamics and pharmacokinetics of levodopa |
| Lee et al. [71] | Genotype association study of COMT genotypes and entacapone efficacy | 65 PD patients with entacapone therapy | After entacapone treatment, the mean of the percentage reduction of daily levo-dopa dose for each individual was significant in patients with HH and HL genotype of COMT |
| Chong et al. [72] | Genotype association study of COMT genotypes and tolcapone efficacy | 24 PD patients who completed tolca-pone clinical trials | no substantial effect of COMT genotype relative to clinical response to the COMT inhibitor tolcapone |
| Trenkwalder et al. [73] | Randomized double-blind crossover multicenter study | 117 German PD patients | Patients with high-activity COMT genotypes Val/Met and Val/Val had a reduced “off” time |
| Liu et al. [74] | Genotyping analysis of UGT1A, UGT2B SNPs | 148 liver samples (125 of European and 23 of African descent) | UGT SNP variants contribute to variability in the metabolism of certain drugs and can lead to adverse effects due to inadequate metabolism |
| Yamanaka et al. [75] | Genotyping analysis of UGT1A9 SNPs | 87 Japanese, 50 Caucasian and 50 African-American participants | The mutant allele with one base insertion in the promoter region of the UGT1A9 gene would alter the level of enzyme expression and the metabolism of those drugs that are substrates of UGT1A9 |
| Ferrari et al. [76] | Genotyping association study of UGT1A9 SNPs and COMT inhibitor induced toxicity | 52 Parkinson’s disease (PD) patients on COMT inhibitors without evidence of adverse reactions and 11 PD patients who had been withdrawn from COMT inhibitors due to adverse reactions | In PD patients UDP-glucuronosyltransferase 1A9 genotypes are associated with adverse reactions to COMT inhibitors |
| Masellis et al. [77] | Genotyping association study of DRD2 SNPs and response to rasagiline | 692 available DNA samples from a placebo-controlled clinical trial of the monoamine oxidase B inhibitor | rs2283265 and rs1076560 were found to be significantly associated with a favourable peak response to rasagiline at 12 weeks in early Parkinson’s disease |
| References | Study Design | Methodology/Specific Mutation Studied | Main Finding |
|---|---|---|---|
| Nalls et al. [92] | Multicenter population-based modelling study | 367 PD patients and 165 controls for the model Model tested on 825 PD patients and 261 controls | The developed model for disease classification could distinguish participants with PD and controls with high sensitivity (0·834, 95% CI 0·711–0·883) and specificity (0·903, 95% CI 0·824–0·946) |
| Shu et al. [93] | Meta-analysis | 66 studies comprising 23,402 PD patients. Association of LRRK2 with clinical features of PD | Clinical heterogeneity in LRRK2-associated PD among different variants, especially for G2019S and G2385R |
| Yahalom et al. [94] | Genotype association study of G2019S LRRK2 mutation with regards to L-dopa induced dyskinesias | 349 Israeli PD patients (222 Askenazi-Jewish) | The prevalence of LID was non-significantly higher among carriers (22/33, 66.7%) than non-carriers (168/316, 53.2%, p = 0.15) |
| Cacabelos et al. [95] | Multivariate association study of atremorin and LRRK2 variants | 183 PD patients split into 2 categories: 135 drug-free patients (DF-PD) who had never before received any anti-parkinsonian medication 48 patients chronically treated with anti-parkinsonian drugs (CT-PD) (>1 y) (n = 48) | LRRK2 associated PD patients had a more robust response to the compound atremorin |
| Nishioka et al. [96] | Genome wide association study | 103 Japanese patients with autosomal dominant PD [43 male and 60 female with a mean age at onset of 50.9613.9 years (6SD)] who had at least one affected individual within one degree of separation, and 71 patients (29 male and 42 female with 37.7613.0 years) with sporadic PD | SNP’s in the SNCA gene are linked to an increased risk of developing PD |
| Kantor et al. [97] | Experimental gene therapy study | Human induced pluripotent stem cell (hiPSC)-derived dopaminergic neurons from a PD patient with the SNCA triplication | DNA hypermethylation at SNCA intron 1 allows an effective and sufficient tight downregulation of SNCA expression levels |
| Jankovic et al. [98] | Multicenter, randomized, double-blind, placebo-controlled, multiple ascending-dose trial | 80 Caucassian PD patients; Effect of PRX002 | PRX002 immunotherapy was capable of engaging peripheral α-synuclein in patients with PD. |
| Silveira et al. [99] | Single center, randomized, double-blind, placebo-controlled trial | 75 individuals with mild to moderate PDD; effect of Ambroxol on GCase | Ambroxol could raise GCase and could therefore be a disease-modifying treatment for PDD |
| Alcalay et al. [100] | Multivariate genotyping analysis of GBA SNPs | 517 PD patients and 252 controls with and without GBA mutations; LRRK2 | Low glucocerebrosidase enzymatic activity may be a risk factor for Parkinson’s disease |
| Lesage et al. [101] | Genotyping analysis of GBA mutations | 525 European (mostly French) PD patients from unrelated multiplex families, 605 patients with apparently sporadic PD and 391 ethnically matched controls | Higher incidence of L-dopa induced dyskinesias in GBA-PD patients |
| Zhang et al. [102] | Genotyping analysis + meta-analysis | 1147 Chinese PD patients for L444P detection; Subsequent comparison between 646 PD patients with GBA mutations and 10344 PD patients without GBA mutations worldwide | Phenotypes of PD patients with GBA mutations are different from GBA non-carriers |
| Kasten et al. [103] | Systematic review | 3652 citations; based on fully curated phenotypic and genotypic data on >1100 patients with recessively inherited PD because of 221 different disease-causing mutations in Parkin, PINK1 or DJ1 | Mutations in the PRKN gene can lead to early onset PD, characterized by a clinically typical form of PD that is often associated with dystonia and dyskinesia |
| Khan et al. [104] | A phenotypic study of a large case series | 24 patients with mutations in the parkin gene | Dyskinesias can occur early on in the course of the disease with very low doses of L-dopa |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuletić, V.; Rački, V.; Papić, E.; Peterlin, B. A Systematic Review of Parkinson’s Disease Pharmacogenomics: Is There Time for Translation into the Clinics? Int. J. Mol. Sci. 2021, 22, 7213. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137213
Vuletić V, Rački V, Papić E, Peterlin B. A Systematic Review of Parkinson’s Disease Pharmacogenomics: Is There Time for Translation into the Clinics? International Journal of Molecular Sciences. 2021; 22(13):7213. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137213
Chicago/Turabian StyleVuletić, Vladimira, Valentino Rački, Eliša Papić, and Borut Peterlin. 2021. "A Systematic Review of Parkinson’s Disease Pharmacogenomics: Is There Time for Translation into the Clinics?" International Journal of Molecular Sciences 22, no. 13: 7213. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137213