
Ipragliflozin Ameliorates Diabetic Nephropathy Associated with Perirenal Adipose Expansion in Mice

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
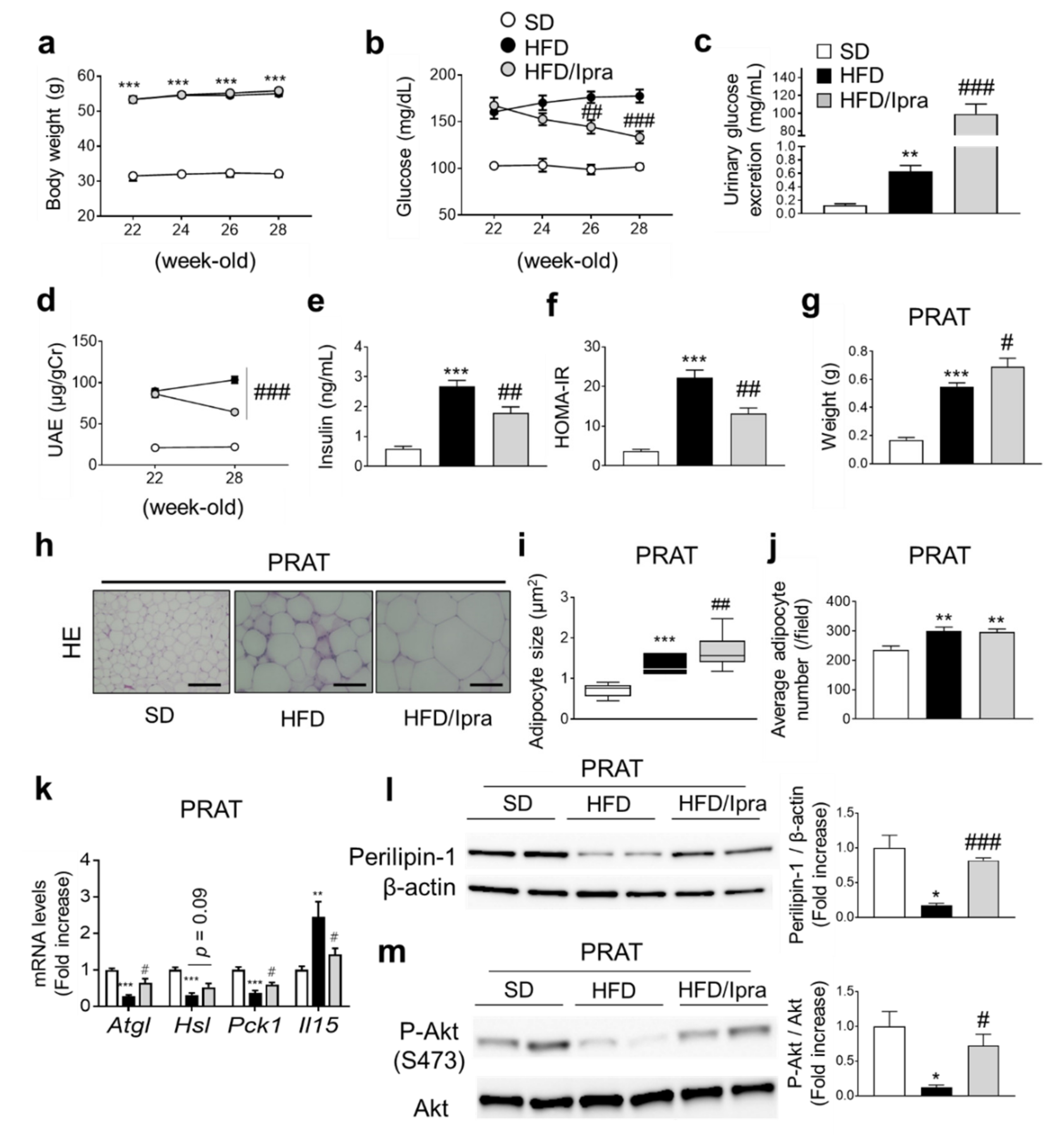
2.1. Ipra Increased Adipocyte Size While Attenuating Insulin Resistance in PRAT of HFD-Fed Mice
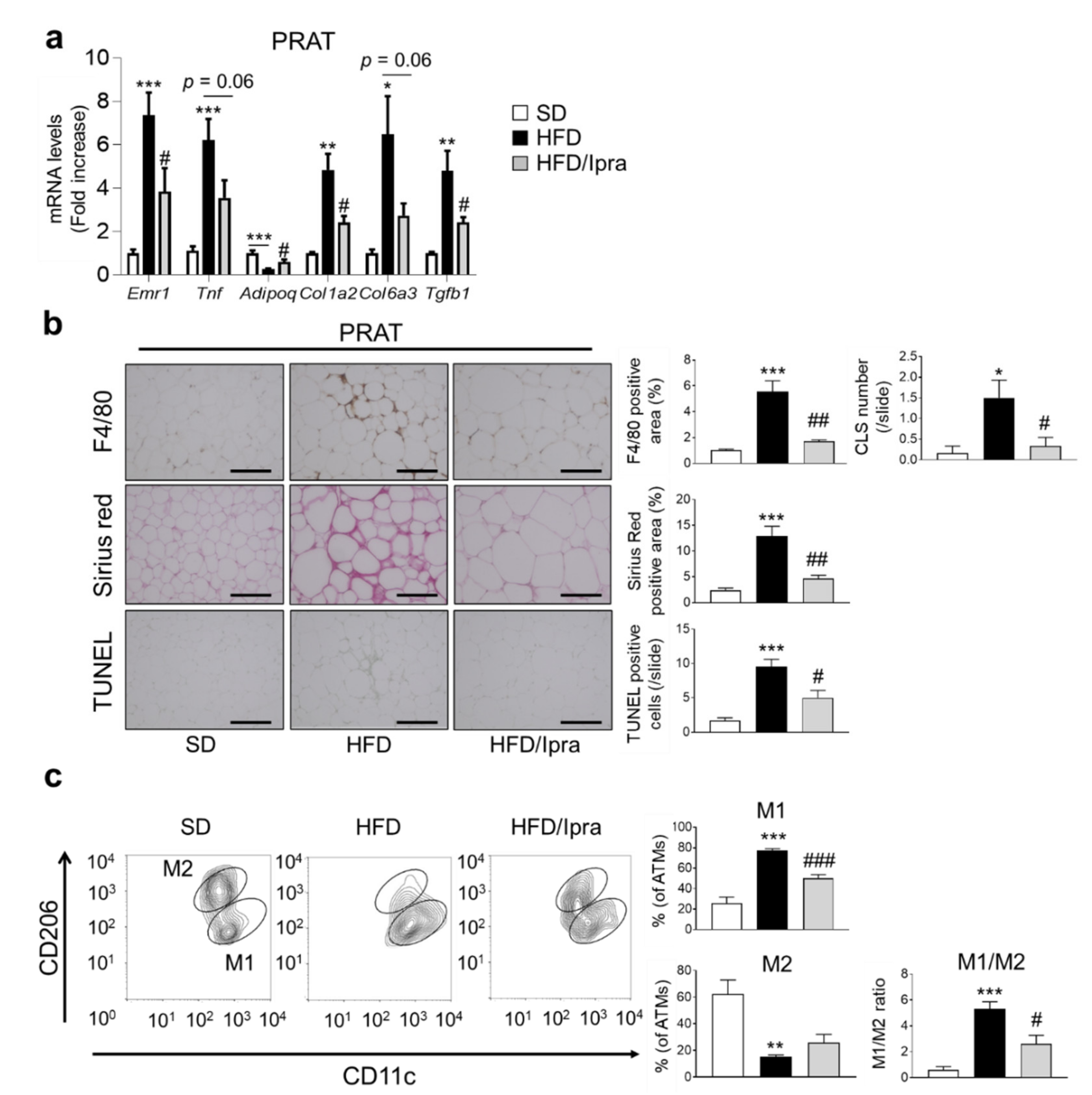
2.2. Ipra Suppressed Inflammation, Fibrosis, and Cell Death in PRAT of HFD-Fed Mice and Shifted Macrophage Properties toward an Anti-Inflammatory State
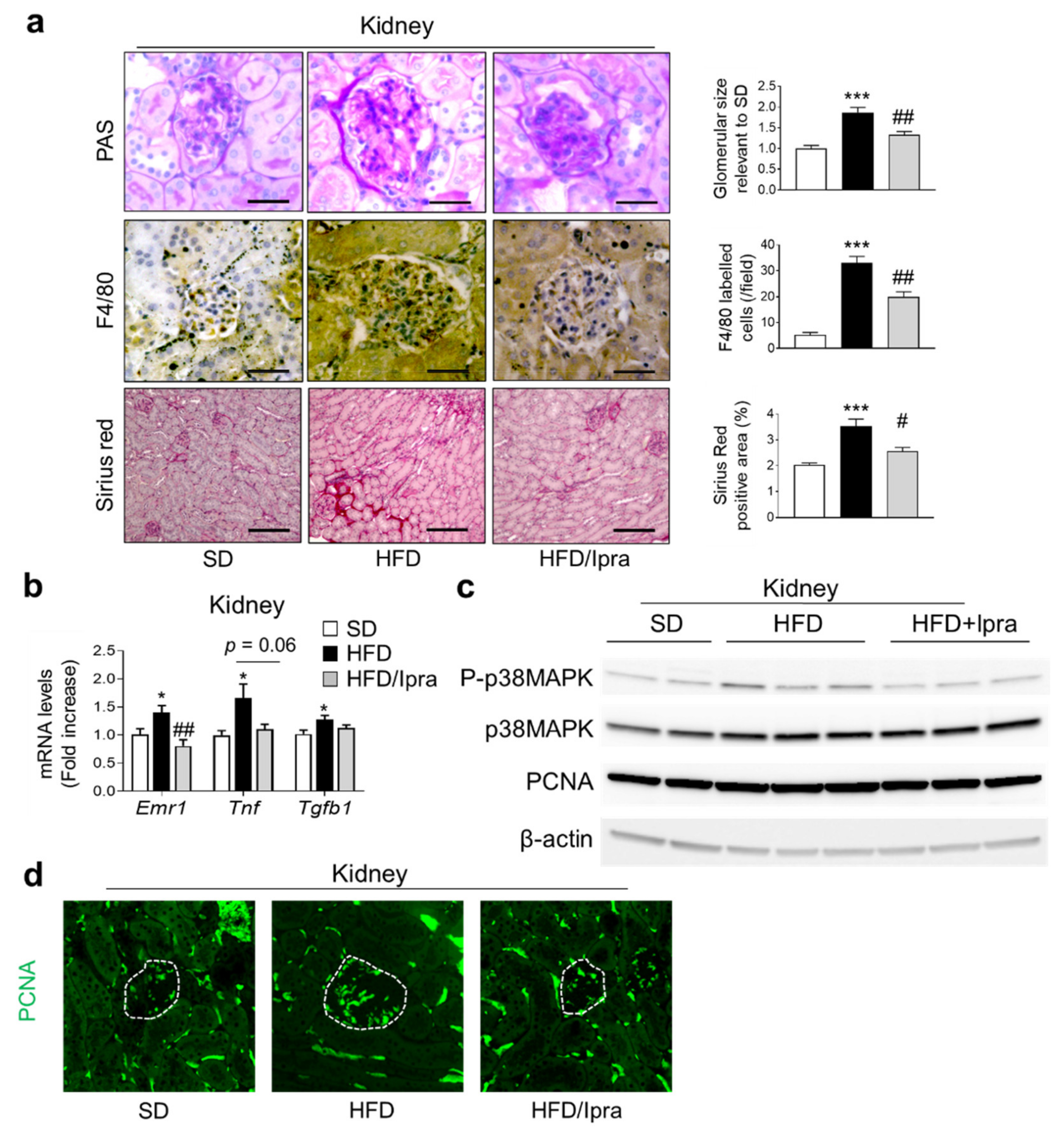
2.3. Ipra Inhibited Macrophage Infiltration and Proliferative Signals in the Kidney
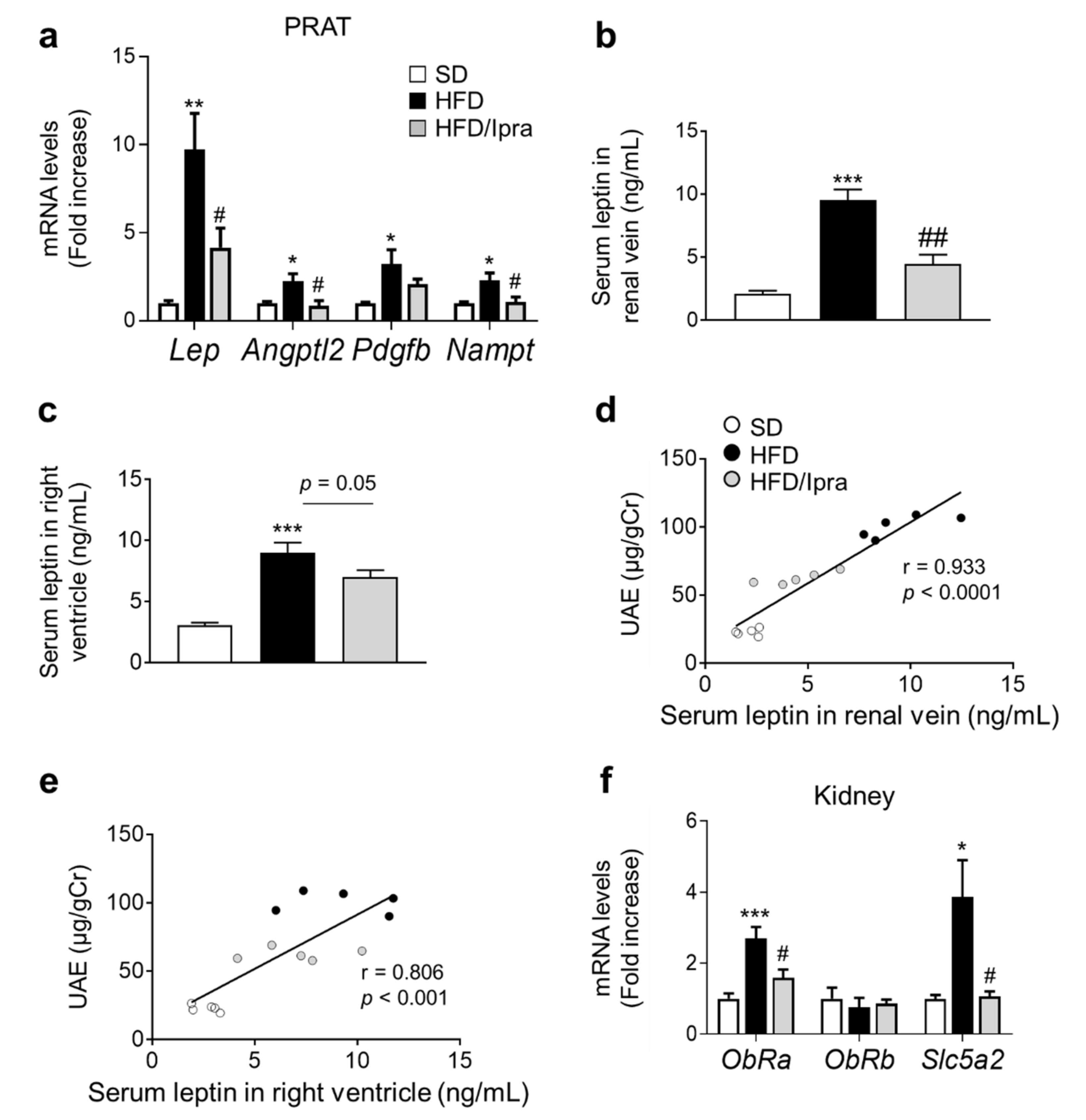
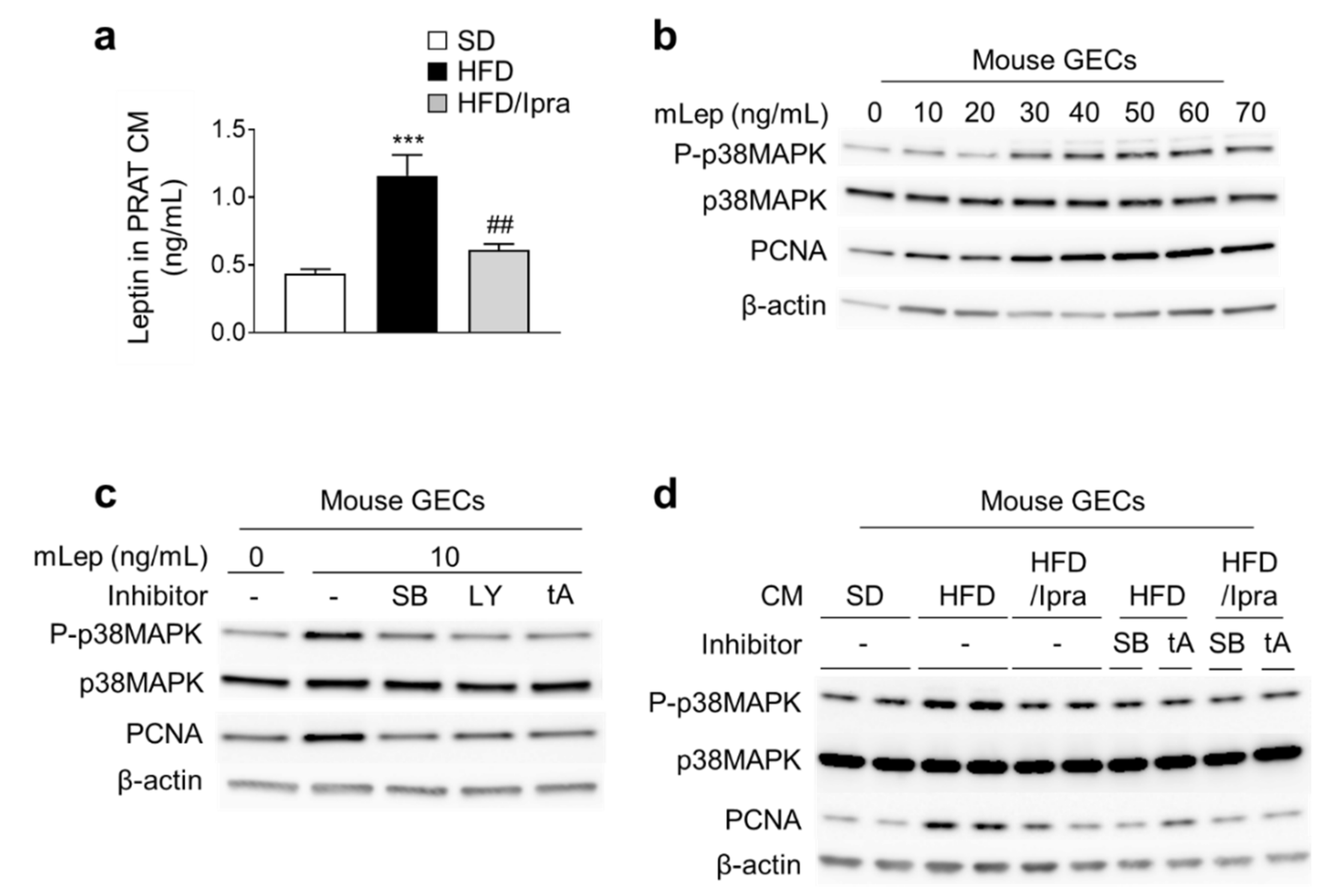
2.4. Reduced Leptin Gene Expression in PRAT and Leptin Concentrations in Renal Veins in Ipra-Treated Mice
2.5. Ipra Suppressed Leptin Secretion from PRAT and Proliferative Signaling in Mouse GECs
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Protocol
4.2. Materials
4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; de Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic kidney disease: A report from an ADA Consensus Conference. Am. J. Kidney Dis. 2014, 64, 510–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.H.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; et al. Renal Hemodynamic Effect of Sodium-Glucose Cotransporter 2 Inhibition in Patients with Type 1 Diabetes Mellitus. Circulation 2014, 129, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Kamezaki, M.; Kusaba, T.; Komaki, K.; Fushimura, Y.; Watanabe, N.; Ikeda, K.; Kitani, T.; Yamashita, N.; Uehara, M.; Kirita, Y.; et al. Comprehensive renoprotective effects of ipragliflozin on early diabetic nephropathy in mice. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, K.; Takata, T.; Sugihara, T.; Matono, T.; Koda, M.; Kanda, T.; Taniguchi, S.; Ida, A.; Mae, Y.; Yamamoto, M.; et al. Ipragliflozin Ameliorates Endoplasmic Reticulum Stress and Apoptosis through Preventing Ectopic Lipid Deposition in Renal Tubules. Int. J. Mol. Sci. 2019, 21, 190. [Google Scholar] [CrossRef] [Green Version]
- Montani, J.-P.; Carroll, J.F.; Dwyer, T.M.; Antic, V.; Yang, Z.; Dulloo, A. Ectopic fat storage in heart, blood vessels and kidneys in the pathogenesis of cardiovascular diseases. Int. J. Obes. 2004, 28, S58–S65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, N.; Han, F.; Wang, M.; Huang, N.; Zhao, J.; Liu, X.; Sun, X. Perirenal fat associated with microalbuminuria in obese rats. Int. Urol. Nephrol. 2014, 46, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Geraci, G.; Zammuto, M.M.; Mattina, A.; Zanoli, L.; Geraci, C.; Granata, A.; Nardi, E.; Fatuzzo, P.M.; Cottone, S.; Mulè, G. Para-perirenal distribution of body fat is associated with reduced glomerular filtration rate regardless of other indices of adiposity in hypertensive patients. J. Clin. Hypertens. 2018, 20, 1438–1446. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, T.M.; Mizelle, H.L.; Cockrell, K.; Buhner, P. Renal sinus lipomatosis and body composition in hypertensive, obese rabbits. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1995, 19, 869–874. [Google Scholar]
- Liu, Y.; Wang, L.; Luo, M.; Chen, N.; Deng, X.; He, J.; Zhang, L.; Luo, P.; Wu, J. Inhibition of PAI-1 attenuates perirenal fat inflammation and the associated nephropathy in high-fat diet-induced obese mice. Am. J. Physiol. Metab. 2019, 316, E260–E267. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, M.; Liu, P.; Wang, Y.; Zhang, H.; Li, H.; Yang, S.; Song, Y.; Yin, Y.; Gao, L.; et al. Telmisartan Ameliorates Nephropathy in Metabolic Syndrome by Reducing Leptin Release from Perirenal Adipose Tissue. Hypertension 2016, 68, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyachi, Y.; Tsuchiya, K.; Shiba, K.; Mori, K.; Komiya, C.; Ogasawara, N.; Ogawa, Y. A reduced M1-like/M2-like ratio of macrophages in healthy adipose tissue expansion during SGLT2 inhibition. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shiba, K.; Tsuchiya, K.; Komiya, C.; Miyachi, Y.; Mori, K.; Shimazu, N.; Yamaguchi, S.; Ogasawara, N.; Katoh, M.; Itoh, M.; et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018, 8, 2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiya, C.; Tsuchiya, K.; Shiba, K.; Miyachi, Y.; Furuke, S.; Shimazu, N.; Yamaguchi, S.; Kanno, K.; Ogawa, Y. Ipragliflozin Improves Hepatic Steatosis in Obese Mice and Liver Dysfunction in Type 2 Diabetic Patients Irrespective of Body Weight Reduction. PLoS ONE 2016, 11, e0151511. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Tsuchiya, K.; Nakamura, S.; Miyachi, Y.; Shiba, K.; Ogawa, Y.; Kitamura, K. Ipragliflozin-induced adipose expansion inhibits cuff-induced vascular remodeling in mice. Cardiovasc. Diabetol. 2019, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.W.; Jiang, W.; Reich, C.F.; Pisetsky, D.S. The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef]
- Johnson, R.J. What mediates progressive glomerulosclerosis? The glomerular endothelium comes of age. Am. J. Pathol. 1997, 151, 1179–1181. [Google Scholar]
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Gal, C.S.-L.; Raufaste, D.; Brossard, G.; Pouzet, B.; Marty, E.; Maffrand, J.-P.; Le Fur, G. Characterization and localization of leptin receptors in the rat kidney. FEBS Lett. 1997, 404, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; Hamann, A.; Han, D.C.; Helmchen, U.; Thaiss, F.; Ziyadeh, F.N.; Stahl, R.A. Leptin stimulates proliferation and TGF-beta expression in renal glomerular endothelial cells: Potential role in glomerulosclerosis [seecomments]. Kidney Int. 1999, 56, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodavarapu, H.; Grobe, N.; Somineni, H.K.; Salem, E.S.B.; Madhu, M.; Elased, K.M. Rosiglitazone Treatment of Type 2 Diabetic db/db Mice Attenuates Urinary Albumin and Angiotensin Converting Enzyme 2 Excretion. PLoS ONE 2013, 8, e62833. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Takashi, Y.; Takahashi, H.; Motonaga, R.; Tanabe, M. Renoprotective Effects of DPP-4 Inhibitors. Antioxidants 2021, 10, 246. [Google Scholar] [CrossRef]
- Wang, C.; Li, L.; Liu, S.; Liao, G.; Li, L.; Chen, Y.; Cheng, J.; Lu, Y.; Liu, J. GLP-1 receptor agonist ameliorates obesity-induced chronic kidney injury via restoring renal metabolism homeostasis. PLoS ONE 2018, 13, e0193473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, M.; Furuichi, K.; Toyama, T.; Kitajima, S.; Hara, A.; Kitagawa, K.; Iwata, Y.; Sakai, N.; Takamura, T.; Yoshimura, M.; et al. Long-Term Outcomes of Japanese Type 2 Diabetic Patients with Biopsy-Proven Diabetic Nephropathy. Diabetes Care 2013, 36, 3655–3662. [Google Scholar] [CrossRef] [Green Version]
- Ballermann, B.J. A role for leptin in glomerulosclerosis? Kidney Int. 1999, 56, 1154–1155. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.X.; Sun, W.; Kong, X.Q. Perirenal Fat: A Unique Fat Pad and Potential Target for Cardiovascular Disease. Angiology 2019, 70, 584–593. [Google Scholar] [CrossRef]
- Lau, W.B.; Ohashi, K.; Wang, Y.; Ogawa, H.; Murohara, T.; Ma, X.-L.; Ouchi, N. Role of Adipokines in Cardiovascular Disease. Circ. J. 2017, 81, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Ndisang, J.F.; Jadhav, A.; Mishra, M. The heme oxygenase system suppresses perirenal visceral adiposity, abates renal inflammation and ameliorates diabetic nephropathy in Zucker diabetic fatty rats. PLoS ONE 2014, 9, e87936. [Google Scholar]
- Morley, T.S.; Xia, J.Y.; Scherer, P.E. Selective enhancement of insulin sensitivity in the mature adipocyte is sufficient for systemic metabolic improvements. Nat. Commun. 2015, 6, 7906. [Google Scholar] [CrossRef]
- Tanaka, M.; Ikeda, K.; Suganami, T.; Komiya, C.; Ochi, K.; Shirakawa, I.; Hamaguchi, M.; Nishimura, S.; Manabe, I.; Matsuda, T.; et al. Macrophage-inducible C-type lectin underlies obesity-induced adipose tissue fibrosis. Nat. Commun. 2014, 5, 4982. [Google Scholar] [CrossRef] [Green Version]
- Okuno, Y.; Fukuhara, A.; Hashimoto, E.; Kobayashi, H.; Kobayashi, S.; Otsuki, M.; Shimomura, I. Oxidative Stress Inhibits Healthy Adipose Expansion Through Suppression of SREBF1-Mediated Lipogenic Pathway. Diabetes 2018, 67, 1113–1127. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef]
- Brasaemle, D.L. Thematic review series: Adipocyte Biology. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 2007, 48, 2547–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Dasu, M.R.; Park, S.H.; Jialal, I.; Devaraj, S.; Dasu, M.R.; Park, S.H.; Jialal, I. Increased levels of ligands of Toll-like receptors 2 and 4 in type 1 diabetes. Diabetologia 2009, 52, 1665–1668. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ma, J.; Wang, P.; Corpuz, T.M.; Panchapakesan, U.; Wyburn, K.R.; Chadban, S.J. HMGB1 Contributes to Kidney Ischemia Reperfusion Injury. J. Am. Soc. Nephrol. 2010, 21, 1878–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Chadban, S.J.; Zhao, C.Y.; Chen, X.; Kwan, T.; Panchapakesan, U.; Pollock, C.A.; Wu, H. TLR4 Activation Promotes Podocyte Injury and Interstitial Fibrosis in Diabetic Nephropathy. PLoS ONE 2014, 9, e97985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Wu, H.; Zhao, C.Y.; Panchapakesan, U.; Pollock, C.; Chadban, S.J. Requirement for TLR2 in the development of albuminuria, inflammation and fibrosis in experimental diabetic nephropathy. Int. J. Clin. Exp. Pathol. 2014, 7, 481–495. [Google Scholar]
- Mudaliar, H.; Pollock, C.; Komala, M.G.; Chadban, S.; Wu, H.; Panchapakesan, U. The role of Toll-like receptor proteins (TLR) 2 and 4 in mediating inflammation in proximal tubules. Am. J. Physiol. Physiol. 2013, 305, F143–F154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ma, J.; Kwan, T.; Stribos, E.G.D.; Messchendorp, A.L.; Loh, Y.W.; Wang, X.; Paul, M.; Cunningham, E.C.; Habib, M.; et al. Blockade of HMGB1 Attenuates Diabetic Nephropathy in Mice. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okuma, H.; Mori, K.; Nakamura, S.; Sekine, T.; Ogawa, Y.; Tsuchiya, K. Ipragliflozin Ameliorates Diabetic Nephropathy Associated with Perirenal Adipose Expansion in Mice. Int. J. Mol. Sci. 2021, 22, 7329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147329
Okuma H, Mori K, Nakamura S, Sekine T, Ogawa Y, Tsuchiya K. Ipragliflozin Ameliorates Diabetic Nephropathy Associated with Perirenal Adipose Expansion in Mice. International Journal of Molecular Sciences. 2021; 22(14):7329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147329
Chicago/Turabian StyleOkuma, Hideyuki, Kentaro Mori, Suguru Nakamura, Tetsuo Sekine, Yoshihiro Ogawa, and Kyoichiro Tsuchiya. 2021. "Ipragliflozin Ameliorates Diabetic Nephropathy Associated with Perirenal Adipose Expansion in Mice" International Journal of Molecular Sciences 22, no. 14: 7329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147329