
How to Obtain a Mega-Intestine with Normal Morphology: In Silico Modelling of Postnatal Intestinal Growth in a Cd97-Transgenic Mouse

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
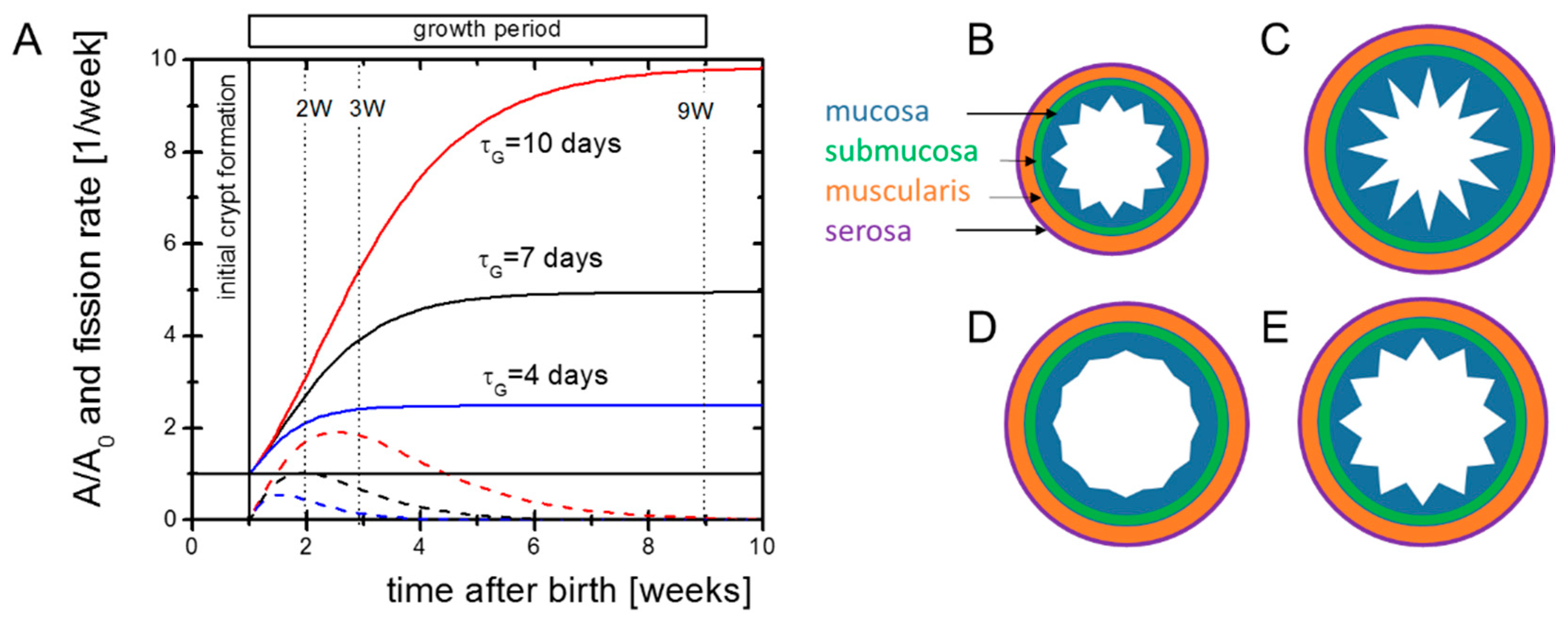
2.1. Properties of Postnatal Intestinal Growth
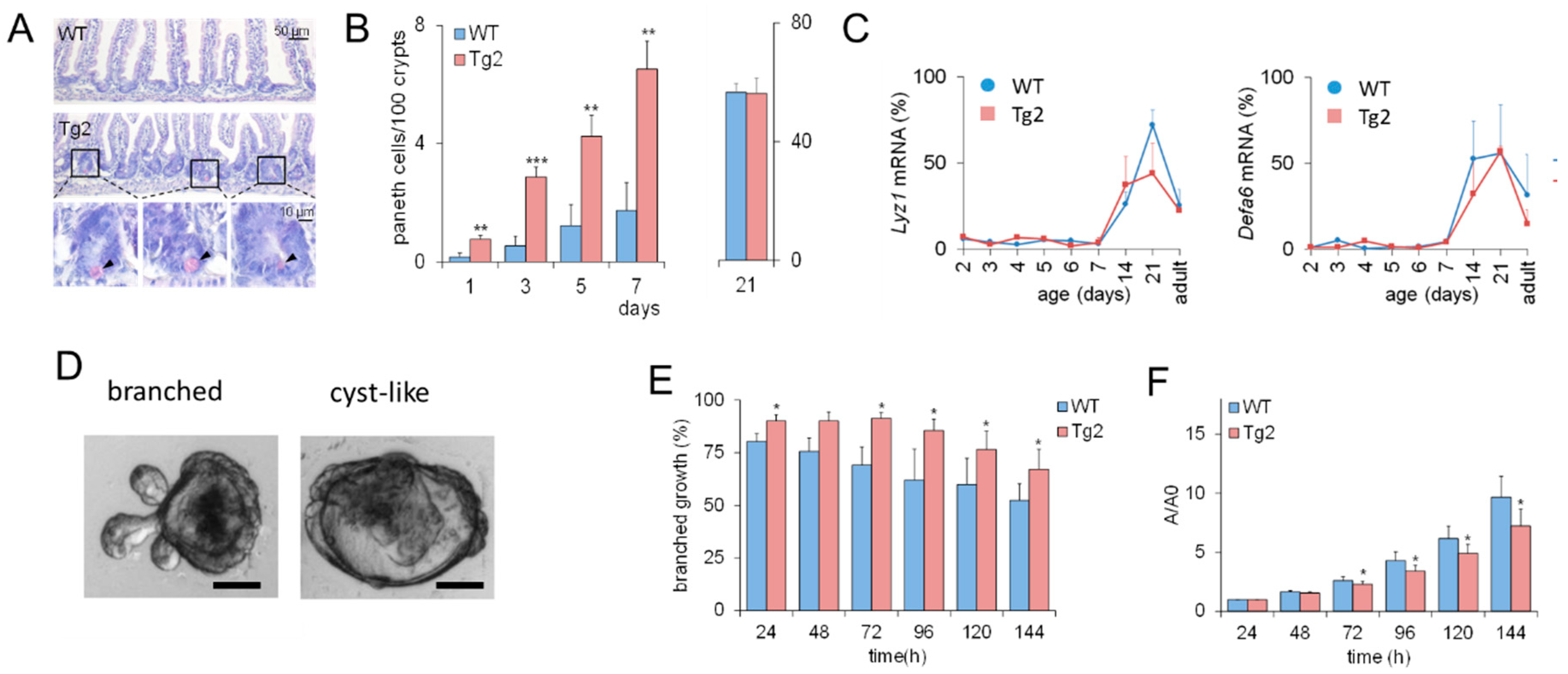
2.2. Accelerated Crypt Formation and Paneth Cell (PC) Specification in Tg2 Mice
2.3. Mature Growth Characteristics of Organoids Derived from 2-Week-Old Tg2 Mice
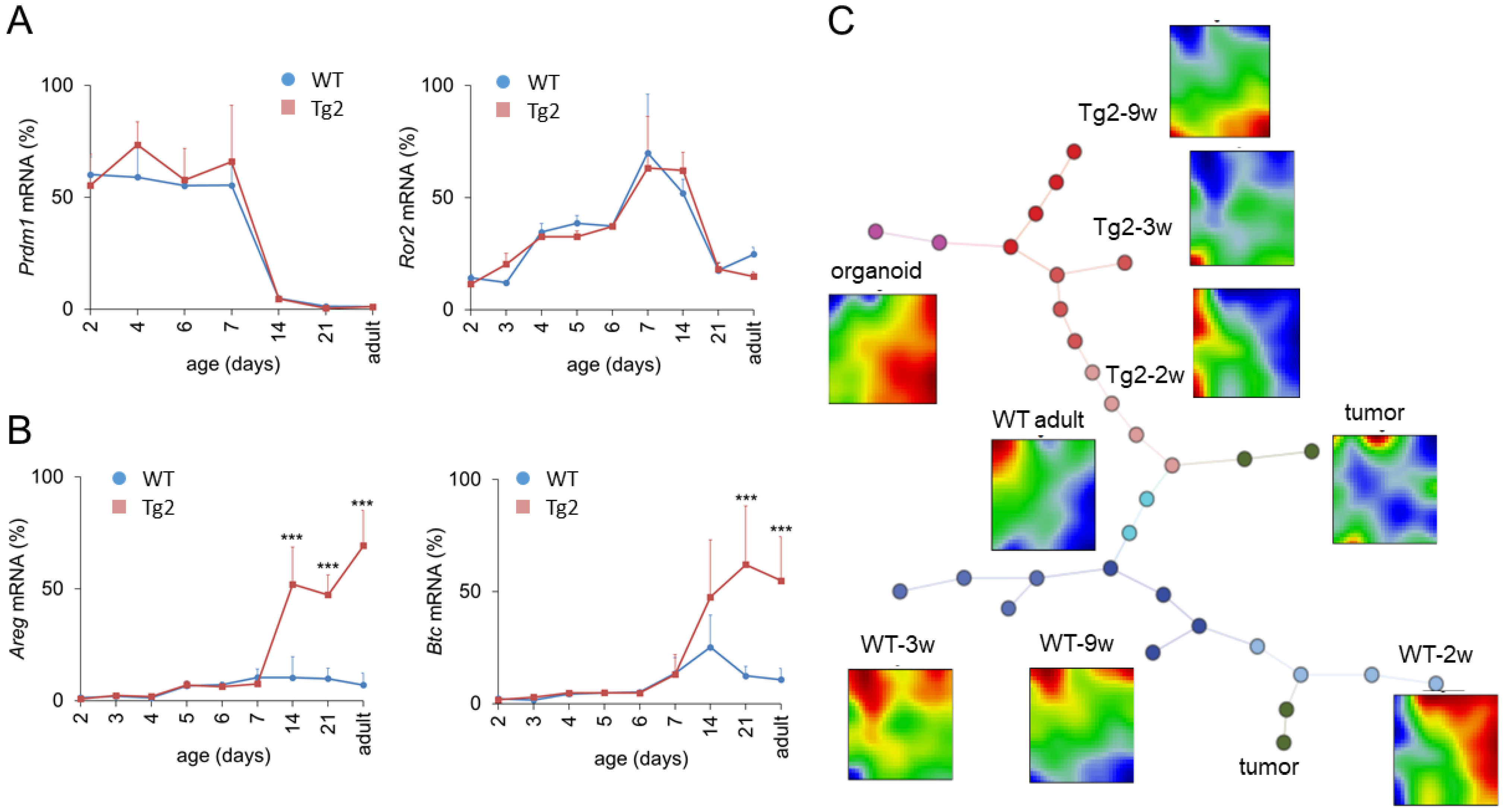
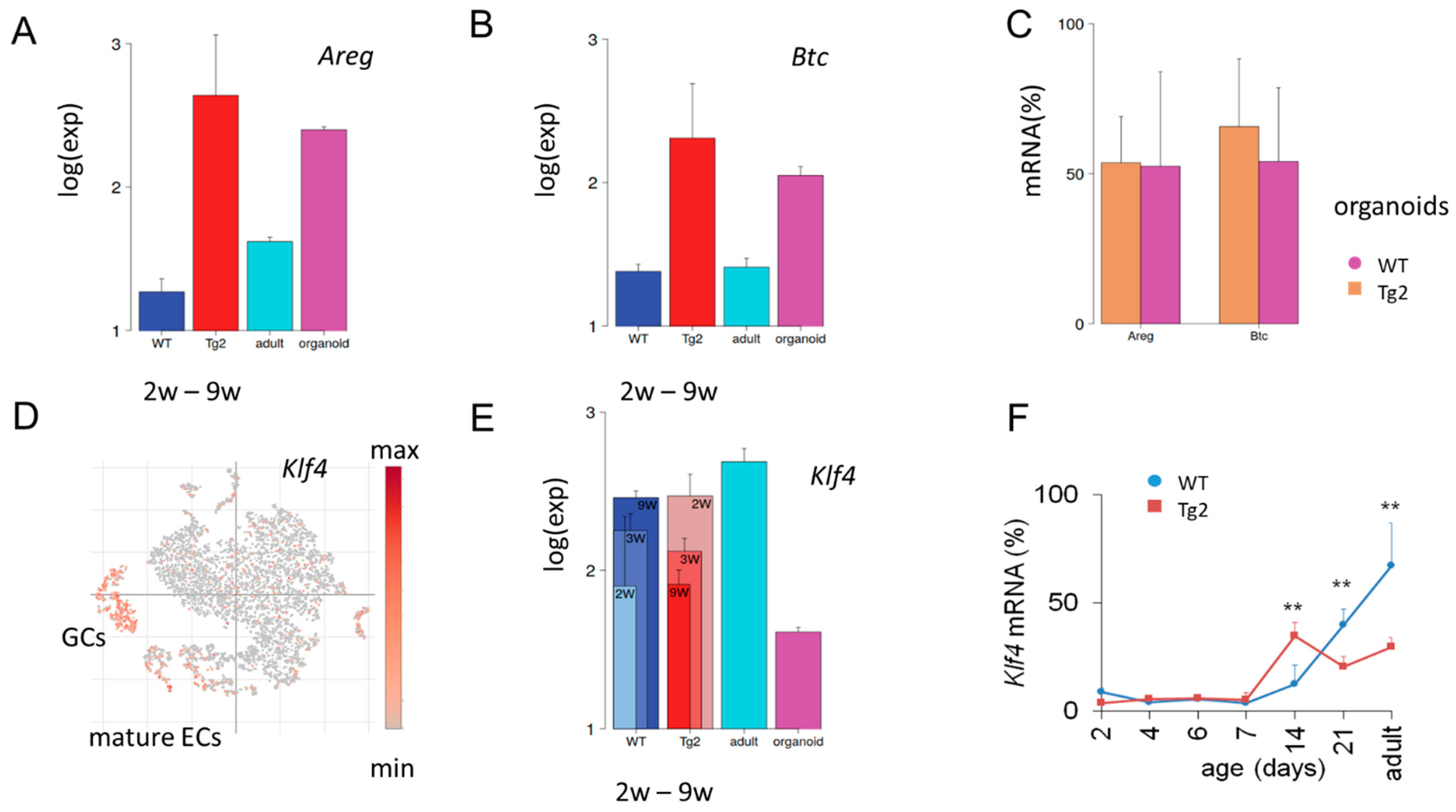
2.4. Changed Expression of Growth Factors in Tg2 Mice after Prdm1 Downregulation
2.5. Global Gene Expression Changes in Tg2 Mice
2.6. Specific Gene Expression in the Intestinal Epithelium in Tg2 Mice
2.7. Early Transient Activation of the Differentiation Marker Klf4 in Tg2 Mice
2.8. A Computational Model of Neonatal Crypt Formation
2.9. Basic Model Simulations of Crypt Formation
2.10. Simulation of Accelerated Crypt Formation
3. Discussion
4. Materials and Methods
4.1. Model of Neonatal Crypt Fission
4.2. Model Computations
4.3. Data Analysis
4.4. Mice and Ethics Statement
4.5. Intestinal Morphology
4.6. Organoid 3D Live Imaging
4.7. Microarray and SOM Analysis
4.8. qRT-PCR
4.9. Statistics Tests
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, S.; Ingber, D. The structural and mechanical complexity of cell-growth control. Nat. Cell Biol. 1999, 1, E131–E138. [Google Scholar] [CrossRef]
- Puliafito, A.; Hufnagel, L.; Neveu, P.; Streichan, S.; Sigal, A.; Fygenson, D.K.; Shraiman, B.I. Collective and single cell behavior in epithelial contact inhibition. Proc. Natl. Acad. Sci. USA 2012, 109, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, R.D.; Sheng, H.M.; Shao, J.Y.; Thompson, E.A.; Ko, T.C. Intestinal cell cycle regulations. Interactions of cyclin D1, Cdk4, and p21Cip1. Ann. Surg. 1996, 223, 620–627. [Google Scholar] [CrossRef]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [Green Version]
- Galle, J.; Hoffmann, M.; Aust, G. From single cells to tissue architecture—A bottom-up approach to modelling the spatio-temporal organisation of complex multi-cellular systems. J. Math. Biol. 2009, 58, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Dehmer, J.J.; Garrison, A.P.; Speck, K.E.; Dekaney, C.M.; Van Landeghem, L.; Sun, X.; Henning, S.J.; Helmrath, M.A. Expansion of intestinal epithelial stem cells during murine development. PLoS ONE 2011, 6, e27070. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Aust, G.; Kerner, C.; Gonsior, S.; Sittig, D.; Schneider, H.; Buske, P.; Scholz, M.; Dietrich, N.; Oldenburg, S.; Karpus, O.N.; et al. Mice overexpressing CD97 in intestinal epithelial cells provide a unique model for mammalian postnatal intestinal cylindrical growth. Mol. Biol. Cell. 2013, 24, 2256–2268. [Google Scholar] [CrossRef]
- Tamura, A.; Kitano, Y.; Hata, M.; Katsuno, T.; Moriwaki, K.; Sasaki, H.; Hayashi, H.; Suzuki, Y.; Noda, T.; Furuse, M.; et al. Megaintestine in claudin-15-deficient mice. Gastroenterology 2008, 134, 523–534. [Google Scholar] [CrossRef]
- Troyer, K.L.; Luetteke, N.C.; Saxon, M.L.; Qiu, T.H.; Xian, C.J.; Lee, D.C. Growth retardation, duodenal lesions, and aberrant ileum architecture in triple null mice lacking EGF, amphiregulin, and TGF-alpha. Gastroenterology 2001, 121, 68–78. [Google Scholar] [CrossRef]
- Riehl, T.E.; Alvarado, D.; Ee, X.; Ciorba, M.A.; Stenson, W.F. Hyaluronic acid promotes Lgr5+ stem cell proliferation and crypt fission through TLR4 and PGE2 transactivation of EGFR. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G63–G73. [Google Scholar] [CrossRef] [PubMed]
- Riehl, T.E.; Santhanam, S.; Foster, L.; Ciorba, M.; Stenson, W.F. CD44 and TLR4 mediate hyaluronic acid regulation of Lgr5+ stem cell proliferation, crypt fission, and intestinal growth in postnatal and adult mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G874–G887. [Google Scholar] [CrossRef] [Green Version]
- Riehl, T.E.; Ee, X.; Stenson, W.F. Hyaluronic acid regulates normal intestinal and colonic growth in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G377–G388. [Google Scholar] [CrossRef] [Green Version]
- Sumigray, K.D.; Terwilliger, M.; Lechler, T. Morphogenesis and Compartmentalization of the Intestinal Crypt. Dev. Cell. 2018, 45, 183–197.e5. [Google Scholar] [CrossRef] [Green Version]
- Wołczuk, K.; Wilczyńska, B.; Jaroszewska, M.; Kobak, J. Morphometric characteristics of the small and large intestines of Mus musculus during postnatal development. Folia Morphol. 2011, 70, 252–259. [Google Scholar]
- Thalheim, T.; Quaas, M.; Herberg, M.; Braumann, U.D.; Kerner, C.; Loeffler, M.; Aust, G.; Galle, J. Linking stem cell function and growth pattern of intestinal organoids. Dev. Biol. 2018, 433, 254–261. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [Green Version]
- Navis, M.; Martins Garcia, T.; Renes, I.B.; Vermeulen, J.L.; Meisner, S.; Wildenberg, M.E.; van den Brink, G.R.; van Elburg, R.M.; Muncan, V. Mouse fetal intestinal organoids: New model to study epithelial maturation from suckling to weaning. EMBO Rep. 2019, 20, e46221. [Google Scholar] [CrossRef]
- Keysselt, K.; Kreutzmann, T.; Rother, K.; Kerner, C.; Krohn, K.; Przybilla, J.; Buske, P.; Löffler-Wirth, H.; Loeffler, M.; Galle, J.; et al. Different in vivo and in vitro transformation of intestinal stem cells in mismatch repair deficiency. Oncogene 2017, 36, 2750–2761. [Google Scholar] [CrossRef]
- Muncan, V.; Heijmans, J.; Krasinski, S.D.; Büller, N.V.; Wildenberg, M.E.; Meisner, S.; Radonjic, M.; Stapleton, K.A.; Lamers, W.H.; Biemond, I.; et al. Blimp1 regulates the transition of neonatal to adult intestinal epithelium. Nat. Commun. 2011, 2, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.; Mould, A.; Andrews, R.M.; Bikoff, E.K.; Robertson, E.J. The transcriptional repressor Blimp1/Prdm1 regulates postnatal reprogramming of intestinal enterocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10585–10590. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Udagawa, J.; Matsumoto, A.; Hashimoto, R.; Hatta, T.; Nishita, M.; Minami, Y.; Otani, H. Ror2 is required for midgut elongation during mouse development. Dev. Dyn. 2010, 239, 941–953. [Google Scholar] [CrossRef]
- Dahlhoff, M.; Horst, D.; Gerhard, M.; Kolligs, F.T.; Wolf, E.; Schneider, M.R. Betacellulin stimulates growth of the mouse intestinal epithelium and increases adenoma multiplicity in Apc+/Min mice. FEBS Lett. 2008, 582, 2911–2915. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Sheng, H. Amphiregulin promotes intestinal epithelial regeneration: Roles of intestinal subepithelial myofibroblasts. Endocrinology 2010, 151, 3728–3737. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.S.; Lee, H.J.; Nishida, M.; Lee, M.S.; Tamura, R.; Yamashita, S.; Matsuzawa, Y.; Lee, I.K.; Koh, G.Y. Betacellulin and amphiregulin induce upregulation of cyclin D1 and DNA synthesis activity through differential signaling pathways in vascular smooth muscle cells. Circ. Res. 2003, 93, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.N.; Yan, J.K.; Zhang, T.; Cai, W.; Yan, W.H. Butyrate promotes the adaptation of intestinal smooth muscle cells through the yes-associated protein (YAP) pathway in a rat model of short bowel syndrome. Am. J. Transl. Res. 2019, 11, 453–462. [Google Scholar] [PubMed]
- Dai, L.N.; Yan, J.K.; Xiao, Y.T.; Wen, J.; Zhang, T.; Zhou, K.J.; Wang, Y.; Cai, W. Butyrate stimulates the growth of human intestinal smooth muscle cells by activation of yes-associated protein. J. Cell Physiol. 2018, 233, 3119–3128. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef]
- Flandez, M.; Guilmeau, S.; Blache, P.; Augenlicht, L.H. KLF4 regulation in intestinal epithelial cell maturation. Exp. Cell Res. 2008, 314, 3712–3723. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Chen, X.; Zhang, W.; Li, J.; Xu, R.; Wang, T.C.; Ai, W.; Liu, C. Krüppel-like factor 4 regulates intestinal epithelial cell morphology and polarity. PLoS ONE 2012, 7, e32492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaleb, A.M.; McConnell, B.B.; Kaestner, K.H.; Yang, V.W. Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the Krüppel-like factor 4 gene. Dev. Biol. 2011, 349, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Dhaliwal, N.K.; Abatti, L.E.; Mitchell, J.A. KLF4 protein stability regulated by interaction with pluripotency transcription factors overrides transcriptional control. Genes Dev. 2019, 33, 1069–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, G.B.; Kim, D. MicroRNA-503-5p Inhibits the CD97-Mediated JAK2/STAT3 Pathway in Metastatic or Paclitaxel-Resistant Ovarian Cancer Cells. Neoplasia 2019, 21, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Buske, P.; Galle, J.; Barker, N.; Aust, G.; Clevers, H.; Loeffler, M. A comprehensive model of the spatio-temporal stem cell and tissue organisation in the intestinal crypt. PLoS Comput. Biol. 2011, 7, e1001045. [Google Scholar] [CrossRef] [Green Version]
- Krndija, D.; Marjou, F.E.; Guirao, B.; Richon, S.; Leroy, O.; Bellaiche, Y.; Hannezo, E.B.; Vignjevic, D.M. Active cell migration is critical for steady-state epithelial turnover in the gut. Science 2019, 365, 705–710. [Google Scholar] [CrossRef]
- Vig, D.K.; Hamby, A.E.; Wolgemuth, C.W. Cellular Contraction Can Drive Rapid Epithelial Flows. Biophys. J. 2017, 113, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.M.; Mao, J.; Taketo, M.M.; Shivdasani, R.A. Phases of canonical Wnt signaling during the development of mouse intestinal epithelium. Gastroenterology 2007, 133, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Langlands, A.J.; Almet, A.A.; Appleton, P.L.; Newton, I.P.; Osborne, J.M.; Näthke, I.S. Paneth Cell-Rich Regions Separated by a Cluster of Lgr5+ Cells Initiate Crypt Fission in the Intestinal Stem Cell Niche. PLoS Biol. 2016, 14, e1002491. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, M.; Fujii, E.; Watanabe, T.; Kato, A.; Suzuki, M. Histopathological evaluation of crypt fission during intestinal development in neonatal mice. J. Toxicol. Pathol. 2020, 33, 39–46. [Google Scholar] [CrossRef]
- Almet, A.A.; Maini, P.K.; Moulton, D.E.; Byrne, H.M. Modeling perspectives on the intestinal crypt, a canonical system for growth, mechanics, and remodeling. Curr. Opin. Biomed. Eng. 2020, 15, 32–39. [Google Scholar] [CrossRef]
- Thalheim, T.; Buske, P.; Przybilla, J.; Rother, K.; Loeffler, M.; Galle, J. Stem cell competition in the gut: Insights from multi-scale computational modelling. J. R. Soc. Interface 2016, 13, 20160218. [Google Scholar] [CrossRef]
- Buske, P.; Przybilla, J.; Loeffler, M.; Sachs, N.; Sato, T.; Clevers, H.; Galle, J. On the biomechanics of stem cell niche formation in the gut—Modelling growing organoids. FEBS J. 2012, 279, 3475–3487. [Google Scholar] [CrossRef]
- Lei, N.Y.; Jabaji, Z.; Wang, J.; Joshi, V.S.; Brinkley, G.J.; Khalil, H.; Wang, F.; Jaroszewicz, A.; Pellegrini, M.; Li, L.; et al. Intestinal subepithelial myofibroblasts support the growth of intestinal epithelial stem cells. PLoS ONE 2014, 9, e84651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Neerven, S.M.; Vermeulen, L. The interplay between intrinsic and extrinsic Wnt signaling in controlling intestinal transformation. Differentiation 2019, 108, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Haramis, A.P.; Begthel, H.; van den Born, M.; van Es, J.; Jonkheer, S.; Offerhaus, G.J.; Clevers, H. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science 2004, 303, 1684–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannezo, E.; Prost, J.; Joanny, J.F. Instabilities of monolayered epithelia: Shape and structure of villi and crypts. Phys. Rev. Lett. 2011, 107, 078104. [Google Scholar] [CrossRef] [Green Version]
- Dekaney, C.M.; Fong, J.J.; Rigby, R.J.; Lund, P.K.; Henning, S.J.; Helmrath, M.A. Expansion of intestinal stem cells associated with long-term adaptation following ileocecal resection in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1013–G1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malipatlolla, D.K.; Patel, P.; Sjöberg, F.; Devarakonda, S.; Kalm, M.; Angenete, E.; Lindskog, E.B.; Grandér, R.; Persson, L.; Stringer, A.; et al. Long-term mucosal injury and repair in a murine model of pelvic radiotherapy. Sci. Rep. 2019, 9, 13803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittelkow, M.R.; Cook, P.W.; Shipley, G.D.; Derynck, R.; Coffey, R.J., Jr. Autonomous growth of human keratinocytes requires epidermal growth factor receptor occupancy. Cell Growth Differ. 1993, 4, 513–521. [Google Scholar]
- Becker, S.; Wandel, E.; Wobus, M.; Schneider, R.; Amasheh, S.; Sittig, D.; Kerner, C.; Naumann, R.; Hamann, J.; Aust, G. Overexpression of CD97 in intestinal epithelial cells of transgenic mice attenuates colitis by strengthening adherens junctions. PLoS ONE 2010, 5, e8507. [Google Scholar] [CrossRef]
- Leedham, S.J.; Wright, N.A. Expansion of a mutated clone: From stem cell to tumour. J. Clin. Pathol. 2008, 61, 164–171. [Google Scholar] [CrossRef]
- Loeffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on Bioconductor. Bioinformatics 2015, 31, 3225–3227, R-package oposSOM, Version 2.2.0. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47, R-package limma, Version 3.42.2. [Google Scholar] [CrossRef]
- Pitzonka, L.; Wang, X.; Ullas, S.; Wolff, D.W.; Wang, Y.; Goodrich, D.W. The THO ribonucleoprotein complex is required for stem cell homeostasis in the adult mouse small intestine. Mol. Cell. Biol. 2013, 33, 3505–3514. [Google Scholar] [CrossRef] [Green Version]
- Galle, J.; Loeffler, M.; Drasdo, D. Modeling the effect of deregulated proliferation and apoptosis on the growth of epithelial cll populations in vitro. Biophys. J. 2005, 88, 62–75. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofmann, F.; Thalheim, T.; Rother, K.; Quaas, M.; Kerner, C.; Przybilla, J.; Aust, G.; Galle, J. How to Obtain a Mega-Intestine with Normal Morphology: In Silico Modelling of Postnatal Intestinal Growth in a Cd97-Transgenic Mouse. Int. J. Mol. Sci. 2021, 22, 7345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147345
Hofmann F, Thalheim T, Rother K, Quaas M, Kerner C, Przybilla J, Aust G, Galle J. How to Obtain a Mega-Intestine with Normal Morphology: In Silico Modelling of Postnatal Intestinal Growth in a Cd97-Transgenic Mouse. International Journal of Molecular Sciences. 2021; 22(14):7345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147345
Chicago/Turabian StyleHofmann, Felix, Torsten Thalheim, Karen Rother, Marianne Quaas, Christiane Kerner, Jens Przybilla, Gabriela Aust, and Joerg Galle. 2021. "How to Obtain a Mega-Intestine with Normal Morphology: In Silico Modelling of Postnatal Intestinal Growth in a Cd97-Transgenic Mouse" International Journal of Molecular Sciences 22, no. 14: 7345. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147345