
Stress-Inducible Gene Atf3 Dictates a Dichotomous Macrophage Activity in Chemotherapy-Enhanced Lung Colonization

 ,
,
Abstract
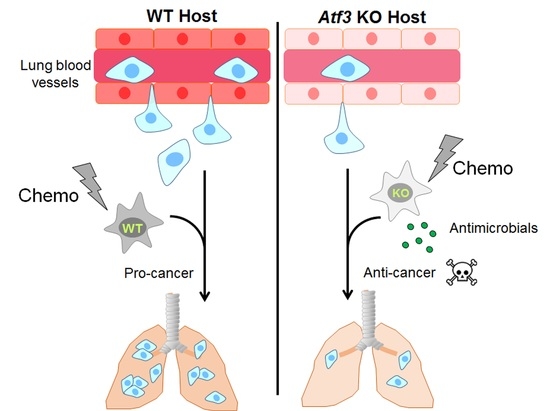
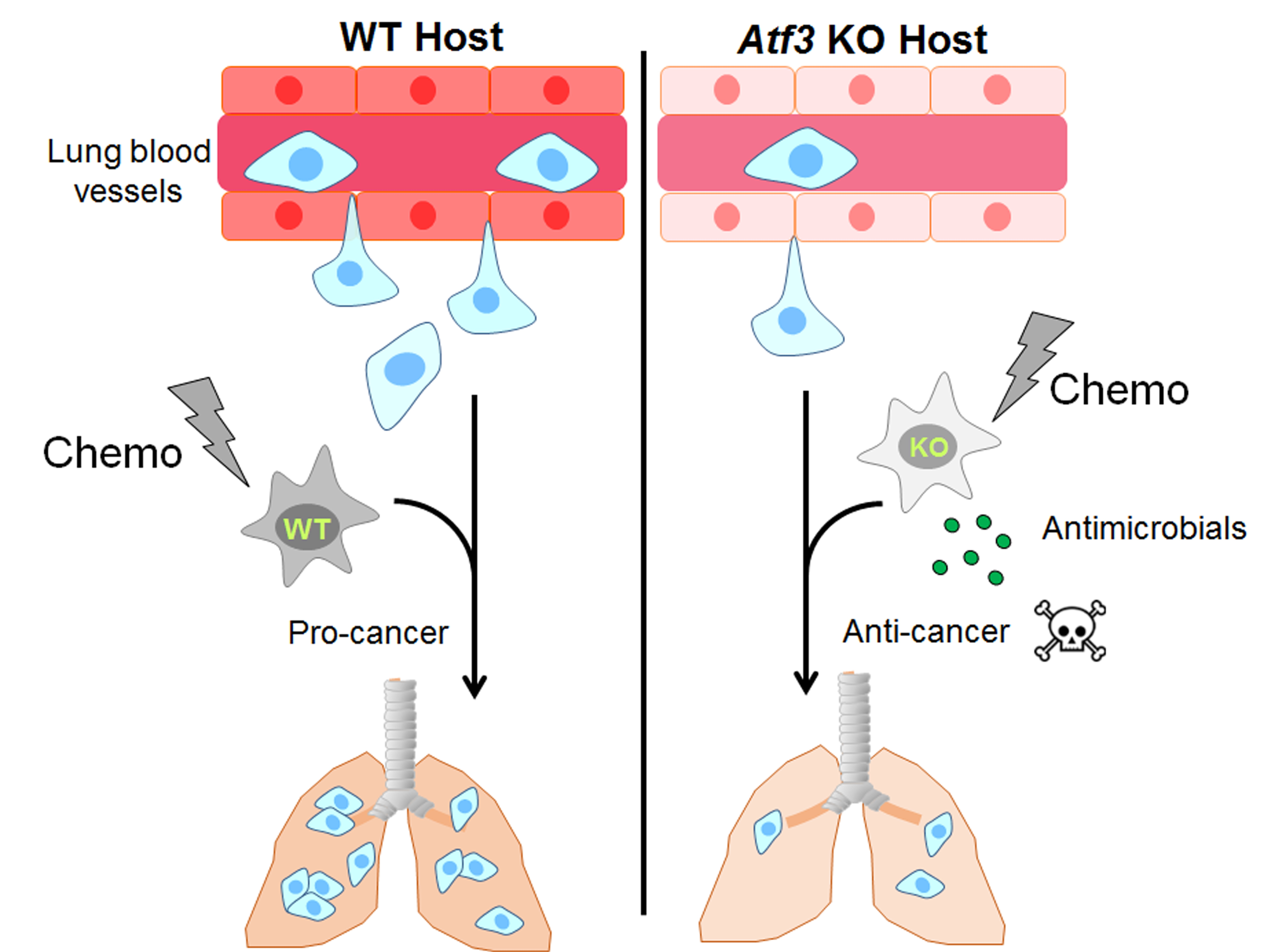
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Host Atf3, Combined with Chemotherapy, Establishes a Cancer-Friendly Tissue Environment at Early Stages of Lung Colonization
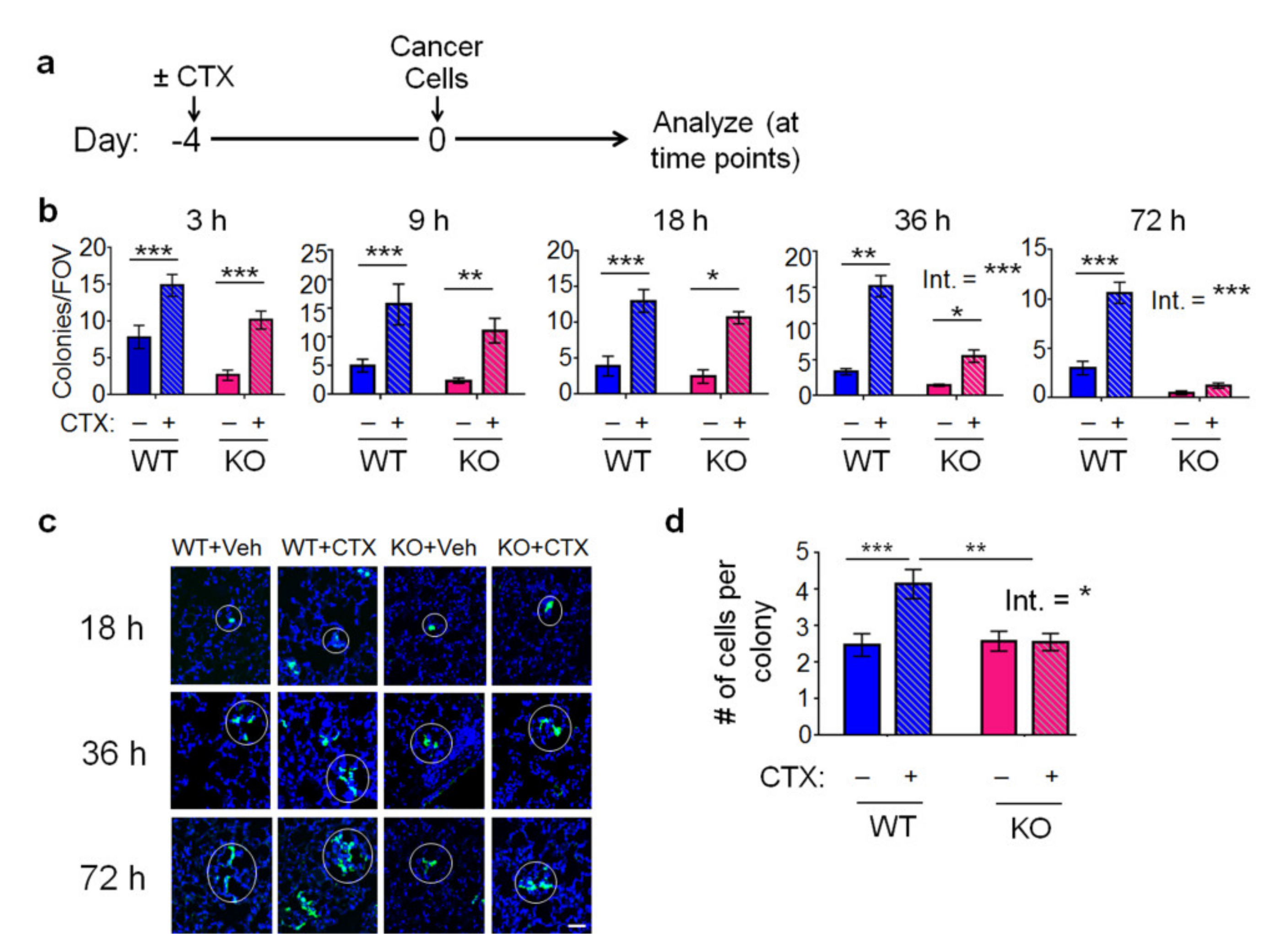
2.2. The Effect of CTX and the Host Atf3 on Cancer Cell Extravasation, Proliferation, and Death
2.3. Macrophage Is a Key Cell Type for the Host Atf3 to Mediate the Pro-Colonization Effect of CTX
2.4. Cancer Cell Transendothelial Migration In Vitro Is Affected by the Genotype of Atf3 in Macrophages, but Not by CTX
2.5. Atf3 Dictates a Dichotomous Role of Macrophage in a Cell-Autonomous Manner
2.6. The Effect of Atf3 Genotype and CTX on Macrophage Gene Expression
2.7. The Antimicrobial Gene Cathelicidin in Cancer Cell Death
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Cell Culture and Treatment
4.3. Cell Isolation from Mouse Tissues
4.4. Macrophage Cytotoxicity Assay
4.5. In Vitro Apoptosis Assay
4.6. Image Analysis
4.7. RNA Isolation and RT-qPCR
4.8. Generation of CTX-Serum and PBS-Serum
4.9. Transendothelial Migration Assay
4.10. Immunofluorescence Microscopy
4.11. Analytical Flow Cytometry and Fluorescence Activated Cell Sorting (FACS)
4.12. Macrophage Depletion Assay
4.13. Microarray
4.14. Public Dataset Analysis
4.15. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emmenegger, U.; Kerbel, R.S. Chemotherapy counteracted. Nat. Cell Biol. 2010, 468, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; Ganesan, A.P.; Rugo, H.S.; Coussens, L.M. Immune microenvironments in solid tumors: New targets for therapy. Genes Dev. 2011, 25, 2559–2572. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Lewis, C.E. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaked, Y. Balancing efficacy of and host immune responses to cancer therapy: The yin and yang effects. Nat. Rev. Clin. Oncol. 2016, 13, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.D.; Stover, D.G.; Hai, T. Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response. Int. J. Mol. Sci. 2018, 19, 3333. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-Induced Metastasis: Molecular Mechanisms, Clinical Manifestations, Therapeutic Interventions. Cancer Res. 2019, 79, 4567–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.C.; Su, Q.B.; Wu, F.X.; Zhang, X.L.; Liu, P.S. Role of TLR4 for paclitaxel chemotherapy in human epithelial ovarian cancer cells. Eur. J. Clin. Investig. 2009, 39, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, M.; Elia, L.; Price, J.H.; Heynen-Genel, S.; Courtneidge, S.A. A Cell-Based High-Content Screening Assay Reveals Activators and Inhibitors of Cancer Cell Invasion. Sci. Signal. 2011, 4, ra49. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Zhou, X.; Yang, J.-J.; Liu, X.; Zhao, X.-H.; Wang, Q.-X.; Han, L.; Song, X.; Zhu, Z.-Y.; Tian, W.-P.; et al. AC1MMYR2 impairs high dose paclitaxel-induced tumor metastasis by targeting miR-21/CDK5 axis. Cancer Lett. 2015, 362, 174–182. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Nakasone, E.S.; Askautrud, H.; Kees, T.; Park, J.-H.; Plaks, V.; Ewald, A.J.; Fein, M.; Rasch, M.G.; Tan, Y.-X.; Qiu, J.; et al. Imaging Tumor-Stroma Interactions during Chemotherapy Reveals Contributions of the Microenvironment to Resistance. Cancer Cell 2012, 21, 488–503. [Google Scholar] [CrossRef] [Green Version]
- Shree, T.; Olson, O.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef] [Green Version]
- Daenen, L.G.; Roodhart, J.M.; Van Amersfoort, M.; Dehnad, M.; Roessingh, W.; Ulfman, L.H.; Derksen, P.W.; Voest, E.E.; Jin, X.; Yin, J.; et al. Chemotherapy Enhances Metastasis Formation via VEGFR-1–Expressing Endothelial Cells. Cancer Res. 2011, 71, 6976–6985. [Google Scholar] [CrossRef] [Green Version]
- Gingis-Velitski, S.; Loven, D.; Benayoun, L.; Munster, M.; Bril, R.; Voloshin, T.; Alishekevitz, D.; Bertolini, F.; Shaked, Y. Host Response to Short-term, Single-Agent Chemotherapy Induces Matrix Metalloproteinase-9 Expression and Accelerates Metastasis in Mice. Cancer Res. 2011, 71, 6986–6996. [Google Scholar] [CrossRef] [Green Version]
- Park, S.I.; Liao, J.; Berry, J.E.; Li, X.; Koh, A.J.; Michalski, M.E.; Eber, M.R.; Soki, F.N.; Sadler, D.; Sud, S.; et al. Cyclophosphamide Creates a Receptive Microenvironment for Prostate Cancer Skeletal Metastasis. Cancer Res. 2012, 72, 2522–2532. [Google Scholar] [CrossRef] [Green Version]
- Alishekevitz, D.; Gingis-Velitski, S.; Kaidar-Person, O.; Gutter-Kapon, L.; Scherer, S.D.; Raviv, Z.; Merquiol, E.; Ben-Nun, Y.; Miller, V.; Rachman-Tzemah, C.; et al. Macrophage-Induced Lymphangiogenesis and Metastasis following Paclitaxel Chemotherapy Is Regulated by VEGFR3. Cell Rep. 2016, 17, 1344–1356. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Jalgaonkar, S.P.; Middleton, J.D.; Hai, T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E7159–E7168. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, eaan0026. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-induced metastasis: Mechanisms and translational opportunities. Clin. Exp. Metastasis 2018, 35, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.D.; Sica, G.L.; Liu, Y.-F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor Microenvironment of Metastasis in Human Breast Carcinoma: A Potential Prognostic Marker Linked to Hematogenous Dissemination. Clin. Cancer Res. 2009, 15, 2433–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.-Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage–Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hai, T.; Wolford, C.C.; Chang, Y.-S. ATF3, a Hub of the Cellular Adaptive-Response Network, in the Pathogenesis of Diseases: Is Modulation of Inflammation a Unifying Component? Gene Expr. 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef]
- Montminy, M. Transcriptional Regulation by Cyclic AMP. Annu. Rev. Biochem. 1997, 66, 807–822. [Google Scholar] [CrossRef]
- Hai, T.; Wolfgang, C.D.; Marsee, D.K.; Allen, A.E.; Sivaprasad, U. ATF3 and Stress Responses. Gene Expr. 1999, 7, 321–335. [Google Scholar]
- Zhao, J.; Li, X.; Guo, M.; Yu, J.; Yan, C. The common stress responsive transcription factor ATF3 binds genomic sites enriched with p300 and H3K27ac for transcriptional regulation. BMC Genom. 2016, 17, 335. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nat. Cell Biol. 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.; Pollard, J.W. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Wolford, C.C.; McConoughey, S.J.; Jalgaonkar, S.P.; Leon, M.; Merchant, A.S.; Dominick, J.L.; Yin, X.; Chang, Y.; Zmuda, E.J.; O’Toole, S.A.; et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J. Clin. Investig. 2013, 123, 2893–2906. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chen, Y.; Qi, F.; Jia, L.; Lu, X.-A.; He, T.; Fu, Y.; Li, L.; Luo, Y. Specific chemotherapeutic agents induce metastatic behaviour through stromal- and tumour-derived cytokine and angiogenic factor signalling. J. Pathol. 2015, 237, 190–202. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Gurley, L.R.; D’Anna, J.A.; Barham, S.S.; Deaven, L.L.; Tobey, R.A. Histone Phosphorylation and Chromatin Structure during Mitosis in Chinese Hamster Cells. JBIC J. Biol. Inorg. Chem. 1978, 84, 1–15. [Google Scholar] [CrossRef]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of Tumor Metastasis by Myeloid-Derived Suppressor Cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Strilic, B.; Offermanns, S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell 2017, 32, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, M.; Thorsson, V.; Li, B.; Rust, A.G.; Korb, M.; Kennedy, K.; Hai, T.; Bolouri, H.; Aderem, A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nat. Cell Biol. 2006, 441, 173–178. [Google Scholar] [CrossRef]
- Van Rooijen, N.; Hendrikx, E. Liposomes for Specific Depletion of Macrophages from Organs and Tissues. Methods Mol. Biol. 2010, 605, 189–203. [Google Scholar] [CrossRef]
- Gil-Bernabé, A.M.; Ferjančič, Š.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [Green Version]
- Bossche, J.V.D.; Bogaert, P.; Van Hengel, J.; Guérin, C.J.; Berx, G.; Movahedi, K.; Bergh, R.V.D.; Pereira-Fernandes, A.; Geuns, J.M.C.; Pircher, H.; et al. Alternatively activated macrophages engage in homotypic and heterotypic interactions through IL-4 and polyamine-induced E-cadherin/catenin complexes. Blood 2009, 114, 4664–4674. [Google Scholar] [CrossRef]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado, J.D.D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-De-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruzel, M.L.; Zimecki, M.; Actor, J.K. Lactoferrin in a Context of Inflammation-Induced Pathology. Front. Immunol. 2017, 8, 1438. [Google Scholar] [CrossRef] [PubMed]
- Golonka, R.; Yeoh, B.S.; Vijay-Kumar, M. The Iron Tug-of-War between Bacterial Siderophores and Innate Immunity. J. Innate Immun. 2019, 11, 249–262. [Google Scholar] [CrossRef]
- Bruns, H.; Büttner, M.; Fabri, M.; Mougiakakos, D.; Bittenbring, J.T.; Hoffmann, M.H.; Beier, F.; Pasemann, S.; Jitschin, R.; Hofmann, A.D.; et al. Vitamin D–Dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci. Transl. Med. 2015, 7, 282ra47. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2008, 9, 239–252. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Roh-Johnson, M.; Bravo-Cordero, J.J.; Patsialou, A.; Sharma, V.P.; Guo, P.; Liu, H.; Hodgson, L.; Condeelis, J. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 2013, 33, 4203–4212. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, J.; Bravo-Cordero, J.J.; Roh-Johnson, M.; Gandhi, S.J.; Wang, Y.; Chen, X.; Eddy, R.J.; Xue, A.; Singer, R.H.; Hodgson, L.; et al. Macrophage-dependent tumor cell transendothelial migration is mediated by Notch1/MenaINV-initiated invadopodium formation. Sci. Rep. 2016, 6, 37874. [Google Scholar] [CrossRef]
- Coffey, J.C.; Wang, J.H.; Smith, M.J.; Bouchier-Hayes, D.; Cotter, T.G.; Redmond, H.P. Excisional surgery for cancer cure: Therapy at a cost. Lancet Oncol. 2003, 4, 760–768. [Google Scholar] [CrossRef]
- El Saghir, N.S.; I Elhajj, I.; Geara, F.B.; Hourani, M.H. Trauma-associated growth of suspected dormant micrometastasis. BMC Cancer 2005, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Rachman-Tzemah, C.; Zaffryar-Eilot, S.; Grossman, M.; Ribero, D.; Timaner, M.; Mäki, J.M.; Myllyharju, J.; Bertolini, F.; Hershkovitz, D.; Sagi, I.; et al. Blocking Surgically Induced Lysyl Oxidase Activity Reduces the Risk of Lung Metastases. Cell Rep. 2017, 19, 774–784. [Google Scholar] [CrossRef] [Green Version]
- Krall, J.A.; Reinhardt, F.; Mercury, O.A.; Pattabiraman, D.; Brooks, M.W.; Dougan, M.; Lambert, A.W.; Bierie, B.; Ploegh, H.L.; Dougan, S.K.; et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 2018, 10, eaan3464. [Google Scholar] [CrossRef] [Green Version]
- Sleeman, J.P. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012, 31, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, K.; Okumura, K.; Isogai, H.; Isogai, E. The Human Cathelicidin Antimicrobial Peptide LL-37 and Mimics are Potential Anticancer Drugs. Front. Oncol. 2015, 5, 144. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.G.; Lu, D.; Kim, M.L.; Kociba, G.J.; Shukri, T.; Buteau, J.; Wang, X.; Frankel, W.L.; Guttridge, D.; Prentki, M.; et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell. Biol. 2004, 24, 5721–5732. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbach, M.L.; Cao, G.; Williams, J.T.; Finklestein, J.M.; Delisser, H.M. Isolation of murine lung endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L1096–L1103. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.-C.; Luscinskas, F.W.; Sean, C.P. Isolation and Culture of Murine Heart and Lung Endothelial Cells for In Vitro Model Systems. Methods Mol. Biol. 2006, 341, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; DeWille, J.W.; Hai, T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 2007, 27, 2118–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hai, T.; Jalgaonkar, S.; Wolford, C.C.; Yin, X. Immunohistochemical detection of Activating Transcription Factor 3, a hub of the cellular adaptive-response network. In Methods in Enzymology; Academic Press: Burlington, MA, USA, 2011; Volume 490, pp. 175–194. [Google Scholar]
- Goswami, C.P.; Nakshatri, H. PROGgene: Gene expression based survival analysis web application for multiple cancers. J. Clin. Bioinform. 2013, 3, 22. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Middleton, J.D.; Fehlman, J.; Sivakumar, S.; Stover, D.G.; Hai, T. Stress-Inducible Gene Atf3 Dictates a Dichotomous Macrophage Activity in Chemotherapy-Enhanced Lung Colonization. Int. J. Mol. Sci. 2021, 22, 7356. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147356
Middleton JD, Fehlman J, Sivakumar S, Stover DG, Hai T. Stress-Inducible Gene Atf3 Dictates a Dichotomous Macrophage Activity in Chemotherapy-Enhanced Lung Colonization. International Journal of Molecular Sciences. 2021; 22(14):7356. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147356
Chicago/Turabian StyleMiddleton, Justin D., Jared Fehlman, Subhakeertana Sivakumar, Daniel G. Stover, and Tsonwin Hai. 2021. "Stress-Inducible Gene Atf3 Dictates a Dichotomous Macrophage Activity in Chemotherapy-Enhanced Lung Colonization" International Journal of Molecular Sciences 22, no. 14: 7356. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147356