
4-Acetyl-Antroquinonol B Improves the Sensitization of Cetuximab on Both Kras Mutant and Wild Type Colorectal Cancer by Modulating the Expression of Ras/Raf/miR-193a-3p Signaling Axis


 , ,
, ,  and
and 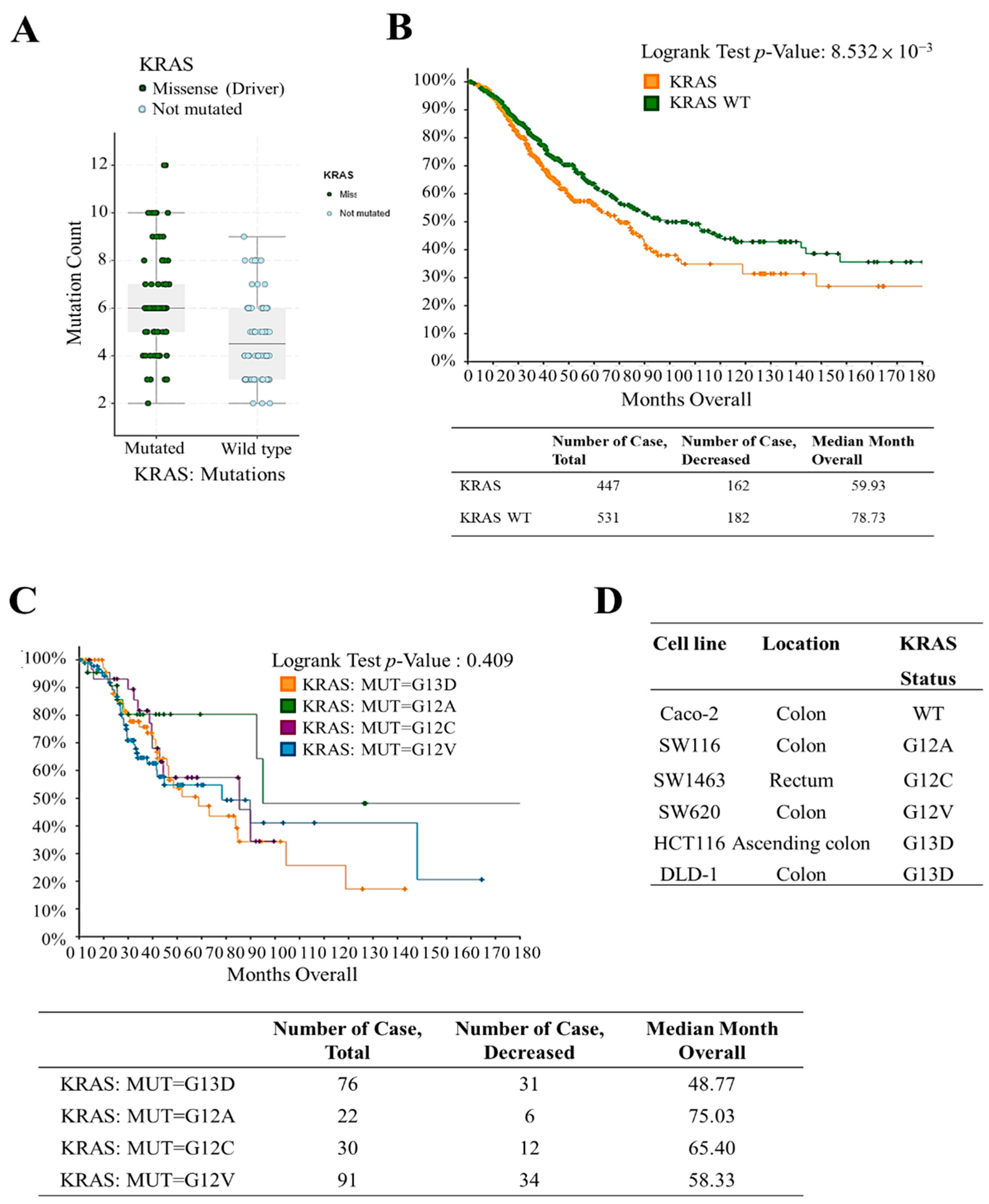{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Screening and Patient Survival Analysis in Patients with KRAS Mutation and Wild-Type Genes
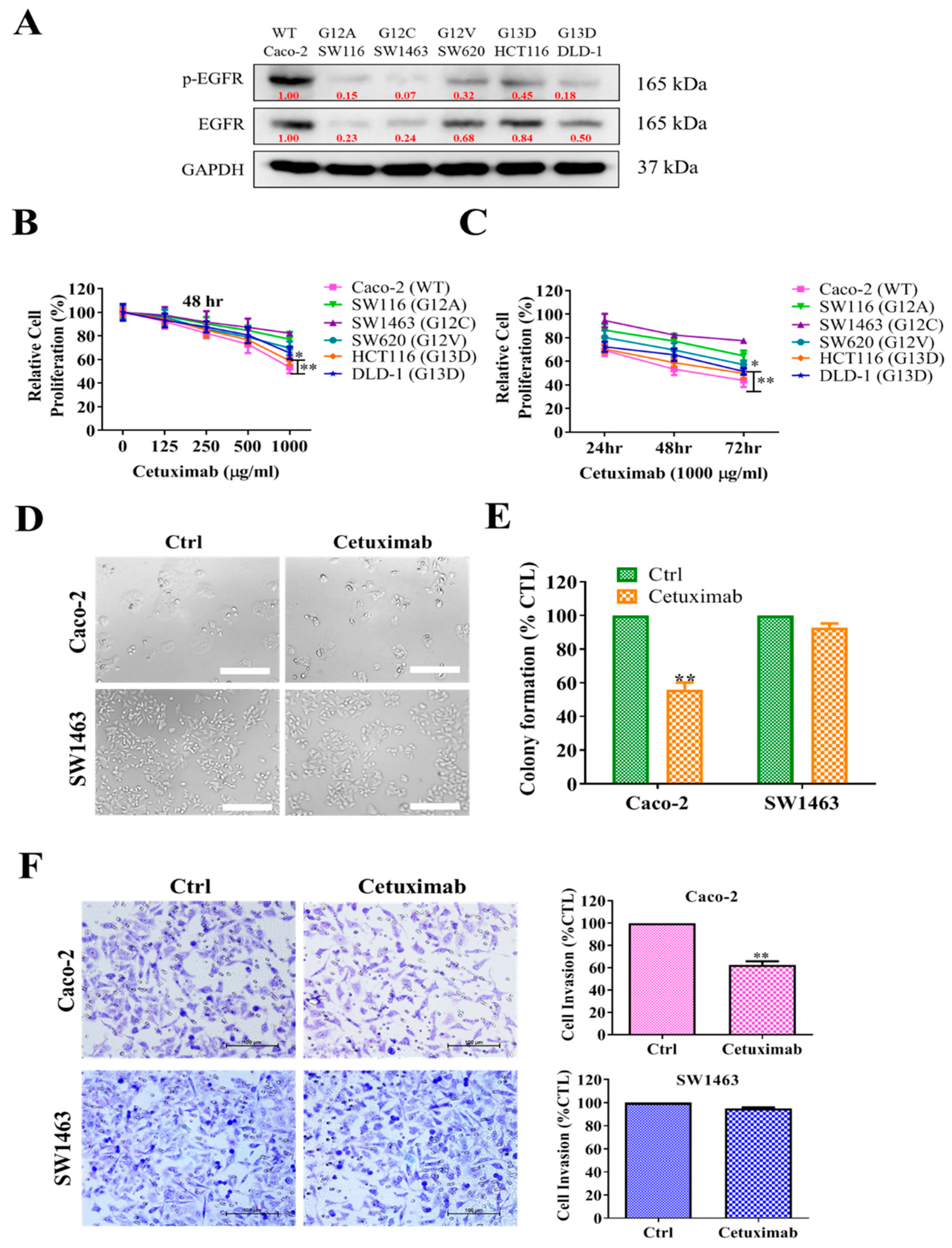
2.2. KRAS-Mutated Colorectal Cells Were Resistant to Cetuximab Treatment
2.3. 4-AAQB Treatment Results in Inhibition of Oncogenic Properties of KRAS-Mutant and Wild-Type Colorectal Cells
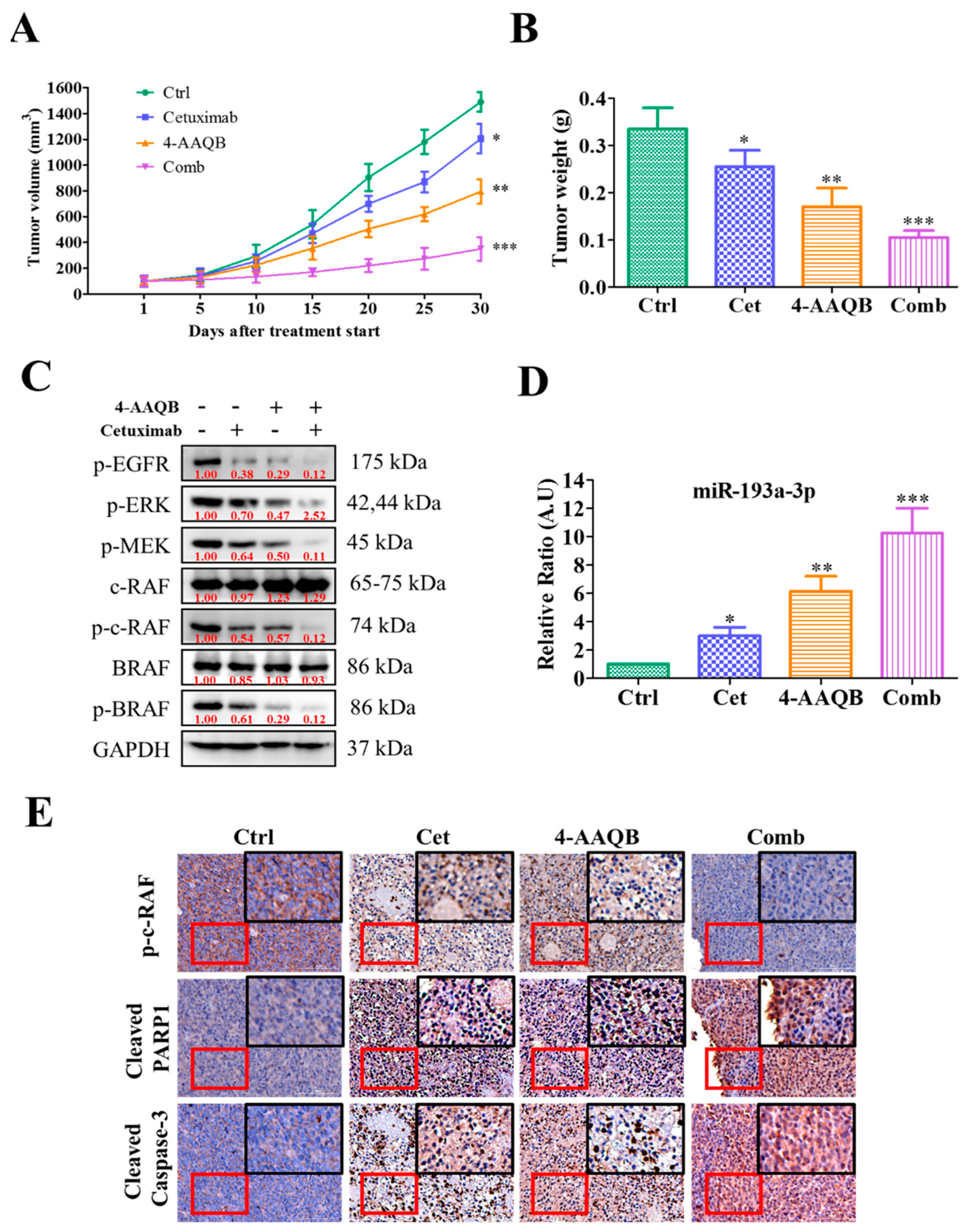
2.4. Combination Treatment with 4-AAQB and Cetuximab Increased Cetuximab Sensitivity in KRAS-Mutant CRC Cells
2.5. Anti-CRC Role of 4-AAQB Was Associated with the Induction of miR-193a-3p and Reductions of Its Oncogenic Targets
2.6. Treatment with 4-AAQB Increased Cetuximab Efficacy In Vivo
3. Discussion
4. Materials and Methods
4.1. Clinical Sample Collection and Preparation
4.2. Drugs, Chemicals, Cell Culture and Media
4.3. Cell Lines
4.4. In Silico Data Acquisition and Analysis
4.5. Colorimetric Assay of Sulforhodamine B (SRB) Cell Proliferation Assay
4.6. Western Blotting
4.7. Transient Oligonucleotide Transfection
4.8. Cell Invasion and Tumorsphere Assay
4.9. Colony Formation Assay
4.10. Detection of Apoptosis through Flow Cytometry
4.11. In Vivo Studies
4.12. Immunostaining
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global burden of 5 major types of gastrointestinal cancer. Gastroenterology 2020, 159, 335–349.e315. [Google Scholar] [CrossRef]
- Pourhoseingholi, M.A.; Vahedi, M.; Baghestani, A.R. Burden of gastrointestinal cancer in asia; an overview. Gastroenterol. Hepatol. Bed Bench 2015, 8, 19–27. [Google Scholar]
- Fassan, M.; Cui, R.; Gasparini, P.; Mescoli, C.; Guzzardo, V.; Vicentini, C.; Munari, G.; Loupakis, F.; Lonardi, S.; Braconi, C.; et al. Mir-224 is significantly upregulated and targets caspase-3 and caspase-7 during colorectal carcinogenesis. Transl. Oncol. 2019, 12, 282–291. [Google Scholar] [CrossRef]
- Seyhan, A.A.; Carini, C. Are innovation and new technologies in precision medicine paving a new era in patients centric care? J. Transl. Med. 2019, 17, 114. [Google Scholar] [CrossRef] [Green Version]
- Dinu, D.; Dobre, M.; Panaitescu, E.; Bîrlă, R.; Iosif, C.; Hoara, P.; Caragui, A.; Boeriu, M.; Constantinoiu, S.; Ardeleanu, C. Prognostic significance of kras gene mutations in colorectal cancer—Preliminary study. J. Med. Life 2014, 7, 581–587. [Google Scholar]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting kras in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.J.N. The clinical kras (g12c) inhibitor amg 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Côté, J.F.; Tomasic, G.; Penna, C.; Ducreux, M. Kras mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of kras, braf, nras, and pik3ca mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. Egfr antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.; Burtness, B. Cetuximab: An epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today 2005, 41, 107–127. [Google Scholar] [CrossRef]
- Giordano, G.; Remo, A.; Porras, A.; Pancione, M. Immune resistance and egfr antagonists in colorectal cancer. Cancers 2019, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.L.; Kuo, Y.H.; Tsai, C.T.; Huang, Y.T.; Chen, S.C.; Chang, H.-W.; Lin, E.; Lin, W.-H.; Hseu, Y.C.J.F.; Toxicology, C. Anti-metastatic activities of antrodia camphorata against human breast cancer cells mediated through suppression of the mapk signaling pathway. Food Chem. Toxicol. 2011, 49, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Chen, S.C.; Chen, H.C.; Liao, J.W.; Yang, H.L.J.F.; Toxicology, C. Antrodia camphorata inhibits proliferation of human breast cancer cells in vitro and in vivo. Food Chem. Toxicol. 2008, 46, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Peng, C.C.; Peng, R.Y.; Su, C.-H.; Chiang, H.S.; Yan, J.H.; Hsieh-Li, H.M. Unique formosan mushroom antrodia camphorata differentially inhibits androgen-responsive lncap and-independent pc-3 prostate cancer cells. Nutr. Cancer 2007, 57, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Yeh, C.T.; Adebayo, B.O.; Lin, Y.C.; Deng, L.; Rao, Y.K.; Huang, C.C.; Lee, W.H.; Wu, A.T.; Hsiao, M.; et al. 4-acetylantroquinonol b inhibits colorectal cancer tumorigenesis and suppresses cancer stem-like phenotype. Toxicol. Appl. Pharm. 2015, 288, 258–268. [Google Scholar] [CrossRef]
- Bamodu, O.A.; Yang, C.K.; Cheng, W.H.; Tzeng, D.T.W.; Kuo, K.T.; Huang, C.C.; Deng, L.; Hsiao, M.; Lee, W.H.; Yeh, C.T. 4-acetyl-antroquinonol b suppresses sod2-enhanced cancer stem cell-like phenotypes and chemoresistance of colorectal cancer cells by inducing hsa-mir-324 re-expression. Cancers 2018, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Lan, Z.; Xiong, X.; Ao, H.; Feng, Y.; Gu, H.; Yu, M.; Cui, Q. The dual role of micrornas in colorectal cancer progression. Int. J. Mol. Sci. 2018, 19, 2791. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Hu, X.; Zhang, H.; Wang, S.; Zhang, H.; You, C.; Zhang, C.Y.; Liang, H.; Chen, X.; Ba, Y. Mir-193a-3p is an important tumour suppressor in lung cancer and directly targets kras. Cell. Physiol. Biochem. 2017, 44, 1311–1324. [Google Scholar] [CrossRef]
- Seviour, E.G.; Sehgal, V.; Mishra, D.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Lee, J.S.; Sood, A.K.; Kim, M.P.; Mills, G.B.; et al. Targeting kras-dependent tumour growth, circulating tumour cells and metastasis in vivo by clinically significant mir-193a-3p. Oncogene 2017, 36, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.F.; Chang, Y.C.; Nieh, S.; Liu, C.L.; Yang, C.Y.; Lin, Y.S. Nonadhesive culture system as a model of rapid sphere formation with cancer stem cell properties. PLoS ONE 2012, 7, e31864. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.J.; Rho, J.K.; Kim, Y.M.; Jung, J.E.; Jin, Y.B.; Ko, Y.G.; Lee, J.S.; Lee, S.J.; Lee, J.C.; Park, M.J. Upregulation of cxcr4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 2013, 32, 209–221. [Google Scholar] [CrossRef]
- Hoda, D.; Simon, G.R.; Garrett, C.R. Targeting colorectal cancer with anti-epidermal growth factor receptor antibodies: Focus on panitumumab. Ther. Clin. Risk Manag. 2008, 4, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Lievre, A.; Bachet, J.-B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouché, O.; Landi, B.; Louvet, C.; et al. Kras mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef]
- Chou, T.C. The combination index (ci <1) as the definition of synergism and of synergy claims. Synergy 2018, 7, 49–50. [Google Scholar] [CrossRef]
- Yang, Y.F.; Wang, G.; He, J.L.; Wu, F.P.; Zhang, Y.N. Overall survival of patients with kras wild-type tumor treated with folfox/forfiri ± cetuximab as the first-line treatment for metastatic colorectal cancer: A meta-analysis. Medicine 2017, 96, e6335. [Google Scholar] [CrossRef] [PubMed]
- Er, T.K.; Chen, C.C.; Bujanda, L.; Herreros-Villanueva, M. Current approaches for predicting a lack of response to anti-egfr therapy in kras wild-type patients. BioMed Res. Int. 2014, 2014, 591867. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-egfr therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, D.; Yang, S. Overcoming resistance to drugs targeting kras(g12c) mutation. Innovation 2020, 1, 100035. [Google Scholar] [PubMed]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting aberrant ras/raf/mek/erk signaling for cancer therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the raf-mek-erk mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.J.; Lai, K.C.; Lee, A.S.; Chang, C.H.; Liu, C.L.; Chung, C.H. Novel antrodia cinnamomea extract reduced cancer stem-like phenotype changes and resensitized kras-mutant colorectal cancer via a microrna-27a pathway. Cancers 2019, 11, 1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Kim, M.; Jung, H.S.; Kim, Y.; Jeoung, D. Targeting autophagy for overcoming resistance to anti-egfr treatments. Cancers 2019, 11, 1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbeau, L.M.O.; Keulers, T.G.H.; Rouschop, K.M.A. Tumors Responsive to Autophagy-Inhibition: Identification and Biomarkers. Cancers 2020, 12, 2463. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bamodu, O.A.; Huang, W.C.; Zucha, M.A.; Lin, Y.K.; Wu, A.T.; Huang, C.C.; Lee, W.-H.; Yuan, C.C.; Hsiao, M.J.T.; et al. 4-acetylantroquinonol b suppresses autophagic flux and improves cisplatin sensitivity in highly aggressive epithelial cancer through the pi3k/akt/mtor/p70s6k signaling pathway. Toxicol. Appl. Pharmacol. 2017, 325, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.H.; Wang, Y.Z.; Tu, J.; Liu, C.-W.; Yuan, Y.J.; Lin, R.; He, W.L.; Cai, S.R.; He, Y.L.; Ye, J.N. Anti-egfr therapy in metastatic colorectal cancer: Mechanisms and potential regimens of drug resistance. Gastroenterol. Rep. 2020, 8, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhang, Z.; Gao, M.; Yu, H.; Sheng, H.; Huang, J. Microrna-193a-3p suppresses the colorectal cancer cell proliferation and progression through downregulating the plau expression. Cancer Manag. Res. 2019, 11, 5353–5363. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.I.; van Lengerich, B.; Eichel, K.; Cha, M.; Patterson, D.M.; Yoon, T.Y.; von Zastrow, M.; Jura, N.; Gartner, Z.J. Phosphorylated egfr dimers are not sufficient to activate ras. Cell Rep. 2018, 22, 2593–2600. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.; Soreide, K. Egfr and downstream genetic alterations in kras/braf and pi3k/akt pathways in colorectal cancer: Implications for targeted therapy. Discov. Med. 2012, 14, 207–214. [Google Scholar]
- Chou, K.C.C.; Yang, S.H.; Wu, H.L.; Lin, P.Y.; Chang, T.L.; Sheu, F.; Chen, K.H.; Chiang, B.H. Biosynthesis of antroquinonol and 4-acetylantroquinonol b via a polyketide pathway using orsellinic acid as a ring precursor in antrodia cinnamomea. J. Agric. Food Chem. 2017, 65, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Takahashi, M.; Ohnuma, S.; Unno, M.; Yoshino, Y.; Ouchi, K.; Takahashi, S.; Yamada, Y.; Shimodaira, H.; Ishioka, C. Microrna-193a-3p is specifically down-regulated and acts as a tumor suppressor in braf-mutated colorectal cancer. BMC Cancer 2017, 17, 723. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro cell migration and invasion assays. J. Vis. Exp. 2014, 51046. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, Y.C.; Tsai, T.-Y.; Yadav, V.K.; Deng, L.; Huang, C.-C.; Tzeng, Y.-M.; Yeh, C.-T.; Chen, M.-Y. 4-Acetyl-Antroquinonol B Improves the Sensitization of Cetuximab on Both Kras Mutant and Wild Type Colorectal Cancer by Modulating the Expression of Ras/Raf/miR-193a-3p Signaling Axis. Int. J. Mol. Sci. 2021, 22, 7508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147508
Chu YC, Tsai T-Y, Yadav VK, Deng L, Huang C-C, Tzeng Y-M, Yeh C-T, Chen M-Y. 4-Acetyl-Antroquinonol B Improves the Sensitization of Cetuximab on Both Kras Mutant and Wild Type Colorectal Cancer by Modulating the Expression of Ras/Raf/miR-193a-3p Signaling Axis. International Journal of Molecular Sciences. 2021; 22(14):7508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147508
Chicago/Turabian StyleChu, Yi Cheng, Tung-Yao Tsai, Vijesh Kumar Yadav, Li Deng, Chun-Chih Huang, Yew-Min Tzeng, Chi-Tai Yeh, and Ming-Yao Chen. 2021. "4-Acetyl-Antroquinonol B Improves the Sensitization of Cetuximab on Both Kras Mutant and Wild Type Colorectal Cancer by Modulating the Expression of Ras/Raf/miR-193a-3p Signaling Axis" International Journal of Molecular Sciences 22, no. 14: 7508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147508