
Biomarkers Associated with Organ-Specific Involvement in Juvenile Systemic Lupus Erythematosus

Abstract
:1. Introduction
2. Methodology
2.1. Search Strategy
2.2. Study Selection
2.3. Data Extraction
3. Autoantibodies as Biomarkers in JSLE
4. Renal Biomarkers
4.1. Classical Renal Biomarkers
4.2. Validated Novel Urinary Renal Biomarkers
4.2.1. NGAL
4.2.2. MCP-1
4.2.3. AGP-1
4.2.4. Ceruloplasmin
4.2.5. L-PGDS
4.2.6. Transferrin
4.3. Other Novel Renal Biomarkers Investigated
4.3.1. Renal Biomarker Combination Panels
4.3.2. Cardiovascular Biomarkers
5. Central Nervous System [CNS] Biomarkers
6. Arthritis Biomarkers
7. Skin Disease Biomarkers
8. Biomarkers for Haematological Manifestations
9. Other Biomarkers
9.1. IFN Signature and IFN Associated Proteins
9.2. Long Non-Coding and Micro RNAs
10. Discussion
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Abulaban, K.M.; Brunner, H.I. Biomarkers for Childhood-Onset Systemic Lupus Erythematosus. Curr. Rheumatol. Rep. 2015, 17, 1–8. [Google Scholar] [CrossRef]
- Rodriguez-Smith, J.; Brunner, H.I. Update on the treatment and outcome of systemic lupus erythematous in children. Curr. Opin. Rheumatol. 2019, 31, 464–470. [Google Scholar] [CrossRef]
- Smith, E.M.D.; Lythgoe, H.; Midgley, A.; Beresford, M.W.; Hedrich, C.M. Juvenile-onset systemic lupus erythematosus: Update on clinical presentation, pathophysiology and treatment options. Clin. Immunol. 2019, 209, 108274. [Google Scholar] [CrossRef] [PubMed]
- Massias, J.S.; Smith, E.M.D.; Al-Abadi, E.; Armon, K.; Bailey, K.; Ciurtin, C.; Davidson, J.; Gardner-Medwin, J.; Haslam, K.; Hawley, D.P.; et al. Clinical and laboratory characteristics in juvenile-onset systemic lupus erythematosus across age groups. Lupus 2020, 29, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M.; Smith, E.; Beresford, M.W. Juvenile-onset systemic lupus erythematosus (jSLE)–Pathophysiological concepts and treatment options. Best Pr. Res. Clin. Rheumatol. 2017, 31, 488–504. [Google Scholar] [CrossRef] [PubMed]
- Oni, L.; Wright, R.; Marks, S.; Beresford, M.W.; Tullus, K. Kidney outcomes for children with lupus nephritis. Pediatr. Nephrol. 2021, 36, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Malattia, C.; Martini, A. Paediatric-onset systemic lupus erythematosus. Best Pr. Res. Clin. Rheumatol. 2013, 27, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kao, A.H.; Manzi, S.; Ahearn, J.M. Biomarkers in systemic lupus erythematosus: Challenges and prospects for the future. Ther. Adv. Musculoskelet. Dis. 2013, 5, 210–233. [Google Scholar] [CrossRef]
- Groot, N.; de Graeff, N.; Avcin, T.; Bader-Meunier, B.; Brogan, P.; Doležalová, P.; Feldman, B.; Kone-Paut, I.; Lahdenne, P.; Marks, S.D.; et al. European evidence-based recommendations for diagnosis and treatment of childhood-onset systemic lupus erythematosus: The SHARE initiative. Ann. Rheum. Dis. 2017, 76, 1788–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.M.D.; Rasul, S.; Ciurtin, C.; Al-Abadi, E.; Armon, K.; Bailey, K.; Brennan, M.; Gardner-Medwin, J.; Haslam, K.; Hawley, D.P.; et al. Limited sensitivity and specificity of the ACR/EULAR-2019 classification criteria for SLE in JSLE?—observations from the UK JSLE Cohort Study. Rheumatology 2021. [Google Scholar] [CrossRef]
- Watson, L.; Leone, V.; Pilkington, C.; Tullus, K.; Rangaraj, S.; McDonagh, J.; Gardner-Medwin, J.; Wilkinson, N.; Riley, P.; Tizard, J.; et al. Disease activity, severity, and damage in the UK juvenile-onset systemic lupus erythematosus cohort. Arthritis Rheum. 2012, 64, 2356–2365. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, N.; Morgan, A.T.; Galloway, J.; Ionnoau, Y.; Beresford, M.W.; Isenberg, A.D. Differences in disease phenotype and severity in SLE across age groups. Lupus 2016, 25, 1542–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.A.; Peng, J.; Dönnes, P.; Coelewij, L.; Naja, M.; Radziszewska, A.; Wincup, C.; Peckham, H.; Isenberg, D.A.; Ioannou, Y.; et al. Disease-associated and patient-specific immune cell signatures in juvenile-onset systemic lupus erythematosus: Patient stratification using a machine-learning approach. Lancet Rheumatol. 2020, 2, e485–e496. [Google Scholar] [CrossRef]
- Binder, E.; Edelbauer, M. Use of Biomarkers in the Management of Children with Lupus. Curr. Rheumatol. Rep. 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Ahearn, J.M. The search for lupus biomarkers. Best Pr. Res. Clin. Rheumatol. 2009, 23, 507–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, J.H.; Parikh, C.R. Biomarkers for Diagnosis and Prognosis of AKI in Children: One Size Does Not Fit All. Clin. J. Am. Soc. Nephrol. 2017, 12, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.; Becker, M.L.; Jones, B.; Clements, M.; Leeder, J.S. Development of biomarkers to optimize pediatric patient management: What makes children different? Biomark. Med. 2011, 5, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.H.; Simard, J.F.; Costenbader, K.; Dore, B.T.; Ward, M.; Fortin, P.R.; Illei, G.G.; Manzi, S.; Mittleman, B.; Buyon, J.; et al. Methodologic Issues in the Validation of Putative Biomarkers and Surrogate Endpoints in Treatment Evaluation for Systemic Lupus Erythematosus. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Illei, G.G.; Lipsky, P.E. Biomarkers in systemic lupus erythematosus. Curr. Rheumatol. Rep. 2004, 6, 382–390. [Google Scholar] [CrossRef]
- Schiffenbauer, J.; Hahn, B.; Weisman, M.H.; Simon, L.S. Biomarkers, surrogate markers, and design of clinical trials of new therapies for systemic lupus erythematosus. Arthritis Rheum. 2004, 50, 2415–2422. [Google Scholar] [CrossRef]
- Utz, P.J. Multiplexed assays for identification of biomarkers and surrogate markers in systemic lupus erythematosus. Lupus 2004, 13, 304–311. [Google Scholar] [CrossRef]
- Petri, M.; Orbai, A.-M.; Alarcón, G.S.; Gordon, C.; Merrill, J.T.; Fortin, P.R.; Bruce, I.N.; Isenberg, D.; Wallace, D.J.; Nived, O.; et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2677–2686. [Google Scholar] [CrossRef]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Catalina, M.D.; Owen, K.A.; Labonte, A.C.; Grammer, A.C.; Lipsky, P.E. The pathogenesis of systemic lupus erythematosus: Harnessing big data to understand the molecular basis of lupus. J. Autoimmun. 2020, 110, 102359. [Google Scholar] [CrossRef] [PubMed]
- Arriens, C.; Wren, J.; Munroe, M.E.; Mohan, C. Systemic lupus erythematosus biomarkers: The challenging quest. Rheumatol. 2016, 56, i32–i45. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, U.; Mayer, G.; Herold, M.; Klotz, W.; Al-Jazrawi, K.S.; Lhotta, K. Sensitivity and specificity of autoantibody tests in the differential diagnosis of lupus nephritis. Lupus 2009, 18, 1276–1280. [Google Scholar] [CrossRef]
- Eriksson, C.; Kokkonen, H.; Johansson, M.; Hallmans, G.; Wadell, G.; Rantapää-Dahlqvist, S. Autoantibodies predate the onset of systemic lupus erythematosus in northern Sweden. Arthritis Res. 2011, 13, R30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of Autoantibodies before the Clinical Onset of Systemic Lupus Erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copple, S.S.; Sawitzke, A.D.; Wilson, A.M.; Tebo, A.E.; Hill, H.R. Enzyme-Linked Immunosorbent Assay Screening Then Indirect Immunofluorescence Confirmation of Antinuclear Antibodies. Am. J. Clin. Pathol. 2011, 135, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Wananukul, S.; Voramethkul, W.; Kaewopas, Y.; Hanvivatvong, O. Prevalence of positive antinuclear antibodies in healthy children. Asian Pac. J. Allergy Immunol. 2005, 23, 153–157. [Google Scholar]
- de Jesus, A.A.; Campos, L.M.A.; Liphaus, B.L.; Carneiro-Sampaio, M.; Mangueira, C.L.P.; Rosseto, E.A.; da Silva, C.A.; Scheinberg, M. Anticorpos anti-C1q, anticromatina/nucleossomo e anti-dsDNA em pacientes com lúpus eritematoso sistêmico juvenil. Rev. Bras. Reumatol. 2012, 52, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Lehman, T.J.; Hanson, V.; Singsen, B.H.; Kornreich, H.K.; Bernstein, B.; King, K. The role of antibodies directed against doublestranded DNA in the manifestations of systemic lupus erythematosus in childhood. J. Pediatr. 1980, 96, 657–661. [Google Scholar] [CrossRef]
- Oshiro, A.C.; Derbes, S.J.; Stopa, A.R.; Gedalia, A. Anti-Ro/SS-A and anti-La/SS-B antibodies associated with cardiac involvement in childhood systemic lupus erythematosus. Ann. Rheum. Dis. 1997, 56, 272–274. [Google Scholar] [CrossRef] [Green Version]
- Shergy, W.J.; Kredich, D.W.; Pisetsky, D.S. The relationship of anticardiolipin antibodies to disease manifestations in pediatric systemic lupus erythematosus. J. Rheumatol. 1988, 15, 1389–1394. [Google Scholar]
- Juřenčák, R.; Fritzler, M.; Tyrrell, P.; Hiraki, L.; Benseler, S.; Silverman, E. Autoantibodies in Pediatric Systemic Lupus Erythematosus: Ethnic Grouping, Cluster Analysis, and Clinical Correlations. J. Rheumatol. 2009, 36, 416–421. [Google Scholar] [CrossRef]
- To, C.H.; Petri, M. Is antibody clustering predictive of clinical subsets and damage in systemic lupus erythematosus? Arthritis Rheum. 2005, 52, 4003–4010. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; Rovin, B.H.; Lipsky, P.E. New Perspectives in Rheumatology: Biomarkers as Entry Criteria for Clinical Trials of New Therapies for Systemic Lupus Erythematosus: The Example of Antinuclear Antibodies and Anti-DNA. Arthritis Rheumatol. 2017, 69, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Petri, M.A.; Van Vollenhoven, R.F.; Buyon, J.; Levy, R.A.; Navarra, S.V.; Cervera, R.; Zhong, Z.J.; Freimuth, W.W. Baseline Predictors of Systemic Lupus Erythematosus Flares: Data from the Combined Placebo Groups in the Phase III Belimumab Trials. Arthritis Rheum. 2013, 65, 2143–2153. [Google Scholar] [CrossRef]
- Wu, H.; Zeng, J.; Yin, J.; Peng, Q.; Zhao, M.; Lu, Q. Organ-specific biomarkers in lupus. Autoimmun. Rev. 2017, 16, 391–397. [Google Scholar] [CrossRef]
- Oni, L.; Beresford, M.W.; Witte, D.; Chatzitolios, A.; Sebire, N.; Abulaban, K.; Shukla, R.; Ying, J.; Brunner, H.I. Inter-observer variability of the histological classification of lupus glomerulonephritis in children. Lupus 2017, 26, 1205–1211. [Google Scholar] [CrossRef]
- Isenberg, D.A.; Rahman, A.; Allen, E.; Farewell, V.; Akil, M.; Bruce, I.N.; D’Cruz, D.; Griffiths, B.; Khamashta, M.; Maddison, P.; et al. BILAG 2004. Development and initial validation of an updated version of the British Isles Lupus Assessment Group’s disease activity index for patients with systemic lupus erythematosus. Rheumatology 2005, 44, 902–906. [Google Scholar] [CrossRef] [Green Version]
- Gladman, D.D.; Ibañez, D.; Urowltz, M.B. Systemic Lupus Erythematosus Disease Activity Index. In Systemic Lupus Erythematosus; Elsevier: Amsterdam, The Netherlands, 2007; p. 524. [Google Scholar]
- Sandhu, V.; Quan, M. SLE and Serum Complement: Causative, Concomitant or Coincidental? Open Rheumatol. J. 2017, 11, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Watson, L.; Tullus, K.; Pilkington, C.; Chesters, C.; Marks, S.D.; Newland, P.; Jones, C.A.; Beresford, M.W. Urine biomarkers for monitoring juvenile lupus nephritis: A prospective longitudinal study. Pediatr. Nephrol. 2014, 29, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.D.; Jorgensen, A.L.; Beresford, M.W. Do classic blood biomarkers of JSLE identify active lupus nephritis? Evidence from the UK JSLE Cohort Study. Lupus 2017, 26, 1212–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, H.I.; Bennett, M.R.; Mina, R.; Suzuki, M.; Petri, M.; Kiani, A.N.; Pendl, J.; Witte, D.P.; Ying, J.; Rovin, B.H.; et al. Association of noninvasively measured renal protein biomarkers with histologic features of lupus nephritis. Arthritis Rheum. 2012, 64, 2687–2697. [Google Scholar] [CrossRef] [Green Version]
- Brunner, H.I.; Bennett, M.R.; Abulaban, K.; Klein-Gitelman, M.S.; O’Neil, K.M.; Tucker, L.B.; Ardoin, S.P.; Rouster-Stevens, K.A.; Onel, K.B.; Singer, N.G.; et al. Development of a Novel Renal Activity Index of Lupus Nephritis in Children and Young Adults. Arthritis Rheum. 2016, 68, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Wiers, K.; Brooks, E.B.; Greis, K.D.; Haines, K.; Klein-Gitelman, M.S.; Brunner, H.I. Initial Validation of a Novel Protein Biomarker Panel for Active Pediatric Lupus Nephritis. Pediatr. Res. 2009, 65, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Watson, L.; Midgley, A.; Pilkington, C.; Tullus, K.; Marks, S.; Holt, R.; Jones, C.; Beresford, M. Urinary monocyte chemoattractant protein 1 and alpha 1 acid glycoprotein as biomarkers of renal disease activity in juvenile-onset systemic lupus erythematosus. Lupus 2012, 21, 496–501. [Google Scholar] [CrossRef]
- Smith, E.M.D.; Yin, P.; Jorgensen, A.L.; Beresford, M.W.; on behalf of the UK JSLE Study Group. Clinical predictors of proteinuric remission following an LN flare-evidence from the UK JSLE cohort study. Pediatr. Rheumatol. 2018, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.; Magder, L.S.; Barr, S.G.; Petri, M. Decreases in anti-double-stranded DNA levels are associated with concurrent flares in patients with systemic lupus erythematosus. Arthritis Rheum. 2001, 44, 2342–2349. [Google Scholar] [CrossRef]
- Hemmelgarn, B.R.; Manns, B.J.; Lloyd, A.; James, M.T.; Klarenbach, S.; Quinn, R.R.; Wiebe, N.; Tonelli, M.; Network, F.T.A.K.D. Relation Between Kidney Function, Proteinuria, and Adverse Outcomes. JAMA 2010, 303, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Brunner, H.I.; Bennett, M.R.; Gulati, G.; Abulaban, K.; Klein-Gitelman, M.S.; Ardoin, S.P.; Tucker, L.B.; Rouster-Stevens, K.A.; Witte, D.; Ying, J.; et al. Urine Biomarkers to Predict Response to Lupus Nephritis Therapy in Children and Young Adults. J. Rheumatol. 2017, 44, 1239–1248. [Google Scholar] [CrossRef]
- Hinze, C.H.; Suzuki, M.; Klein-Gitelman, M.; Passo, M.H.; Olson, J.; Singer, N.; Haines, K.A.; Onel, K.; O’Neil, K.; Silverman, E.D.; et al. Neutrophil gelatinase-associated lipocalin is a predictor of the course of global and renal childhood-onset systemic lupus erythematosus disease activity. Arthritis Rheum. 2009, 60, 2772–2781. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Mueller, M.; Rutherford, C.; Passo, M.H.; Witte, D.; Grom, A.; Mishra, J.; Devarajan, P. Urinary neutrophil gelatinase–associated lipocalin as a biomarker of nephritis in childhood-onset systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Abd El Baky, A.N.E.D.; Assal, H.; Farid, T.; Rasheed, I.; Thabet, E.; Gheita, T. Serum cystatin C, urinary neutrophil gelatinase-associated lipocalin and N-acetyl-beta-D-glucosaminidase in juvenile and adult patients with systemic lupus erythematosus: Correlation with clinical manifestations, disease activity and damage. Saudi J. Kidney Dis. Transpl. 2015, 26, 497. [Google Scholar] [CrossRef] [PubMed]
- Hammad, A.; Mosaad, Y.; Elhanbly, S.; Youssef, H.; El Refaaey, A.; ElHusseini, F.; Bakr, A. Urinary neutrophil gelatinase-associated lipocalin as a marker of severe lupus nephritis in children. Lupus 2013, 22, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Eleuteri, A.; Goilav, B.; Lewandowski, L.; Phuti, A.; Rubinstein, T.; Wahezi, D.; Jones, C.; Marks, S.; Corkhill, R.; et al. A Markov Multi-State model of lupus nephritis urine biomarker panel dynamics in children: Predicting changes in disease activity. Clin. Immunol. 2019, 198, 71–78. [Google Scholar] [CrossRef]
- Smith, E.M.D.; Lewandowski, L.B.; Jorgensen, A.L.; Phuti, A.; Nourse, P.; Scott, C.; Beresford, M.W. Growing international evidence for urinary biomarker panels identifying lupus nephritis in children–verification within the South African Paediatric Lupus Cohort. Lupus 2018, 27, 2190–2199. [Google Scholar] [CrossRef]
- Schrezenmeier, E.V.; Barasch, J.; Budde, K.; Westhoff, T.; Schmidt-Ott, K.M. Biomarkers in acute kidney injury-pathophysiological basis and clinical performance. Acta Physiol. 2017, 219, 556–574. [Google Scholar] [CrossRef] [PubMed]
- Zappitelli, M.; Washburn, K.K.; Arikan, A.A.; Loftis, L.; Ma, Q.; Devarajan, P.; Parikh, C.R.; Goldstein, S.L. Urine neutrophil gelatinase-associated lipocalin is an early marker of acute kidney injury in critically ill children: A prospective cohort study. Crit. Care 2007, 11, R84. [Google Scholar] [CrossRef] [Green Version]
- Haase-Fielitz, A.; Bellomo, R.; Devarajan, P.; Story, D.; Matalanis, G.; Dragun, D.; Haase, M. Novel and conventional serum biomarkers predicting acute kidney injury in adult cardiac surgery—A prospective cohort study*. Crit. Care Med. 2009, 37, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Wiers, K.M.; Klein-Gitelman, M.S.; Haines, K.A.; Olson, J.; Onel, K.B.; O’Neil, K.; Passo, M.H.; Singer, N.G.; Tucker, L.; et al. Neutrophil gelatinase-associated lipocalin as a biomarker of disease activity in pediatric lupus nephritis. Pediatr. Nephrol. 2008, 23, 403–412. [Google Scholar] [CrossRef]
- Viedt, C.; Dechend, R.; Fei, J.; Hänsch, G.M.; Kreuzer, J.; Orth, S.R. MCP-1 Induces Inflammatory Activation of Human Tubular Epithelial Cells: Involvement of the Transcription Factors, Nuclear Factor-κB and Activating Protein-1. J. Am. Soc. Nephrol. 2002, 13, 1534–1547. [Google Scholar] [CrossRef] [Green Version]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Liu, X.H.; Donadelli, R.; Abbate, M.; Testa, D.; Corna, D.; Taraboletti, G.; Vecchi, A.; Dong, Q.G.; Rollins, B.J.; et al. Renal expression of monocyte chemoattractant protein-1 in lupus autoimmune mice. J. Am. Soc. Nephrol. 1997, 8, 720–729. [Google Scholar] [CrossRef]
- Tesch, G.H.; Maifert, S.; Schwarting, A.; Rollins, B.J.; Kelley, V.R. Monocyte Chemoattractant Protein 1–Dependent Leukocytic Infiltrates Are Responsible for Autoimmune Disease in Mrl-Faslpr Mice. J. Exp. Med. 1999, 190, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Song, H.; Birmingham, D.J.; Hebert, L.A.; Yu, C.-Y.; Nagaraja, H.N. Urine Chemokines as Biomarkers of Human Systemic Lupus Erythematosus Activity. J. Am. Soc. Nephrol. 2004, 16, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, T.; Mejdoubi, N.; Monnet, D.; Durand, G.; Porquet, D. Phenobarbital induction of α1-acid glycoprotein in primary rat hepatocyte cultures. Hepatology 1994, 20, 1584–1588. [Google Scholar] [CrossRef]
- Fournier, T.; Medjoubi-N, N.; Porquet, D. Alpha-1-acid glycoprotein. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2000, 1482, 157–171. [Google Scholar] [CrossRef]
- Kalmovarin, N.; Friedrichs, W.E.; O’Brien, H.V.; Linehan, L.A.; Bowman, B.H.; Yang, F. Extrahepatic expression of plasma protein genes during inflammation. Inflammation 1991, 15, 369–379. [Google Scholar] [CrossRef]
- Shibata, Y.; Tamura, K.; Ishida, N. Cultured Human Monocytes, Granulocytes and a Monoblastoid Cell Line (THP-1) Synthesize and Secrete Immunosuppressive Acidic Protein (a Type of α1-Acid Glycoprotein). Microbiol. Immunol. 1984, 28, 99–111. [Google Scholar] [CrossRef]
- Sorensen, C.J.; Butler-Dawson, J.; Dally, M.; Krisher, L.; Griffin, B.; Johnson, R.J.; Lemery, J.; Asensio, C.; Tenney, L.; Newman, L.S. Risk Factors and Mechanisms Underlying Cross-Shift Decline in Kidney Function in Guatemalan Sugarcane Workers. J. Occup. Environ. Med. 2019, 61, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Komori, H.; Watanabe, H.; Shuto, T.; Kodama, A.; Maeda, H.; Watanabe, K.; Hirofumi, K.; Otagiri, M.; Maruyama, T. α1-acid glycoprotein up-regulates CD163 via TLR4/CD14 protein pathway: Possible protection against hemolysis-induced oxidative stress. J. Biol. Chem. 2012, 287, 30688–30700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Watanabe, H.; Fujimura, R.; Nishida, K.; Nakamura, R.; Oshiro, S.; Imafuku, T.; Komori, H.; Miyahisa, M.; Tanaka, M.; et al. A downstream molecule of 1,25-dihydroxyvitamin D3, alpha-1-acid glycoprotein, protects against mouse model of renal fibrosis. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.; Walter, S.J.; Wolfs, T.G.A.M.; Hochepied, T.; Räbinä, J.; Heeringa, P.; Parkkinen, J.; Libert, C.; Buurman, W. Exogenous alpha-1-acid glycoprotein protects against renal ischemia-reperfusion injury by inhibition of inflammation and apoptosis. Transplantation 2004, 78, 1116–1124. [Google Scholar] [CrossRef]
- Gitlin, J.D. Transcriptional regulation of ceruloplasmin gene expression during inflammation. J. Biol. Chem. 1988, 263, 6281–6287. [Google Scholar] [CrossRef]
- Orzheshkovskyi, V.V.; Trishchynska, M.A. Ceruloplasmin: Its Role in the Physiological and Pathological Processes Neurophysiology; Springer: New York, NY, USA, 2019; Volume 51, pp. 141–149. [Google Scholar]
- Jiang, B.; Liu, G.; Zheng, J.; Chen, M.; Maimaitiming, Z.; Chen, M.; Liu, S.; Jiang, R.; Fuqua, B.K.; Dunaief, J.L.; et al. Hephaestin and ceruloplasmin facilitate iron metabolism in the mouse kidney. Sci. Rep. 2016, 6, 39470. [Google Scholar] [CrossRef]
- Urade, Y.; Eguchi, N. Lipocalin-type and hematopoietic prostaglandin D synthases as a novel example of functional convergence. Prostaglandins Other Lipid Mediat. 2002, 68–69, 375–382. [Google Scholar] [CrossRef]
- Kannaian, B.; Sharma, B.; Phillips, M.; Chowdhury, A.; Manimekalai, M.S.S.; Adav, S.S.; Ng, J.T.Y.; Kumar, A.; Lim, S.; Mu, Y.; et al. Abundant neuroprotective chaperone Lipocalin-type prostaglandin D synthase (L-PGDS) disassembles the Amyloid-β fibrils. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Mase, M.; Yamada, K.; Shimazu, N.; Seiki, K.; Oda, H.; Nakau, H.; Inui, T.; Li, W.; Eguchi, N.; Urade, Y. Lipocalin-type prostaglandin D synthase (beta-trace) in cerebrospinal fluid: A useful marker for the diagnosis of normal pressure hydrocephalus. Neurosci. Res. 2003, 47, 455–459. [Google Scholar] [CrossRef]
- Eguchi, Y.; Eguchi, N.; Oda, H.; Seiki, K.; Kijima, Y.; Matsu-Ura, Y.; Urade, Y.; Hayaishi, O. Expression of lipocalin-type prostaglandin D synthase (-trace) in human heart and its accumulation in the coronary circulation of angina patients. Proc. Natl. Acad. Sci. USA 1997, 94, 14689–14694. [Google Scholar] [CrossRef] [Green Version]
- Hirawa, N.; Uehara, Y.; Yamakado, M.; Toya, Y.; Gomi, T.; Ikeda, T.; Eguchi, Y.; Takagi, M.; Oda, H.; Seiki, K.; et al. Lipocalin-Type Prostaglandin D Synthase in Essential Hypertension. Hypertension 2002, 39, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Eguchi, N.; Eguchi, Y.; Numabe, A.; Nakajima, H.; Oda, H.; Seiki, K.; Hakamada-Taguchi, R.; Urade, Y.; Uehara, Y. Lipocalin-Type Prostaglandin D Synthase in Urine in Adriamycin-Induced Nephropathy of Mice. Nephron Physiol. 2004, 96, 42–51. [Google Scholar] [CrossRef]
- Rao, P.S.; Cavanagh, D.; Dietz, J.R.; Marsden, K.A.; O’Brien, W.F.; Spaziani, E. Dose-dependent effects of prostaglandin D2 on hemodynamics, renal function, and blood gas analyses. Am. J. Obstet. Gynecol. 1987, 156, 843–851. [Google Scholar] [CrossRef]
- Fraij, B.M. Transferrin and albumin excretion as a measure of glomerular function. Clin. Physiol. Biochem. 1989, 7, 296–302. [Google Scholar]
- Zhang, D.; Meyron-Holtz, E.; Rouault, T.A. Renal Iron Metabolism: Transferrin Iron Delivery and the Role of Iron Regulatory Proteins. J. Am. Soc. Nephrol. 2007, 18, 401–406. [Google Scholar] [CrossRef]
- Kazumi, T.; Hozumi, T.; Ishida, Y.; Ikeda, Y.; Kishi, K.; Hayakawa, M.; Yoshino, G. Increased urinary transferrin excretion predicts microalbuminuria in patients with type 2 diabetes. Diabetes Care 1999, 22, 1176–1180. [Google Scholar] [CrossRef]
- Bajema, I.M.; Wilhelmus, S.; Alpers, C.E.; Bruijn, J.A.; Colvin, R.B.; Cook, H.T.; D’Agati, V.D.; Ferrario, F.; Haas, M.; Jennette, J.C.; et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: Clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 2018, 93, 789–796. [Google Scholar] [CrossRef]
- Hill, G.S.; Delahousse, M.; Nochy, D.; Tomkiewicz, E.; Rémy, P.; Mignon, F.; Méry, J.-P. A new morphologic index for the evaluation of renal biopsies in lupus nephritis. Kidney Int. 2000, 58, 1160–1173. [Google Scholar] [CrossRef] [Green Version]
- Donohue, S.; Midgley, A.; Davies, J.; Wright, R.; Bruce, I.; Beresford, M.; Hedrich, C. Differential analysis of serum and urine S100 proteins in juvenile-onset systemic lupus erythematosus (jSLE). Clin. Immunol. 2020, 214, 108375. [Google Scholar] [CrossRef]
- Turnier, J.L.; Fall, N.; Thornton, S.; Witte, D.; Bennett, M.R.; Appenzeller, S.; Klein-Gitelman, M.S.; Grom, A.A.; Brunner, H.I. Urine S100 proteins as potential biomarkers of lupus nephritis activity. Arthritis Res. 2017, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hassan, W.; Behiry, E.; Mahgoub, M. Urinary Soluble Alpha Chain of the Interleukin-2 Receptor as a Biomarker of Active Lupus Nephritis in Egyptian Children with Juvenile Systemic Lupus Erythematosus. Arch. Rheumatol. 2020, 36, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; Abdel-Rahim, M.; Elsayh, K.I.; Hassanien, M.M.; Mahran, S.A.; Hetta, H.F. Natural Killer and Natural Killer T Cells in Juvenile Systemic Lupus Erythematosus: Relation to Disease Activity and Progression. Arch. Immunol. Ther. Exp. 2019, 67, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Wuthrich, R.P.; Snyder, T.L. Vascular cell adhesion molecule-1 (VCAM-1) expression in murine lupus nephritis. Kidney Int. 1992, 42, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Wu, T.; Xie, C.; Vanarsa, K.; Han, J.; Mahajan, T.; Oei, H.B.; Ahn, C.; Zhou, X.J.; Putterman, C.; et al. Urine VCAM-1 as a marker of renal pathology activity index in lupus nephritis. Arthritis Res. Ther. 2012, 14, R164. [Google Scholar] [CrossRef] [Green Version]
- Kawano, J.; Arora, R. The Role of Adiponectin in Obesity, Diabetes, and Cardiovascular Disease. J. CardioMetabolic Syndr. 2009, 4, 44–49. [Google Scholar] [CrossRef]
- Rovin, B.H.; Song, H.; Hebert, L.A.; Nadasdy, T.; Nadasdy, G.; Birmingham, D.J.; Yu, C.-Y.; Nagaraja, H.N. Plasma, urine, and renal expression of adiponectin in human systemic lupus erythematosus. Kidney Int. 2005, 68, 1825–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Johnson, A.C.M.; Becker, K. Renal cortical hemopexin accumulation in response to acute kidney injury. Am. J. Physiol. Physiol. 2012, 303, F1460–F1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T.; Nemeth, E. Iron Balance and the Role of Hepcidin in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Yu, J.; Prayogo, G.W.; Cao, W.; Wu, Y.; Jia, Z.; Zhang, A. Understanding kidney injury molecule 1: A novel immune factor in kidney pathophysiology. Am. J. Transl. Res. 2019, 11, 1219–1229. [Google Scholar]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.; Shah, R.; Biser, A.; Staicu, S.A.; Niranjan, T.; Garcia, A.M.; Gruenwald, A.; Thomas, D.B.; Shatat, I.F.; Supe, K.; et al. The Role of Osteopontin in the Development of Albuminuria. J. Am. Soc. Nephrol. 2008, 19, 884–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykjaer, A.; Dragun, D.; Walther, D.; Vorum, H.; Jacobsen, C.; Herz, J.; Melsen, F.; Christensen, E.I.; Willnow, T.E. An Endocytic Pathway Essential for Renal Uptake and Activation of the Steroid 25-(OH) Vitamin D3. Cell 1999, 96, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xie, Y.; Shao, X.; Ni, Z.; Mou, S. L-FABP: A novel biomarker of kidney disease. Clin. Chim. Acta 2015, 445, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kessel, C.; Holzinger, D.; Foell, D. Phagocyte-derived S100 proteins in autoinflammation: Putative role in pathogenesis and usefulness as biomarkers. Clin. Immunol. 2013, 147, 229–241. [Google Scholar] [CrossRef]
- Kar, S.; Paglialunga, S.; Islam, R. Cystatin C Is a More Reliable Biomarker for Determining eGFR to Support Drug Development Studies. J. Clin. Pharmacol. 2018, 58, 1239–1247. [Google Scholar] [CrossRef]
- Zhang, R.J.; Zhang, X.; Chen, J.; Shao, M.; Yang, Y.; Balaubramaniam, B.; Sun, X.L.; Ambrus, J.J.L.; He, J.; Li, Z.G. Serum soluble CD25 as a risk factor of renal impairment in systemic lupus erythematosus—A prospective cohort study. Lupus 2018, 27, 1100–1106. [Google Scholar] [CrossRef]
- Gupta, R.; Yadav, A.; Misra, R.; Aggarwal, A. Urinary sCD25 as a biomarker of lupus nephritis disease activity. Lupus 2015, 24, 273–279. [Google Scholar] [CrossRef]
- Chan, A.; Hong, D.-L.; Atzberger, A.; Kollnberger, S.; Filer, A.D.; Buckley, C.D.; McMichael, A.; Enver, T.; Bowness, P. CD56bright Human NK Cells Differentiate into CD56dim Cells: Role of Contact with Peripheral Fibroblasts. J. Immunol. 2007, 179, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.C.; Carlsson, E.; Midgley, A.; Smith, E.M.D.; Bruce, I.N.; Beresford, M.W.; Hedrich, C.M. A panel of urinary proteins predicts active lupus nephritis and response to rituximab treatment. Rheumatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.; Kari, J.; Pilkington, C.; Deanfield, J.; Shroff, R.; Marks, S.D.; Tullus, K. The vascular phenotype of children with systemic lupus erythematosus. Pediatr. Nephrol. 2015, 30, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Canpolat, N.; Kasapcopur, O.; Çalışkan, S.; Gokalp, S.; Bor, M.; Tasdemir, M.; Sever, L.; Arisoy, N. Ambulatory blood pressure and subclinical cardiovascular disease in patients with juvenile-onset systemic lupus erythematosus. Pediatr. Nephrol. 2012, 28, 305–313. [Google Scholar] [CrossRef]
- Huang, Y.-L.; Chung, H.-T.; Chang, C.-J.; Yeh, K.-W.; Chen, L.-C.; Huang, J.-L. Lymphopenia is a risk factor in the progression of carotid intima-media thickness in juvenile-onset systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 3766–3775. [Google Scholar] [CrossRef] [PubMed]
- Salomão, R.G.; De Carvalho, L.M.; Izumi, C.; Czernisz, É.S.; Rosa, J.C.; Antonini, S.R.R.; Bueno, A.C.; Almada, M.O.R.D.V.; Coelho-Landell, C.D.A.; Jordão, A.A.; et al. Homocysteine, folate, hs-C-reactive protein, tumor necrosis factor alpha and inflammatory proteins: Are these biomarkers related to nutritional status and cardiovascular risk in childhood-onset systemic lupus erythematosus? Pediatr. Rheumatol. 2018, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.A.; Waddington, K.E.; Coelewij, L.; Peng, J.; Naja, M.; Wincup, C.; Radziszewska, A.; Peckham, H.; Isenberg, D.A.; Ioannou, Y.; et al. Increased apolipoprotein-B:A1 ratio predicts cardiometabolic risk in patients with juvenile onset SLE. EBioMedicine 2021, 65, 103243. [Google Scholar] [CrossRef]
- Al, M.; Ng, L.; Tyrrell, P.; Bargman, J.; Bradley, T.; Silverman, E. A dipokines as novel biomarkers in paediatric systemic lupus erythematosus. Rheumatology 2009, 48, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, P.A.; Kumar, V.; Rodriguez, A.E.; Li, C.T.; Sanchez-Alvarez, C.; Thanarajasingam, U.; Zalewski, N.L.; Flanagan, E.P. MOG-IgG myelitis coexisting with systemic lupus erythematosus in the post-partum setting. Mult. Scler. J. 2020, 26, 97–1000. [Google Scholar] [CrossRef]
- Mostafa, G.A.; Ibrahim, D.H.; Shehab, A.A.; Mohammed, A.K. The role of measurement of serum autoantibodies in prediction of pediatric neuropsychiatric systemic lupus erythematosus. J. Neuroimmunol. 2010, 227, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Moraitis, E.; Stathopoulos, Y.; Hong, Y.; Al-Obaidi, M.; Mankad, K.; Hacohen, Y.; Sen, D.; Hemingway, C.; Eleftheriou, D. Aquaporin-4 IgG antibody-related disorders in patients with juvenile systemic lupus erythematosus. Lupus 2019, 28, 1243–1249. [Google Scholar] [CrossRef]
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific Incidence Rates of Myocardial Infarction and Angina in Women with Systemic Lupus Erythematosus: Comparison with the Framingham Study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Bernatsky, S.; Boivin, J.-F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Barsalou, J.; Bradley, T.J.; Silverman, E.D. Cardiovascular risk in pediatric-onset rheumatological diseases. Arthritis Res. Ther. 2013, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hersh, A.O.; Trupin, L.; Yazdany, J.; Panopalis, P.; Julian, L.; Katz, P.; Criswell, L.A.; Yelin, E. Childhood-onset disease as a predictor of mortality in an adult cohort of patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 1152–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, I.N. ‘Not only…but also’: Factors that contribute to accelerated atherosclerosis and premature coronary heart disease in systemic lupus erythematosus. Rheumatology 2005, 44, 1492–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanberg, L.E.; Sandborg, C.; Barnhart, H.X.; Ardoin, S.P.; Yow, E.; Evans, G.W.; Mieszkalski, K.L.; Ilowite, N.; Eberhard, A.; Levy, D.M.; et al. Premature atherosclerosis in pediatric systemic lupus erythematosus: Risk factors for increased carotid intima-media thickness in the atherosclerosis prevention in pediatric lupus erythematosus cohort. Arthritis Rheum. 2009, 60, 1496–1507. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.; Tyrell, P.; Ng, L.; Slorach, C.; Beyene, J.; Schneider, R.; Feldman, B.; Silverman, E. 6.3 DETERMINING EARLY CARDIOVASCULAR RISK PROFILES IN PAEDIATRIC RHEUMATIC DISEASE. Artery Res. 2009, 3, 157–158. [Google Scholar] [CrossRef]
- Su-Angka, N.; Khositseth, A.; Vilaiyuk, S.; Tangnararatchakit, K.; Prangwatanagul, W. Carotid intima-media thickness and arterial stiffness in pediatric systemic lupus erythematosus. Lupus 2017, 26, 989–995. [Google Scholar] [CrossRef]
- Nascif, A.K.S.; Hilário, M.O.E.; Terrer, M.T.R.A.; Ajzen, S.A.; D’Almeida, V.; Plavnik, F.L.; Christofalo, D.M.D.J. Endothelial function analysis and atherosclerotic risk factors in adolescents with systemic lupus erythematosus. Int. J. Adolesc. Med. Health 2007, 19. [Google Scholar] [CrossRef]
- Guo, Q.; Wu, L.M.; Wang, Z.; Shen, J.Y.; Su, X.; Wang, C.Q.; Gong, X.R.; Yan, Q.R.; He, Q.; Zhang, W.; et al. Early Detection of Silent Myocardial Impairment in Drug-Naive Patients with New-Onset Systemic Lupus Erythematosus: A Three-Center Prospective Study. Arthritis Rheumatol. 2018, 70, 2014–2024. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.A.; Woodard, P.K.; Dávila-Román, V.G.; Waggoner, A.D.; Gutierrez, F.R.; Zheng, J.; Eisen, S.A. Cardiac magnetic resonance imaging abnormalities in systemic lupus erythematosus: A preliminary report. Lupus 2005, 14, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Khoshdel, A.R.; Carney, S.L.; Nair, B.R.; Gillies, A. Better Management of Cardiovascular Diseases by Pulse Wave Velocity: Combining Clinical Practice with Clinical Research using Evidence-Based Medicine. Clin. Med. Res. 2007, 5, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Falaschi, F.; Ravelli, A.; Martignoni, A.; Migliavacca, D.; Sartori, M.; Pistorio, A.; Perani, G.; Martini, A. Nephrotic-range proteinuria, the major risk factor for early atherosclerosis in juvenile-onset systemic lupus erythematosus. Arthritis Rheum. 2000, 43, 1405–1409. [Google Scholar] [CrossRef]
- Yu, H.-H.; Chen, P.-C.; Yang, Y.-H.; Wang, L.-C.; Lee, J.-H.; Lin, Y.-T.; Chiang, B.-L. Statin reduces mortality and morbidity in systemic lupus erythematosus patients with hyperlipidemia: A nationwide population-based cohort study. Atherosclerosis 2015, 243, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Limon, P.; Barbarroja, N.; Perez-Sanchez, C.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Almadén, Y.; Segui, P.; Khraiwesh, H.; et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: Effects of in vivo statin treatment. Ann. Rheum. Dis. 2015, 74, 1450–1458. [Google Scholar] [CrossRef]
- Petri, M.A.; Kiani, A.N.; Post, W.; Christopher-Stine, L.; Magder, L.S. Lupus Atherosclerosis Prevention Study (LAPS). Ann. Rheum. Dis. 2010, 70, 760–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanberg, L.E.; Sandborg, C.; Barnhart, H.X.; Ardoin, S.P.; Yow, E.; Evans, G.W.; Mieszkalski, K.L.; Ilowite, N.T.; Eberhard, A.; Imundo, L.F.; et al. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum. 2011, 64, 285–296. [Google Scholar] [CrossRef]
- Robinson, A.; Tangpricha, V.; Yow, E.; Gurion, R.; Schanberg, L.E.; McComsey, G. A31: Vitamin D Status is a Determinant of the Effect of Atorvastatin on Carotid Intima Medial Thickening Progression Rate in Children with Lupus: An Atherosclerosis Prevention in Pediatric Lupus Erythematosus Substudy. Arthritis Rheumatol. 2014, 66, S47–S48. [Google Scholar] [CrossRef]
- Ardoin, S.P.; Schanberg, L.E.; Sandborg, C.I.; Barnhart, H.X.; Evans, G.W.; Yow, E.; Mieszkalski, K.L.; Ilowite, N.; Eberhard, A.; Imundo, L.F.; et al. Secondary analysis of APPLE study suggests atorvastatin may reduce atherosclerosis progression in pubertal lupus patients with higher C reactive protein. Ann. Rheum. Dis. 2013, 73, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Tyrrell, P.N.; Beyene, J.; Benseler, S.M.; Sarkissian, T.; Silverman, E.D. Predictors of lipid abnormalities in children with new-onset systemic lupus erythematosus. J. Rheumatol. 2007, 34, 2112–2119. [Google Scholar]
- Posadas-Romero, C.; Torres-Tamayo, M.; Zamora-González, J.; Aguilar-Herrera, B.E.; Posadas-Sánchez, R.; Cardoso-Saldaña, G.; De Guevara, G.L.; Solis-Vallejo, E.; El Hafidi, M. High insulin levels and increased low-density lipoprotein oxidizability in pediatric patients with systemic lupus erythematosus. Arthritis Rheum. 2004, 50, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Ardoin, S.P.; Sandborg, C.; Schanberg, L.E. Review: Management of dyslipidemia in children and adolescents with systemic lupus erythematosus. Lupus 2007, 16, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Soep, J.B.; Mietus-Snyder, M.; Malloy, M.J.; Witztum, J.L.; Von Scheven, E. Assessment of atherosclerotic risk factors and endothelial function in children and young adults with pediatric-onset systemic lupus erythematosus. Arthritis Rheum. 2004, 51, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Croca, S.; Waddington, K.; Sofat, R.; Griffin, M.; Nicolaides, A.; Isenberg, D.A.; Torra, I.P.; Rahman, A.; Jury, E.C. Cross-talk between iNKT cells and monocytes triggers an atheroprotective immune response in SLE patients with asymptomatic plaque. Sci. Immunol. 2016, 1, eaah4081. [Google Scholar] [CrossRef] [PubMed]
- Coelewij, L.; Waddington, K.E.; Robinson, G.A.; Chocano, E.; McDonnell, T.; Farinha, F.; Peng, J.; Dönnes, P.; Smith, E.; Croca, S.; et al. Serum Metabolomic Signatures Can Predict Subclinical Atherosclerosis in Patients With Systemic Lupus Erythematosus. Arter. Thromb. Vasc. Biol. 2021, 41, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Ardoin, S.P.; Schanberg, L.E.; Sandborg, C.; Yow, E.; Barnhart, H.X.; Mieszkalski, K.L.; Ilowite, N.T.; Scheven, E.; Eberhard, A.; Levy, D.M.; et al. Laboratory markers of cardiovascular risk in pediatri SLE: The APPLE baseline cohort. Lupus. 2010, 19, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.; Barsalou, J.; Bradley, T.; Slorach, C.; Reynolds, J.; Hasni, S.; Thompson, B.; Ng, L.; Levy, D.M.; Silverman, E.D.; et al. Brief Report: Endothelial Progenitor Cell Phenotype and Function Are Impaired in Childhood-Onset Systemic Lupus Erythematosus. Arthritis Rheumatol. 2015, 67, 2257–2262. [Google Scholar] [CrossRef]
- Gattorno, M.; Vignola, S.; Barbano, G.; Sormani, M.P.; Sabatini, F.; Buoncompagni, A.; Picco, P.; Pistoia, V. Tumor necrosis factor induced adhesion molecule serum concentrations in Henoch-Schönlein purpura and pediatric systemic lupus erythematosus. J. Rheumatol. 2000, 27, 2251–2255. [Google Scholar]
- Rubinstein, T.B.; Putterman, C.; Goilav, B. Biomarkers for CNS involvement in pediatric lupus. Biomark. Med. 2015, 9, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Steens, S.C.A.; Bosma, G.P.T.; Cate, R.T.; Doornbos, J.; Kros, J.M.; Van Der Laan, L.J.W.; Steup-Beekman, G.M.; Van Buchem, M.A.; Huizinga, T.W.J. A neuroimaging follow up study of a patient with juvenile central nervous system systemic lupus erythematosus. Ann. Rheum. Dis. 2003, 62, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Hanly, J.G.; Urowitz, M.B.; Su, L.; Bae, S.-C.; Gordon, C.; Clarke, A.; Bernatsky, S.; Vasudevan, A.; Isenberg, D.; Rahman, A.; et al. Autoantibodies as biomarkers for the prediction of neuropsychiatric events in systemic lupus erythematosus. Ann. Rheum. Dis. 2011, 70, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.H.; Corzillius, M.; Bae, S.; Lew, R.A.; Fortin, P.R.; Gordon, C.; Isenberg, D.; Alarcon, G.; Straaton, K.V.; Denburg, J.; et al. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999, 42, 599–608. [Google Scholar]
- Muscal, E.; Myones, B.L. The role of autoantibodies in pediatric neuropsychiatric systemic lupus erythematosus. Autoimmun. Rev. 2007, 6, 215–217. [Google Scholar] [CrossRef]
- Kaleda, M.I.; Nikishina, I.P. Neuropsychiatric involvement in juvenile-onset systemic lupus erythematosus. Rheumatol. Sci. Pr. 2020, 58, 437–442. [Google Scholar] [CrossRef]
- Jeltsch-David, H.; Muller, S. Neuropsychiatric systemic lupus erythematosus: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2014, 10, 579–596. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef] [Green Version]
- Aldar, H.; Lapa, A.; Belini, B.; Sinicato, N.; Postal, M.; Fernandes, P.; Marini, R.; Appenzeller, S. AB1200 Cognitive impairment associated with S100β protein in childhood-onset systemic lupus erythematosus. Ann. Rheum. Dis. 2013, 71, 706. [Google Scholar] [CrossRef]
- Sibbitt, W.L.; Brandt, J.R.; Johnson, C.R.; Maldonado, M.E.; Patel, S.R.; Ford, C.C.; Bankhurst, A.D.; Brooks, W.M. The incidence and prevalence of neuropsychiatric syndromes in pediatric onset systemic lupus erythematosus. J. Rheumatol. 2002, 29, 1536–1542. [Google Scholar]
- Lee, W.-F.; Wu, C.-Y.; Yang, H.-Y.; Lee, W.-I.; Chen, L.-C.; Ou, L.-S.; Huang, J.-L. Biomarkers associating endothelial dysregulation in pediatric-onset systemic lupus erythematous. Pediatr. Rheumatol. 2019, 17, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.-H.; Lee, J.-H.; Wang, L.-C.; Yang, Y.-H.; Chiang, B.-L. Neuropsychiatric manifestations in pediatric systemic lupus erythematosus: A 20-year study. Lupus 2006, 15, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.; Amissah-Arthur, M.B.; Gayed, M.; Brown, S.; Bruce, I.N.; D’Cruz, D.; Empson, B.; Griffiths, B.; Jayne, D.; Khamashta, M.; et al. The British Society for Rheumatology guideline for the management of systemic lupus erythematosus in adults. Rheumatology 2018, 57, e1–e45. [Google Scholar] [CrossRef] [Green Version]
- Gazar, Y.; Rashed, S. AB0978 Neurological Manifestations of Pediatric Systemic Lupus Erythemtosus in Egyption patients. Ann. Rheum. Dis. 2019, 78, 1955–1956. [Google Scholar]
- Duzova, A.; Bakkaloglu, A. Central Nervous System Involvement in Pediatric Rheumatic Diseases: Current Concepts in Treatment. Curr. Pharm. Des. 2008, 14, 1295–1301. [Google Scholar] [CrossRef]
- Joseph, F.G.; Scolding, N.J. Neurolupus. Pract. Neurol. 2010, 10, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Harel, L.; Sandborg, C.; Lee, T.; Von Scheven, E. Neuropsychiatric manifestations in pediatric systemic lupus erythematosus and association with antiphospholipid antibodies. J. Rheumatol. 2006, 33, 1873–1877. [Google Scholar] [PubMed]
- Singh, S.; Gupta, M.K.; Ahluwalia, J.; Singh, P.; Malhi, P. Neuropsychiatric manifestations and antiphospholipid antibodies in pediatric onset lupus: 14 years of experience from a tertiary center of North India. Rheumatol. Int. 2009, 29, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Avčin, T.; Benseler, S.M.; Tyrrell, P.N.; Čučnik, S.; Silverman, E.D. A followup study of antiphospholipid antibodies and associated neuropsychiatric manifestations in 137 children with systemic lupus erythematosus. Arthritis Rheum. 2008, 59, 206–213. [Google Scholar] [CrossRef]
- Press, J.; Palayew, K.; Laxer, R.M.; Elkon, K.; Eddy, A.; Rakoff, D.; Silverman, E.D. Antiribosomal P antibodies in pediatric patients with systemic lupus erythematosus and psychosis. Arthritis Rheum. 1996, 39, 671–676. [Google Scholar] [CrossRef]
- Abrol, E.; Coutinho, E.; Chou, M.; Hart, M.; Vincent, A.; Howard, R.; Zandi, M.S.; Isenberg, D. Psychosis in systemic lupus erythematosus (SLE): 40-year experience of a specialist centre. Rheumatology 2021, keab160. [Google Scholar] [CrossRef] [PubMed]
- Vega-Fernandez, P.; White, S.V.; Zelko, F.; Ruth, N.M.; Levy, D.M.; Muscal, E.; Klein-Gitelman, M.S.; Huber, A.M.; Tucker, L.B.; Roebuck-Spencer, T.; et al. Cognitive Performance Scores for the Pediatric Automated Neuropsychological Assessment Metrics in Childhood-Onset Systemic Lupus Erythematosus. Arthritis Rheum. 2015, 67, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- De Amorim, J.; Kishimoto, S.; Fernandes, P.; Silva, C.; Marini, R.; Appenzeller, S. 243 Validation of pedanam as an instrument of cognitive evaluation in neuropsychiatric SLE. Lupus Sci. Med. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- AlE’Ed, A.; Vega-Fernandez, P.; Muscal, E.; Hinze, C.H.; Tucker, L.B.; Appenzeller, S.; Bader-Meunier, B.; Roth, J.; Torrente-Segarra, V.; Klein-Gitelman, M.S.; et al. Challenges of Diagnosing Cognitive Dysfunction with Neuropsychiatric Systemic Lupus Erythematosus in Childhood. Arthritis Rheum. 2017, 69, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, N.; Bargalló, N.; Cervera, R. Advanced and conventional magnetic resonance imaging in neuropsychiatric lupus [version 2; referees: 3 approved]. F1000Research 2015, 4, 162. [Google Scholar]
- Silverman, E. Pediatric systemic lupus erythematosus. Futur. Rheumatol. 2007, 2, 23–50. [Google Scholar] [CrossRef]
- DiFrancesco, M.W.; Holland, S.K.; Ris, M.D.; Adler, C.M.; Nelson, S.; DelBello, M.P.; Altaye, M.; Brunner, H.I. Functional magnetic resonance imaging assessment of cognitive function in childhood-onset systemic lupus erythematosus: A pilot study. Arthritis Rheum. 2007, 56, 4151–4163. [Google Scholar] [CrossRef]
- Barraclough, M.; Elliott, R.; McKie, S.; Parker, B.; Bruce, I. Cognitive dysfunction and functional magnetic resonance imaging in systemic lupus erythematosus. Lupus 2015, 24, 1239–1247. [Google Scholar] [CrossRef]
- DiFrancesco, M.W.; Gitelman, D.R.; Klein-Gitelman, M.S.; Sagcal-Gironella, A.C.P.; Zelko, F.; Beebe, D.; Parrish, T.; Hummel, J.; Ying, J.; I Brunner, H. Functional neuronal network activity differs with cognitive dysfunction in childhood-onset systemic lupus erythematosus. Arthritis Res. Ther. 2013, 15, R40. [Google Scholar] [CrossRef] [Green Version]
- Navallas, M.; Clemente, E.J.I.; Iglesias, E.; Rebollo-Polo, M.; Antón, J.; Navarro, O. Connective Tissue Disorders in Childhood: Are They All the Same? Radiographics 2019, 39, 229–250. [Google Scholar] [CrossRef] [PubMed]
- Tani, C.; Carli, L.; Stagnaro, C.; Elefante, E.; Signorini, V.; Balestri, F.; Sedie, A.D.; Mosca, M. Imaging of joints in systemic lupus erythematosus. Clin. Exp. Rheumatol. 2018, 36, 68–73. [Google Scholar]
- Di Matteo, A.; Isidori, M.; Corradini, D.; Cipolletta, E.; McShane, A.; De Angelis, R.; Filippucci, E.; Grassi, W. Ultrasound in the assessment of musculoskeletal involvement in systemic lupus erythematosus: State of the art and perspectives. Lupus. 2019, 28, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Ruano, C.A.; Malheiro, R.; Oliveira, J.F.; Pinheiro, S.; Vieira, L.S.; Moraes-Fontes, M.F. Ultrasound detects subclinical joint inflammation in the hands and wrists of patients with systemic lupus erythematosus without musculoskeletal symptoms. Lupus Sci. Med. 2017, 4, e000184. [Google Scholar] [CrossRef] [Green Version]
- Gabba, A.; Piga, M.; Vacca, A.; Porru, G.; Garau, P.; Cauli, A.; Mathieu, A. Joint and tendon involvement in systemic lupus erythematosus: An ultrasound study of hands and wrists in 108 patients. Rheumatology 2012, 51, 2278–2285. [Google Scholar] [CrossRef] [Green Version]
- Witte, T.; Hartung, K.; Sachse, C.; Matthias, T.; Fricke, M.; Kalden, J.R.; Lakomek, H.J.; Peter, H.H.; Schmidt, R.E. Rheumatoid factors in systemic lupus erythematosus: Association with clinical and laboratory parameters. Rheumatol. Int. 2000, 19, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Fedrigo, A.; Santos, T.A.F.G.D.; Nisihara, R.; Skare, T. The lupus patient with positive rheumatoid factor. Lupus 2018, 27, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, J.; Li, X.-X.; Li, C.; Li, L.; Li, Z.-G. What can we learn from the presence of anti-cyclic citrullinated peptide antibodies in systemic lupus erythematosus? Jt. Bone Spine 2009, 76, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Qing, Y.-F.; Zhang, Q.-B.; Zhou, J.-G.; Yuan, G.-H.; Wei, J.; Xing, Y.; Liu, J.-P.; Jiang, L.; Chen, J.-P. The detecting and clinical value of anti-cyclic citrullinated peptide antibodies in patients with systemic lupus erythematosus. Lupus 2009, 18, 713–717. [Google Scholar] [CrossRef]
- Yoon, H.-S.; Kim, K.-J.; Baek, I.-W.; Park, Y.-J.; Kim, W.-U.; Yoon, C.-H.; Cho, C.-S. Ultrasonography is useful to detect subclinical synovitis in SLE patients without musculoskeletal involvement before symptoms appear. Clin. Rheumatol. 2014, 33, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Iagnocco, A.; Ceccarelli, F.; Rizzo, C.; Truglia, S.; Massaro, L.; Spinelli, F.R.; Vavala, C.; Valesini, G.; Conti, F. Ultrasound evaluation of hand, wrist and foot joint synovitis in systemic lupus erythematosus. Rheumatology 2013, 53, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Matteo, A.; Satulu, I.; Di Carlo, M.; Lato, V.; Filippucci, E.; Grassi, W. Entheseal involvement in systemic lupus erythematosus: Are we missing something? Lupus 2017, 26, 320–328. [Google Scholar] [CrossRef]
- Mahmoud, K.; Zayat, A.S.; Yusof, M.Y.M.; Dutton, K.; Teh, L.S.; Yee, C.-S.; D’Cruz, D.; Ng, N.; Isenberg, D.; Ciurtin, C.; et al. Ultrasound to identify SLE patients with musculoskeletal symptoms who respond best to therapy: The USEFUL longitudinal multicentre study. Rheumatology 2021, keab288. [Google Scholar] [CrossRef]
- Demirkaya, E.; Özçakar, L.; Türker, T.; Haghari, S.; Ayaz, N.A.; Bakkaloǧlu, A.; Ozen, S. Musculoskeletal sonography in juvenile systemic lupus erythematosus. Arthritis Care Res. 2009, 61, 58–60. [Google Scholar] [CrossRef] [Green Version]
- Grönhagen, C.; Fored, C.; Granath, F.; Nyberg, F. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: A population-based cohort of 1088 patients in Sweden. Br. J. Dermatol. 2011, 164, 1335–1341. [Google Scholar] [CrossRef]
- Dickey, B.; Holland, K.; Drolet, B.; Galbraith, S.; Lyon, V.; Siegel, D.; Chiu, Y. Demographic and clinical characteristics of cutaneous lupus erythematosus at a paediatric dermatology referral centre. Br. J. Dermatol. 2013, 169, 428–433. [Google Scholar] [CrossRef]
- AlKharafi, N.N.A.H.; Alsaeid, K.; Alsumait, A.; Al-Sabah, H.; Al-Ajmi, H.; Rahim, J.A.; Al-Enezi, H.; Nanda, A. Cutaneous Lupus Erythematosus in Children: Experience from a Tertiary Care Pediatric Dermatology Clinic. Pediatr. Dermatol. 2016, 33, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Chiewchengchol, D.; Murphy, R.; Edwards, S.W.; Beresford, M.W. Mucocutaneous manifestations in juvenile-onset systemic lupus erythematosus: A review of literature. Pediatr. Rheumatol. 2015, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aringer, M.; Johnson, S.R. Classifying and diagnosing systemic lupus erythematosus in the 21st century. Rheumatology 2020, 59, v4–v11. [Google Scholar] [CrossRef] [PubMed]
- Wananukul, S.; Watana, D.; Pongprasit, P. Cutaneous manifestations of childhood systemic lupus erythematosus. Pediatr. Dermatol. 1998, 15, 342–346. [Google Scholar] [CrossRef]
- AlE’Ed, A.; Aydin, P.O.A.; Al Mutairi, N.; Alsaleem, A.; Sonmez, H.E.; Henrickson, M.; Huggins, J.L.; Ozen, S.; Al-Mayouf, S.M.; Brunner, H.I. Validation of the Cutaneous Lupus Erythematosus Disease Area and Severity Index and pSkindex27 for use in childhood-onset systemic lupus erythematosus. Lupus Sci. Med. 2018, 5, e000275. [Google Scholar] [CrossRef]
- Chiewchengchol, D.; Murphy, R.; Morgan, T.; Edwards, S.W.; Leone, V.; Friswell, M.; Pilkington, C.; Tullus, K.; Rangaraj, S.; McDonagh, J.; et al. Mucocutaneous manifestations in a UK national cohort of juvenile-onset systemic lupus erythematosus patients. Rheumatology 2014, 53, 1504–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, A.M.; Su, J.; Mussani, F.; Siddha, S.K.; Gladman, D.D.; Urowitz, M.B. Prognostic implications of active discoid lupus erythematosus and malar rash at the time of diagnosis of systemic lupus erythematosus: Results from a prospective cohort study. Lupus 2016, 25, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Chottawornsak, N.; Rodsaward, P.; Suwannachote, S.; Rachayon, M.; Rattananupong, T.; Deekajorndech, T.; Asawanonda, P.; Chiewchengchol, D.; Rerknimitr, P. Skin signs in juvenile- and adult-onset systemic lupus erythematosus: Clues to different systemic involvement. Lupus 2018, 27, 2069–2075. [Google Scholar] [CrossRef]
- Arkin, L.M.; Ansell, L.; Rademaker, A.; Curran, M.L.; Miller, M.L.; Wagner, A.; Kenner-Bell, B.M.; Chamlin, S.L.; Mancini, A.J.; Klein-Gitelman, M.; et al. The natural history of pediatric-onset discoid lupus erythematosus. J. Am. Acad. Dermatol. 2015, 72, 628–633. [Google Scholar] [CrossRef]
- Fenniche, S.; Triki, S.; Benmously, R.; Ben Ammar, H.M.M.F.; Mokhtar, I. Lupus erythematosus in children: A report of six cases. Dermatol. Online J. 2005, 11. [Google Scholar] [CrossRef]
- Walling, H.W.; Sontheimer, R.D. Cutaneous Lupus Erythematosus. Am. J. Clin. Dermatol. 2009, 10, 365–381. [Google Scholar] [CrossRef]
- Werth, V.P. Clinical manifestations of cutaneous lupus erythematosus. Autoimmun. Rev. 2005, 4, 296–302. [Google Scholar] [CrossRef]
- Zhu, J.L.; Black, S.M.; Chong, B.F. Role of biomarkers in the diagnosis and prognosis of patients with cutaneous lupus erythematosus. Ann. Transl. Med. 2021, 9, 429. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Henault, J.; Karnell, J.L.; Parker, M.L.; Riggs, J.M.; Sinibaldi, D.; Taylor, D.K.; Ettinger, R.; Grant, E.P.; Sanjuan, M.A.; et al. SLE Plasma Profiling Identifies Unique Signatures of Lupus Nephritis and Discoid Lupus. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogunsanya, M.E.; Cho, S.K.; Hudson, A.; Chong, B.F. Factors associated with quality of life in cutaneous lupus erythematosus using the Revised Wilson and Cleary Model. Lupus 2020, 29, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.C. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997, 40, 1725. [Google Scholar] [CrossRef] [PubMed]
- Fayyaz, A.; Igoe, A.; Kurien, B.T.; Danda, D.; James, J.A.; Stafford, H.; Scofield, R.H. Haematological manifestations of lupus. Lupus Sci. Med. 2015, 2, e000078. [Google Scholar] [CrossRef] [Green Version]
- Isenberg, D.; Garton, M.; Reichlin, M.W. Long-term follow-up of autoantibody profiles in black female lupus patients and clinical comparison with Caucasian and Asian patients. Rheumatology 1997, 36, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Vilá, L.M.; Molina, M.J.; Mayor, A.M.; Peredo, R.A.; Santaella, M.L.; Vilá, S. Clinical and prognostic value of autoantibodies in puerto Ricans with systemic lupus erythematosus. Lupus 2006, 15, 892–898. [Google Scholar] [CrossRef]
- Yu, H.-H.; Wang, L.-C.; Lee, J.-H.; Lee, C.-C.; Yang, Y.-H.; Chiang, B.-L. Lymphopenia is associated with neuropsychiatric manifestations and disease activity in paediatric systemic lupus erythematosus patients. Rheumatology 2007, 46, 1492–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilá, L.M.; Alarcón, G.S.; McGwin, G.; Bastian, H.M.; Fessler, B.J.; Reveille, J.D. LUMINA Study Group Systemic lupus erythematosus in a multiethnic US cohort, XXXVII: Association of lymphopenia with clinical manifestations, serologic abnormalities, disease activity, and damage accrual. Arthritis Rheum. 2006, 55, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.I.; Silverman, E.D.; To, T.; Bombardier, C.; Feldman, B.M. Risk factors for damage in childhood-onset systemic lupus erythematosus: Cumulative disease activity and medication use predict disease damage. Arthritis Rheum. 2002, 46, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.O.; Corrente, J.E.; Saad-Magalhães, C. Chronic active disease pattern predicts early damage in juvenile systemic lupus erythematosus. Lupus 2015, 24, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Kirou, K.A.; Lee, C.; George, S.; Louca, K.; Peterson, M.G.E.; Crow, M.K. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005, 52, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Landolt-Marticorena, C.; Bonventi, G.; Lubovich, A.; Ferguson, C.; Unnithan, T.; Su, J.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Wither, J. Lack of association between the interferon-α signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann. Rheum. Dis. 2008, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Fu, W.; Ranger, A.; Allaire, N.; Cullen, P.; Magder, L.S.; Zhang, Y. Association between changes in gene signatures expression and disease activity among patients with systemic lupus erythematosus. BMC Med. Genom. 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Rönnblom, L.; Eloranta, M.-L.; Alm, G.V. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 408–420. [Google Scholar] [CrossRef]
- Niewold, T.B.; Clark, D.N.; Salloum, R.; Poole, B.D. Interferon Alpha in Systemic Lupus Erythematosus. J. Biomed. Biotechnol. 2010, 2010, 948364. [Google Scholar] [CrossRef] [Green Version]
- Oka, H.; Hirohata, S. Erratum: Regulation of human B cell responsiveness by interferon-α: Interferon-α-mediated suppression of B cell function is reversed through direct interactions between monocytes and B cells. Cell. Immunol. 1993, 146, 238–248. [Google Scholar] [CrossRef]
- Wahadat, M.J.; Bodewes, I.L.A.; Maria, N.I.; Van Helden-Meeuwsen, C.G.; Van Dijk-Hummelman, A.; Steenwijk, E.C.; Kamphuis, S.; Versnel, M.A. Type I IFN signature in childhood-onset systemic lupus erythematosus: A conspiracy of DNA- and RNA-sensing receptors? Arthritis Res. 2018, 20, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesser, A.; de Carvalho, L.M.; Sandrin-Garcia, P.; Pin, A.; Pastore, S.; Taddio, A.; Roberti, L.R.; Queiroz, R.G.D.P.; Ferriani, V.P.L.; Crovella, S.; et al. Higher interferon score and normal complement levels may identify a distinct clinical subset in children with systemic lupus erythematosus. Arthritis Res. 2020, 22, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, W.P.; Maciuca, R.; Wolslegel, K.; Tew, W.; Abbas, A.R.; Chaivorapol, C.; Morimoto, A.; McBride, J.M.; Brunetta, P.; Richardson, B.C.; et al. Association of the interferon signature metric with serological disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci. Med. 2015, 2, e000080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, T.; Grützkau, A.; Hirseland, H.; Huscher, D.; Dähnrich, C.; Dzionek, A.; Ozimkowski, T.; Schlumberger, W.; Enghard, P.; Radbruch, A.; et al. IFNα and its response proteins, IP-10 and SIGLEC-1, are biomarkers of disease activity in systemic lupus erythematosus. Ann. Rheum. Dis. 2012, 72, 1639–1645. [Google Scholar] [CrossRef]
- Oliveira, J.J.; Karrar, S.; Rainbow, D.B.; Pinder, C.L.; Clarke, P.; García, A.R.; Al-Assar, O.; Burling, K.; Morris, S.; Stratton, R.; et al. The plasma biomarker soluble SIGLEC-1 is associated with the type I interferon transcriptional signature, ethnic background and renal disease in systemic lupus erythematosus. Arthritis Res. 2018, 20, 152. [Google Scholar] [CrossRef] [Green Version]
- El-Sherbiny, Y.M.; Yusof, Y.M.; Psarras, A.; Hensor, E.M.A.; Kabba, K.Z.; Dutton, K.; Mohamed, A.A.A.; Elewaut, D.; McGonagle, D.; Tooze, R.; et al. B Cell Tetherin: A Flow Cytometric Cell-Specific Assay for Response to Type I Interferon Predicts Clinical Features and Flares in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 72, 769–779. [Google Scholar] [CrossRef]
- Webb, K.; Peckham, H.; Radziszewska, A.; Menon, M.; Oliveri, P.; Simpson, F.; Deakin, C.T.; Lee, S.; Ciurtin, C.; Butler, G.; et al. Sex and Pubertal Differences in the Type 1 Interferon Pathway Associate With Both X Chromosome Number and Serum Sex Hormone Concentration. Front. Immunol. 2019, 9, 3167. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.A.; Morand, E.F.; Bruce, I.N.; Manzi, S.; Kalunian, K.C.; Vital, E.M.; Ford, T.L.; Gupta, R.; Hiepe, F.; Santiago, M.; et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): A randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019, 1, e208–e219. [Google Scholar] [CrossRef]
- Xue, M.; Zhuo, Y.; Shan, B. MicroRNAs, Long Noncoding RNAs, and Their Functions in Human Disease. Methods Mol. Biol. 2017, 1617, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-C.; Pan, H.-F.; Leng, R.-X.; Wang, D.-G.; Li, X.-P.; Li, X.-M.; Ye, D. Emerging role of long noncoding RNAs in autoimmune diseases. Autoimmun. Rev. 2015, 14, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Ji, Y.; Wang, X.; Hu, J.; Zhang, Q.; Xu, J.; Liu, W.; Wang, A. Relationship of miRNA-146a to systemic lupus erythematosus: A PRISMA-compliant meta-analysis. Medicine 2020, 99. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, C.; Zhang, J.; Tan, X.; Deng, J.; Jiang, R.; Li, Y.; Piao, Y.; Li, C.; Yang, W.; et al. Expression profile of long noncoding RNAs in children with systemic lupus erythematosus: A microarray analysis. Clin. Exp. Rheumatol. 2018, 37, 156–163. [Google Scholar]
- Ye, D.Q.; Wu, G.C.; Hu, Y.; Guan, S.Y.; Pan, H.F. Differential plasma expression profiles of long non-coding rnas reveal potential biomarkers for systemic lupus erythematosus. Biomolecules 2019, 9, 206. [Google Scholar]
- Liao, Z.; Ye, Z.; Xue, Z.; Wu, L.; Ouyang, Y.; Yao, C.; Cui, C.; Xu, N.; Ma, J.; Hou, G.; et al. Identification of Renal Long Non-coding RNA RP11-2B6.2 as a Positive Regulator of Type I Interferon Signaling Pathway in Lupus Nephritis. Front. Immunol. 2019, 10, 975. [Google Scholar] [CrossRef]
- Cai, B.; Cai, J.; Yin, Z.; Jiang, X.; Yao, C.; Ma, J.; Xue, Z.; Miao, P.; Xiao, Q.; Cheng, Y.; et al. Long non-coding RNA expression profiles in neutrophils revealed potential biomarker for prediction of renal involvement in SLE patients. Rheumatology 2021, 60, 1734–1746. [Google Scholar] [CrossRef]
- Chiaro, T.R.; Davis, K.W.; Wilson, A.; Suh-Lailam, B.; Tebo, A.E. Significant differences in the analytic concordance between anti-dsDNA IgG antibody assays for the diagnosis of systemic lupus erythematosus—Implications for inter-laboratory testing. Clin. Chim. Acta 2011, 412, 1076–1080. [Google Scholar] [CrossRef]
- Lakos, G.; Gonzalez, M.; Flaherty, D.; Bentow, C.; Ibarra, C.; Stimson, D.; Dervieux, T. Detection of anti-dsDNA antibodies by computer-aided automated immunofluorescence analysis. J. Immunol. Methods 2016, 433, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]


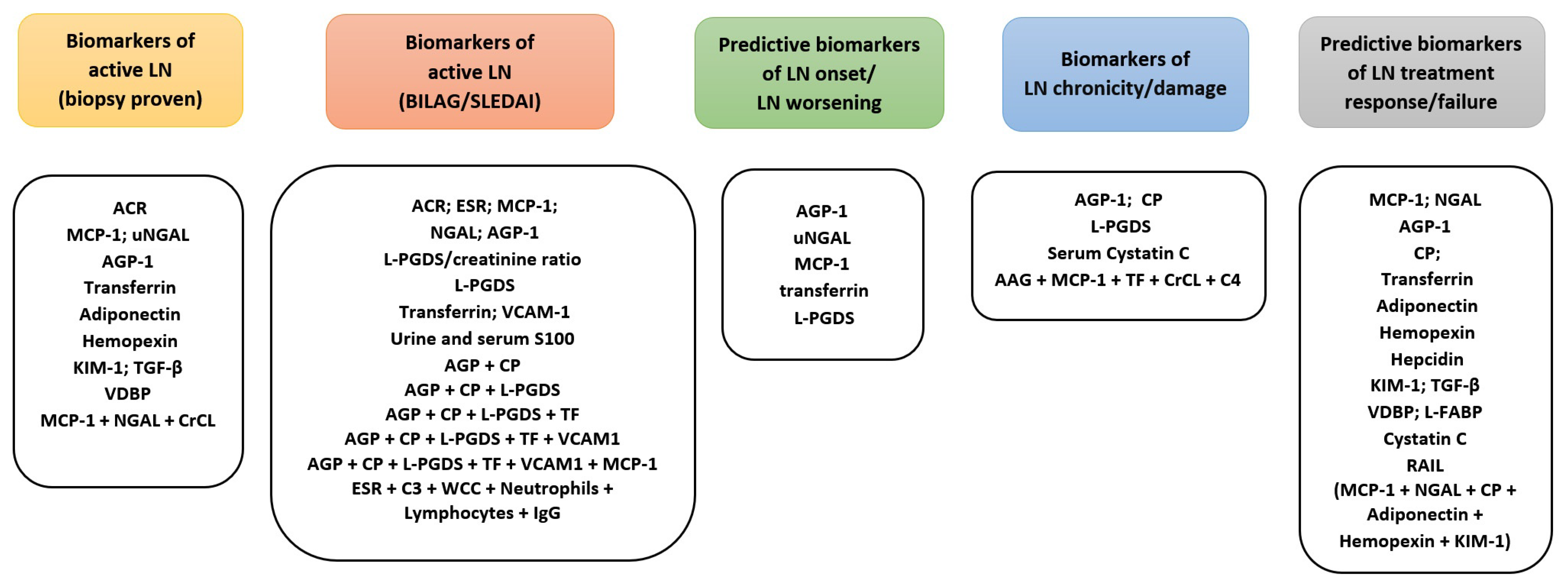{kind=link}
| Biomarker | Study | Definition of Active Nephritis | Patient Number | Findings |
|---|---|---|---|---|
| C3/C4 | Smith 2017 [44] Watson 2013 [45] Smith 2017 [46] Brunner 2012 [47] Brunner 2016 [48] Suzuki 2009 [49] Watson 2012 [50] | BILAG BILAG BILAG SLEDAI, Biopsy (BAI) Biopsy (NIH-AI and TIAI) SLEDAI, BILAG BILAG | 91 64 370 76 47 98 60 | No significant difference in C3 levels in active LN in the UK and US cohorts. C3 is a poor predictor of active LN (AUC 0.65 UK, 0.64 US). C4 was not a predictor of active LN (AUC 0.59 UK, 0.48 US). C3 and C4 significantly lower in active LN (p < 0.001 and p 0.009, respectively). C3 is an independent predictor of active LN (β −0.459, CI −0.663 to 0.254, p < 0.001). Negative correlation between MCP-1 and serum C3 (Rho −0.22, p = 0.002). C3 and C4 significantly lower in active LN (p < 0.001 and p = 0.017, respectively). C3 weakly correlated with SLEDAI LN (r −0.34, p < 0.0043). C4 not correlated with SLEDAI LN (r < 0.2). No significant difference in C3/C4 levels in active BAI LN. C3 and C4 were significantly lower in active NIH-AI LN (p = 0.005 and 0.015, respectively) but not in active TIAI LN (p = 0.931 and 0.981, respectively). No significant difference in C3 or C4 levels in active LN (SLEDAI or BILAG). C3 was not a predictor of active SLEDAI LN (AUC 0.58), was a poor predictor of active BILAG LN (AUC 0.63) and a good predictor of SDI LN chronicity (AUC 0.75). C4 was a poor predictor of active SLEDAI LN (AUC 0.60), not a predictor of active BILAG LN (AUC 0.49) and a poor predictor of SDI LN chronicity (AUC 0.64). Significantly lower in active LN (p = 0.02). |
| dsDNA | Smith 2017 [44] Watson 2014 [45] Smith 2017 [46] Brunner 2016 [48] Watson 2012 [50] | BILAG BILAG BILAG Biopsy (NIH-AI and TIAI) BILAG | 91 64 370 47 60 | No significant difference in active LN in the UK and US cohorts. Poor predictor of active LN (AUC 0.62 UK, 0.64 US). Significantly higher in active renal disease (p < 0.001). Not a predictor of worsening LN (AUC 0.44; p = 0.62). No significant difference in active LN (p = 0.068). No significant difference in active NIH-AI or TIAI LN (p = 0.353 and 0.849, respectively). No significant difference in active LN (p = 0.34). |
| Proteinuria | Smith 2017 [44] Watson 2014 [45] Smith 2017 [46] Brunner 2012 [48] Brunner 2016 [48] Suzuki 2009 [49] Watson 2012 [50] Smith 2018 [51] | BILAG BILAG BILAG SLEDAI, Biopsy (BAI) Biopsy (NIH-AI and TIAI) SLEDAI, BILAG BILAG BILAG, Biopsy | 91 64 370 76 47 98 60 64 | uACR significantly higher in active LN in the UK and US cohorts (p < 0.01 and p = 0.03, respectively). uACR significantly higher in active LN (p < 0.001). Not a predictor of worsening LN (AUC 0.46; p = 0.76). uACR and uPCR significantly higher in active LN (both p < 0.001). uPCR weakly correlated with SLEDAI LN (r = 0.40, p < 0.0004). Significantly higher in active BAI LN (p = 0.008). Good predictor of active BAI LN (AUC 0.76). uPCR was not significantly different in active LN. uPCR significantly higher in active SLEDAI and BILAG LN (both p < 0.0001). Excellent predictor of SLEDAI LN activity (AUC 0.91), a good predictor of BILAG LN activity (AUC 0.85), and a fair predictor of SDI LN chronicity (AUC 0.76). uACR significantly higher in active LN (p < 0.001). An elevated uACR or uPCR persists after active LN flare in 61% of patients. Age at time of LN flare (p = 0.007), eGFR (p = 0.035), and haematological involvement at the time of LN onset (p = 0.016) were significantly associated with time to normalisation of uACR and uPCR. |
| Creatinine | Smith 2017 [44] Watson 2014 [45] Smith 2017 [46] Brunner 2012 [47] | BILAG BILAG BILAG SLEDAI, Biopsy (BAI) | 91 64 370 76 | No significant difference in active LN in the UK and US cohorts. Significantly higher in active LN (p = 0.026). Poor predictor of worsening LN (AUC 0.52; p = 0.79). No significant difference in active LN (p = 1.0). Weakly correlated with SLEDAI LN (r = 0.29, p < 0.01). No significant difference in active BAI LN. |
| eGFR | Smith 2017 [44] Watson 2013 [45] Smith 2017 [46] Brunner 2012 [47] Brunner 2016 [48] Suzuki 2009 [49] Watson 2012 [50] | BILAG BILAG BILAG SLEDAI, Biopsy (BAI) Biopsy (NIH-AI and TIAI) SLEDAI, BILAG BILAG | 91 64 370 76 47 98 60 | No significant difference in active LN in the UK and US cohorts. No significant difference in active LN (p = 0.085). Poor predictor of worsening LN (AUC 0.54; p = 0.75) No significant difference in active LN (p = 1.0). No correlation with SLEDAI LN (r < 0.2). No significant difference in active BAI LN. Significantly higher in active NIH-AI and TIAI LN (p = 0.003). No significant difference in active LN. Not a predictor of SLEDAI or BILAG LN activity (AUC 0.45 and 0.50, respectively). Not a predictor of SDI LN chronicity (AUC 0.39). No significant difference in active LN (p = 0.58). |
| CRP | Smith 2017 [46] | BILAG | 370 | No significant difference in active LN (p = 1.0). |
| ESR | Smith 2017 [44] Watson 2013 [45] Smith 2017 [46] Watson 2012 [50] | BILAG BILAG BILAG BILAG | 91 64 370 60 | Significantly higher in active LN in the UK cohort (p < 0.01). Fair predictor of active LN (AUC 0.796) in the UK cohort. Not measured in the US cohort. Significantly higher in active LN (p 0.018). Not a predictor of worsening LN (AUC 0.42; p 0.52). Significantly higher in active LN (p < 0.001). No significant difference in active LN (p = 0.07). |
| Biomarker | Study | Definition of Active Nephritis | Patient Number | Findings |
|---|---|---|---|---|
| MCP-1 | Smith 2017 [44] Watson 2014 [45] Brunner 2012 [47] Brunner 2016 [48] Watson 2012 [50] Brunner 2017 [54] | BILAG BILAG SLEDAI, Biopsy (BAI) Biopsy (NIH-AI andTIAI) BILAG SLEDAI | 91 64 76 47 60 87 | Significantly higher in active LN in the UK (p = 0.028) and US cohorts (p < 0.001). MCP-1 and MCP-1-crea are significantly higher in active LN (p < 0.001 and p = 0.006, respectively). An independent predictor of active LN (β 0.183, CI 0.60–0.305, p = 0.004). MCP-1 is a good predictor of improved renal disease over time (AUC 0.81, p = 0.013). Good predictor of BAI activity (AUC 0.82). Weakly correlated with SLEDAI LN activity (r = 0.23, p < 0.07). Significantly higher in presence of mesangial proliferation (p 0.008) and capillary proliferation (p = 0.014) on biopsy. Significantly higher in active LN on NIH-AI (p = 0.000) + TIAI (p = 0.035). Significantly higher in biopsies which had endocapillary hypercellularity, leukocyte infiltrates, subendothelial deposits, tubular cell flattening and tubular cell necrosis (all p < 0.05). Fair predictor for NIH-AI LN activity (AUC > 0.7). Significantly higher in active LN (p = 0.005). At 3 and 6 months after therapy, MCP-1 was significantly higher in non-responders to therapy compared to responders (p < 0.05 and p < 0.005, respectively), although there was no significant difference at baseline. |
| NGAL | Smith 2017 [44] Watson 2014 [45] Brunner 2016 [48] Suzuki 2008 [49] Hinze 2009 [55] Brunner 2006 [56] Gheita 2015 [57] Hammad 2013 [58] Brunner 2017 [54] | BILAG BILAG Biopsy (NIH-AI and TIAI), SLEDAI BILAG, SLEDAI, PA SLEDAI, biopsy Biopsy SLEDAI SLEDAI BILAG | 91 64 47 85 111 35 28 33 87 | No significant difference in active LN in the UK and US cohorts (p = 1.0) NGAL and NGAL-crea are significantly higher in active LN (p = 0.001 and 0.02, respectively). Fair predictor of worsened renal disease activity over time (AUC 0.76, p = 0.04). Significantly higher in NIH-AI active LN (p = 0.017) but not TIAI active LN (p = 0.094). Significantly higher in biopsies which had endocapillary hypercellularity (p < 0.05) and epithelial cells in tubular lumen (p < 0.05). Fair predictor of NIH-AI LN activity (AUC > 0.7). NGAL-crea was significantly higher in active LN (p = 0.02) and weakly correlated with LN activity (r = 0.4, p < 0.008). Worsening LN was associated with an increase in NGAL (380%) and NGAL-crea (125%) (p < 0.01). Significant increase in NGAL 3–6 months before flare of LN (+104% BILAG [p = 0.01] +70% SLEDAI [p = 0.03], +70% PA [p = 0.04]). Good predictor of BILAG LN activity (AUC 0.8 [sensitivity 81.8%, specificity 82.4%, PPV 60.7%, NPV 93.2%]), and fair predictor of SLEDAI LN activity (AUC 0.78 [sensitivity 82.6%, specificity 70.9%, PPV 48.6%, NPV 92.4%]). NGAL-crea was significantly higher in active LN (p < 0.0005) and moderately correlated with LN activity (r > 0.59 p < 0.0001). NGAL-crea was weakly correlated with SDI LN chronicity (r >0.47 p < 0.001). NGAL-crea was 16-fold higher in biopsy-proven LN than in those without LN (p = 0.0002) and strongly correlated with biopsy LN activity (r = 0.73, p < 0.001) and chronicity (r > 0.58 p = 0.004). Excellent predictor of biopsy-proven LN (AUC 0.944 [sensitivity 90%, specificity 100%]). Significantly higher in patients with LN on biopsy (p = 0.019) and weakly correlated with the presence of LN (r = 0.3 p = 0.02). No significant difference in NGAL in active LN. However, NGAL-crea was significantly higher in active LN (p < 0.001) and moderately correlated with LN activity (r = 0.5 p = 0.02). NGAL was significantly predictive of proliferative LN (class III and IV) (p = 0.005, 91% sensitivity, 70% specificity. NGAL was significantly higher in non-responders to therapy compared to responders (p < 0.005) 6 months after therapy, although there was no significant difference at baseline and at 3 months. |
| AGP-1 | Smith 2017 [44] Brunner 2012 [47] Brunner 2016 [48] Suzuki 2009 [49] Watson 2012 [50] Smith 2019 [59] Smith 2018 [60] Brunner 2017 [54] | BILAG SLEDAI Biopsy (BAI) Biopsy (NIH-AI and TIAI) SLEDAI, BILAG BILAG BILAG BILAG SLEDAI | 91 76 47 98 60 80 23 87 | Significantly higher in active LN in the UK (p < 0.001) and US cohorts (p < 0.001). Weakly correlated with SLEDAI LN activity (r = 0.35, p < 0.003). Fair predictor of BAI activity (AUC 0.76). Significantly higher when mesangial proliferation (p < 0.005) and cellular crescents (p < 0.003) present on biopsy. Significantly higher in active TIAI LN (p = 0.043) but not NIH-AI active LN (p = 0.377). Significantly higher when tubular cell necrosis (p < 0.05) and macrophages in tubular lumen (p < 0.05) present on biopsy. Urinary AGP-1: Significantly higher in JSLE vs. JIA (p < 0.0001). Significantly higher in active SLEDAI (p = 0.005) and BILAG (p < 0.0001) LN. uAGP-1 significantly increased at least 3 months before diagnosis of worsening LN activity (p < 0.009). uAGP-1-crea was significantly higher in active LN (p < 0.0001) but there was no significant difference in uAGP-1-prot. uAGP-1 is a fair predictor of SLEDAI LN activity (AUC 0.76), a good predictor of BILAG LN activity (AUC 0.81) and a fair predictor of SDI LN damage (AUC 0.73). Serum AGP-1: No difference in JSLE vs. JIA. No difference in active LN. Significantly higher in active LN (p = 0.027). Predictive of LN flare 3, 9 and 12 months before (HR 1.49 [95% CI 1.10–2.02]). Best predictor of LN activity on Markov Multi-State model (AICc 139.81). Significantly higher in active LN (p < 0.001). AGP-1 was significantly higher in non-responders to therapy compared to responders at baseline (p = 0.023) and 3, 6 and 12 months after therapy (p < 0.005 for all). Excellent predictor of non-responders to therapy at 3 months (AUC 0.98). |
| CP | Smith 2017 [44] Brunner 2016 [48] Suzuki 2009 [49] Smith 2019 [59] Smith 2018 [60] Brunner 2017 [54] | BILAG Biopsy (NIH-AI and TIA) SLEDAI, BILAG BILAG BILAG SLEDAI | 91 47 98 80 23 87 | Significantly higher in active LN in the UK (p < 0.001) and US cohorts (p < 0.001). Significantly higher in active NIH-AI LN (p = 0.015) but not active TIAI LN (p = 0.138). Urinary CP: Significantly higher in JSLE vs. JIA (p < 0.0001) and significantly higher in active SLEDAI (p = 0.004) and BILAG (p = 0.003) LN. There was no significance in uCP-crea or uCP-prot in active LN. uCP is a poor predictor of SLEDAI LN activity (AUC 0.68), a good predictor of BILAG LN activity (AUC 0.80) and a good predictor of SDI LN damage (AUC 0.84). Serum CP: No difference in JSLE vs. JIA. No difference in active LN. Predictive of LN remission 3, 9 and 12 months before (HR 0.60 [95% CI 0.39–0.93]. Second best predictor of LN activity on Markov Multi-State model after AGP-1 (AICc 141.40). Significantly higher in active LN (p < 0.001) CP was significantly higher in non-responders to therapy compared to responders at baseline (p = 0.006), 3 months (p < 0.005) and 6 months after therapy (p < 0.005). Good predictor of non-responders to therapy at 3 months (AUC 0.83). |
| L-PGDS | Smith 2017 [44] Brunner 2012 [47] Brunner 2016 [48] Suzuki 2009 [49] Smith 2018 [60] Brunner 2017 [54] | BILAG SLEDAI Biopsy (BAI) SLEDAI, BILAG BILAG SLEDAI | 91 76 47 98 23 87 | Significantly higher in active LN in the UK (p < 0.001) and US cohorts (p = 0.021). Weakly correlated with SLEDAI LN activity (r = 0.28, p < 0.016). Not associated with any histological features. No significant difference based on NIH-AI (p = 0.061) or TIAI (p = 0.081) LN activity. Significantly higher when endocapillary hypercellularity present on biopsy (p < 0.05). Urinary L-PGDS: Significantly higher in JSLE vs. JIA (p < 0.0025) and significantly higher in active SLEDAI (p < 0.0001) and BILAG (p = 0.004) LN. uL-PGDS significantly increased at least 3 months before clinical diagnosis of worsening LN activity (p < 0.009). L-PGDS-crea was significantly higher in active LN (p < 0.0005) but L-PGDS-protein was significantly lower in active LN (p < 0.009). uL-PGDS is a fair predictor of SLEDAI LN activity (AUC 0.71) and BILAG LN activity (AUC 0.73) and a good predictor of SDI LN damage (AUC 0.84). Serum L-PGDS: No difference in JSLE vs. JIA. No difference in active LN. Significantly higher in active LN (p = 0.018). L-PGDS was significantly higher in non-responders to therapy compared to responders at baseline (p = 0.044) and 3 months (p < 0.05). Excellent predictor of non-responders to therapy at 3 months (AUC 0.96). |
| Transferrin | Smith 2017 [44] Brunner 2012 [47] Brunner 2016 [48] Suzuki 2009 [49] Smith 2018 [60] Brunner 2017 [54] | BILAG SLEDAI, Biopsy (BAI) Biopsy (NIH-AI and TIA) SLEDAI, BILAG BILAG SLEDAI | 91 76 47 98 23 87 | No significant difference in active LN in the UK cohort (p = 0.063), significantly higher in active LN group in the US cohort (p < 0.001). No correlation to SLEDAI LN activity (r < 0.2). Fair predictor of BAI LN activity (AUC 0.76). Significantly higher when mesangial proliferation (p = 0.024), capillary proliferation (p = 0.017) and cellular crescents present on biopsy (p = 0.024) Significantly higher in NIH-AI active LN (p = 0.029) but not in TIA active LN (p = 0.478) Urinary TF: Significantly higher in JSLE vs. JIA (p ≤ 0.0001) and significantly higher in active SLEDAI (p < 0.0001) and BILAG (p < 0.0001) LN. Levels significantly increased at least 3 months before clinical diagnosis of active flare (p < 0.0009). uTF-crea and uTF-prot were significantly higher in active LN (p <0.0001 and p < 0.05, respectively). uTF is a good predictor of SLEDAI LN activity (AUC 0.80), BILAG LN activity (AUC 0.81) and SDI LN damage (AUC 0.84). Serum TF: No difference in JSLE vs. JIA. No difference in active LN. Significantly higher in active LN than inactive LN (p < 0.05). Transferrin was significantly higher in non-responders to therapy compared to responders at baseline (p = 0.012) and 3 months, 6 and 12 months (p < 0.05). Excellent predictor of non-responders to therapy at 3 months (AUC 0.95). |
| Biomarker | Study | Definition of Active Nephritis | Patient Number | Findings |
|---|---|---|---|---|
| VCAM-1 | Smith 2017 [44] Smith 2018 [60] | BILAG BILAG | 91 23 | Significantly higher in active LN in the UK (p = 0.007) and US cohorts (p < 0.001). Significantly higher in active LN (p = 0.010). |
| Adiponectin | Brunner 2016 [48] Brunner 2017 [54] | Biopsy SLEDAI | 47 87 | Significantly higher in NIH-AI and TIAI active LN (p = 0.023 and 0.024, respectively). Significantly higher in non-responders to therapy compared to responders at 3, 6 and 9 months (p < 0.05). Excellent predictor of non-responders to therapy at 3 months (AUC 0.90). |
| Hemopexin | Brunner 2016 [48] Brunner 2017 [54] | Biopsy SLEDAI | 47 87 | Significantly higher in TIAI active LN (p 0.010) but not in NIH-AI active LN (p = 0.138). Significantly higher in non-responders to therapy compared to responders at 3 and 6 months (p < 0.01). Good predictor of non-responders to therapy at 3 months (AUC 0.8). |
| Hepcidin | Brunner 2016 [48] Brunner 2017 [54] | Biopsy SLEDAI | 47 87 | No significant difference in NIH-AI and TIAI active LN (p = 0.753 and 0.802, respectively). Significantly higher in non-responders to therapy compared to responders at baseline (p = 0.037). |
| KIM-1 | Brunner 2016 [48] Brunner 2017 [54] | Biopsy SLEDAI | 47 87 | Significantly higher in NIH-AI active LN (p 0.000) but not TIAI active LN (p = 0.140). Significantly higher in non-responders to therapy compared to responders at 3, 6 and 9 months (p < 0.05, p < 0.05, and p < 0.05, respectively). Good predictor of non-responders to therapy at 3 months (AUC 0.8). |
| TGF-β | Brunner 2016 [48] Brunner 2017 [54] | Biopsy SLEDAI | 47 87 | Significantly higher in NIH-AI and TIAI active LN (p = 0.013 and 0.030, respectively). Significantly higher in non-responders to therapy compared to responders at 3, 6 and 9 months (p < 0.005, p < 0.05, and p < 0.01, respectively). Good predictor of non-responders to therapy at 3 months (AUC 0.81). |
| VDBP | Brunner 2016 [48] Brunner 2017 [54] | Biopsy SLEDAI | 47 87 | Significantly higher in TIAI active LN (p = 0.844) but not NIH-AI active LN (p = 0.030). Significantly higher in non-responders to therapy compared to responders at baseline, 3, 6 and 12 months (p = 0.027, p < 0.005, p < 0.005, and p < 0.05, respectively). Excellent predictor of non-responder to therapy at 3 months (AUC 0.9). |
| L-FABP | Brunner 2016 [48] Brunner 2017 [54] | Biopsy SLEDAI | 47 87 | No significant difference in NIH-AI and TIAI active LN (p = 0.454 and 0.099, respectively). Significantly higher in non-responders to therapy compared to responders at baseline, 3, 6 and 12 months (p < 0.05, p < 0.005, and p < 0.05, respectively). Good predictor of non-responders to therapy at 3 months (AUC 0.8). |
| Cystatin C | Brunner 2016 [48] Brunner 2017 [54] Gheita 2015 [61] | Biopsy SLEDAI Biopsy | 47 87 28 | Urine: No significant difference in NIH-AI and TIAI active LN (p = 0.352 and = 0.138, respectively). Urine: Significantly higher in non-responders to therapy compared to responders at 6 months (p < 0.05). Fair predictor of non-responders at 3 months (AUC 0.7). Serum: Weakly correlated with SDI LN damage (r = 0.38, p 0.003). |
| Osteopontin | Brunner 2017 [54] | SLEDAI | 87 | No significant difference in non-responders to therapy compared to responders. |
| S100 | Donohue 2020 [93] Turnier 2017 [94] | BILAG SLEDAI | 64 96 | Serum: Significantly higher levels of S100A8/A9 (p < 0.001) and S100A12 (p = 0.035) in JSLE patients when compared to healthy controls. Significantly higher levels of S100A8/A9 (p = 0.043) and S100A12 (p = 0.021) in JSLE patients with active LN compared to those with inactive/no renal disease. No differences in levels of S100A4. Urine: Significantly higher levels of S100A12 (p = 0.029) in JSLE patients when compared to healthy controls. Significantly higher levels of S00A12 (p = 0.0095) in JSLE patients with active LN compared to those with inactive/no renal disease. No difference in levels of S100A4 or S100A8/A9. Serum: No significant difference in S100A4, S100A6, S100A8/9 and S100A12 in active LN. Urine: significantly higher levels of S100A4 (p < 0.0001), S100A6 (p = 0.0008), S100A8/9 (p = 0.0235) and S100A12 (p = 0.018) in active LN. Levels of all S100 decreased with improvement of LN; S100A4 (p < 0.0001), S100A6 (p = 0.0075), S100A8/9 (p = 0.019), and S100A12 (p = 0.0143). S100A4 was higher in proliferative (class III/IV) LN than in membranous (class V) LN (p = 0.03). |
| IP-10 | Watson 2012 [50] | BILAG | 60 | No significant difference in active LN (p = 0.55) or between JSLE vs. healthy controls (p = 0.13). |
| sCD25 | Hassan 2021 [95] | SLEDAI, SLICC renal activity score | 53 | Urine: JSLE patients had higher normalised sCD25 levels compared to the healthy controls (p = 0.001). JSLE patients with active LN had significantly higher sCD25 levels than active JSLE patients without LN (p = 0.002) and JSLE patients with inactive disease (p < 0.001). Serum: CD25 levels were significantly higher in active SLEDAI LN (p < 0.001, AUC 0.88 [sensitivity 68.7%, specificity 91.9%]), and active SLICC renal activity score LN (p < 0.001). |
| NK cells | Zahran 2021 [96] | Biopsy | 35 | Peripheral blood: NK strongly negatively correlation with LN activity (r −0.6, p = 0.001). CD56bright NK cells strongly correlated with LN activity (r = 0.6, p < 0.0001) and moderately correlated with LN chronicity (r = 0.4 p = 0.01). NKT moderately negative correlation with LN activity (r = −0.5, p = 0.001) |
| Biomarker | Study | Findings |
|---|---|---|
| AGP-1 + CP AGP-1 + CP + L-PGDS AGP-1 + CP + L-PGDS + TF AGP-1 + CP + L-PGDS + TF + VCAM1 AGP-1 + CP + L-PGDS + TF + VCAM1 + MCP-1 AGP-1 + CP + MCP-1 + PCR AGP-1 + MCP-1 + TF + CrCL + C4 MCP-1 + NGAL + CrCL RAIL (MCP-1 + NGAL + CP + adiponectin + hemopexin + KIM-1) RAIL (MCP-1 + NGAL + CP + adiponectin + hemopexin + KIM-1)- creatinine adjusted ESR + C3 + WCC + Neutrophils + Lymphocytes + IgG | Smith 2017 [44] Smith 2018 [60] Smith 2017 [44] Smith 2018 [60] Smith 2017 [44] Smith 2018 [60] Smith 2017 [44] Smith 2017 [44] Brunner 2012 [47] Brunner 2012 [47] Brunner 2012 [47] Brunner 2016 [48] Brunner 2017 [54] Brunner 2017 [54] Smith 2017 [46] | Good predictor of active BILAG LN in UK (AUC = 0.881) cohort and excellent in US cohort (0.982) Excellent predictor of active BILAG LN (AUC = 0.992 [95% CI = 0.970-1]) Excellent predictor of active BILAG LN in UK (AUC = 0.900) and US cohort (0.982) Excellent predictor of active BILAG LN (AUC 0.992 [95% CI = 0.970-1]) Excellent predictor of active BILAG LN in UK (AUC = 0.920) and US cohort (0.991) Excellent predictor of active BILAG LN (AUC = 1 [95% CI = 1-1]) Excellent predictor of active BILAG LN in UK (AUC = 0.920) and US cohort (0.987) Excellent predictor of active BILAG LN in UK cohort (AUC = 0.920) Good predictor of BAI LN activity (AUC = 0.85 [sensitivity 72%, specificity 66%]). Fair predictor of membranous LN (AUC = 0.75 [sensitivity 75%, specificity 48%]). Good predictor of BCI (AUC = 0.83 [sensitivity 73%, specificity 67%]) Excellent predictor of NIH-AI active LN (AUC = 0.92 [sensitivity 90%, specificity 86%]), 0.94 when corrected for chronicity [sensitivity 90%, specificity 90%]) and good for predicting TIAI active LN (AUC = 0.80 [sensitivity 80%, specificity 68%], 0.83 when corrected for chronicity [sensitivity 90%, specificity 68%]) Fair predictor of response to therapy at baseline (AUC = 0.72), excellent for predicting response to therapy after 3 months (AUC = 0.92) and good after 6 months (AUC = 0.84). Fair predictor of response to therapy at baseline (AUC = 0.74), excellent for predicting response to therapy after 3 (AUC = 0.92) and 6 (AUC = 0.91) months. Fair predictor of active BILAG LN (AUC = 0.724) |
| Biomarker | Study | Findings/Clinical Associations |
|---|---|---|
| CVR biomarkers | ||
| CIMT CIMT, PWV Lymphopenia CRP hsCRP ApoB:ApoA1 ratio Adiponectin | [114] [115] [116] [116,117] [118] [119] [119] | Higher CIMT was detected in JSLE patients compared to matched HCs (0.45 vs. 0.37 mm, respectively, p < 0.0001) Increased CIMT (in 48% vs. 17%, p value N/A) and PWV (p 0.011) in JSLE patients compared to age and sex-matched HCs Lymphopenia at baseline and at diagnosis were consistently associated with progression of CIMT (p = 0.012 and p = 0.045, respectively). CRP at baseline positively associated with CTIMP (p = 0.049) hsCRP was able to differentiate between matched HCs, and JSLE patients clustered in two groups: with the best and the worst metabolic profiles (defined by a combination of homocysteine, folate, hsCRP, TNF-alpha parameters) (p < 0.05 for all comparisons) Strong predictor of CVR (defined by a metabolic signature cross-validated in an adult SLE cohort with atherosclerosis plaques on vascular ultrasound) in JSLE when accounted for BMI (specificity 96.2% and sensitivity 96.7%). Adiponectin correlated with total cholesterol for both JSLE males (r = 0.296; p = 0.01) and females patients (r = 0.554; p = 0.008); with HDL in females (r = 0.700; p = 0.0009) but not males (p = 0.32); with LDL in males (r −0.288; p = 0.01) but not in females (p = 0.22); and with lipoprotein A for males (r = 0.862; p < 0.001) but not for females (p = 0.18) |
| CNS manifestations | ||
| APLA Anti-ganglioside M1 antibody Anti-aquaporin 4 antibodies Anti-dsDNA, Anti-U1RNP, and Anti-Sm antibodies | [120] [121] [122] [36] |
Anticardiolipin antibodies were seen more frequently in children with NPSLE as compared to those without NPSLE (57.8 vs. 23%), lupus anticoagulant was more frequent in children without NPSLE (53.8 vs. 34.7%) (but p > 0.05) Significant positive association between anti-ganglioside M1 seropositivity and cognitive dysfunction (p < 0.001), as well as with a significant risk for association with cognitive dysfunction (odds ratio: 36; 95% CI: 4.3–302.8) Increased likelihood to experience neurological symptoms (p 0.002) and to have received anti-epileptic (p = 0.023) and anti-coagulant (p = 0.007) Predicted a group of JSLE patients (clustered based on the analysis of the most common autoantibodies found in JSLE in general) with highest prevalence of neuropsychiatric disease (p = 0.036) |
| Skin manifestations | ||
| Anti-ds-DNA antibodies | [36] | Increased prevalence of JSLE malar rash in one group of JSLE patients clustered based on frequency of various JSLE autoantibodies (85.3% compared to the other two clusters (53.3%; p < 0.001) or cluster 3 (70.7%; p = 0.046)) |
| Haematological manifestations | ||
| Anti-Ro and anti-ribosomal P antibodies | [36] | Increased prevalence of haemolytic anaemia in one group of JSLE patients clustered based on frequency of various JSLE autoantibodies (40%) compared to the other two clusters (17.7%; p = 0.018 and 15.5%; p = 0.011, respectively). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greenan-Barrett, J.; Doolan, G.; Shah, D.; Virdee, S.; Robinson, G.A.; Choida, V.; Gak, N.; de Gruijter, N.; Rosser, E.; Al-Obaidi, M.; et al. Biomarkers Associated with Organ-Specific Involvement in Juvenile Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2021, 22, 7619. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147619
Greenan-Barrett J, Doolan G, Shah D, Virdee S, Robinson GA, Choida V, Gak N, de Gruijter N, Rosser E, Al-Obaidi M, et al. Biomarkers Associated with Organ-Specific Involvement in Juvenile Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2021; 22(14):7619. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147619
Chicago/Turabian StyleGreenan-Barrett, James, Georgia Doolan, Devina Shah, Simrun Virdee, George A. Robinson, Varvara Choida, Nataliya Gak, Nina de Gruijter, Elizabeth Rosser, Muthana Al-Obaidi, and et al. 2021. "Biomarkers Associated with Organ-Specific Involvement in Juvenile Systemic Lupus Erythematosus" International Journal of Molecular Sciences 22, no. 14: 7619. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147619