
HBsAg Dampened STING Associated Activation of NK Cells in HBeAg-Negative CHB Patients

Abstract
:1. Introduction
2. Result
2.1. STING Expression Was Suppressed in NK Cells from HBeAg-Negative CHB Patients
2.2. Dampened Expression of STING Correlated with NK Cell Dysfunction in HBeAg-Negative CHB Patients
2.3. HBsAg Downregulated STING Expression in NK Cells
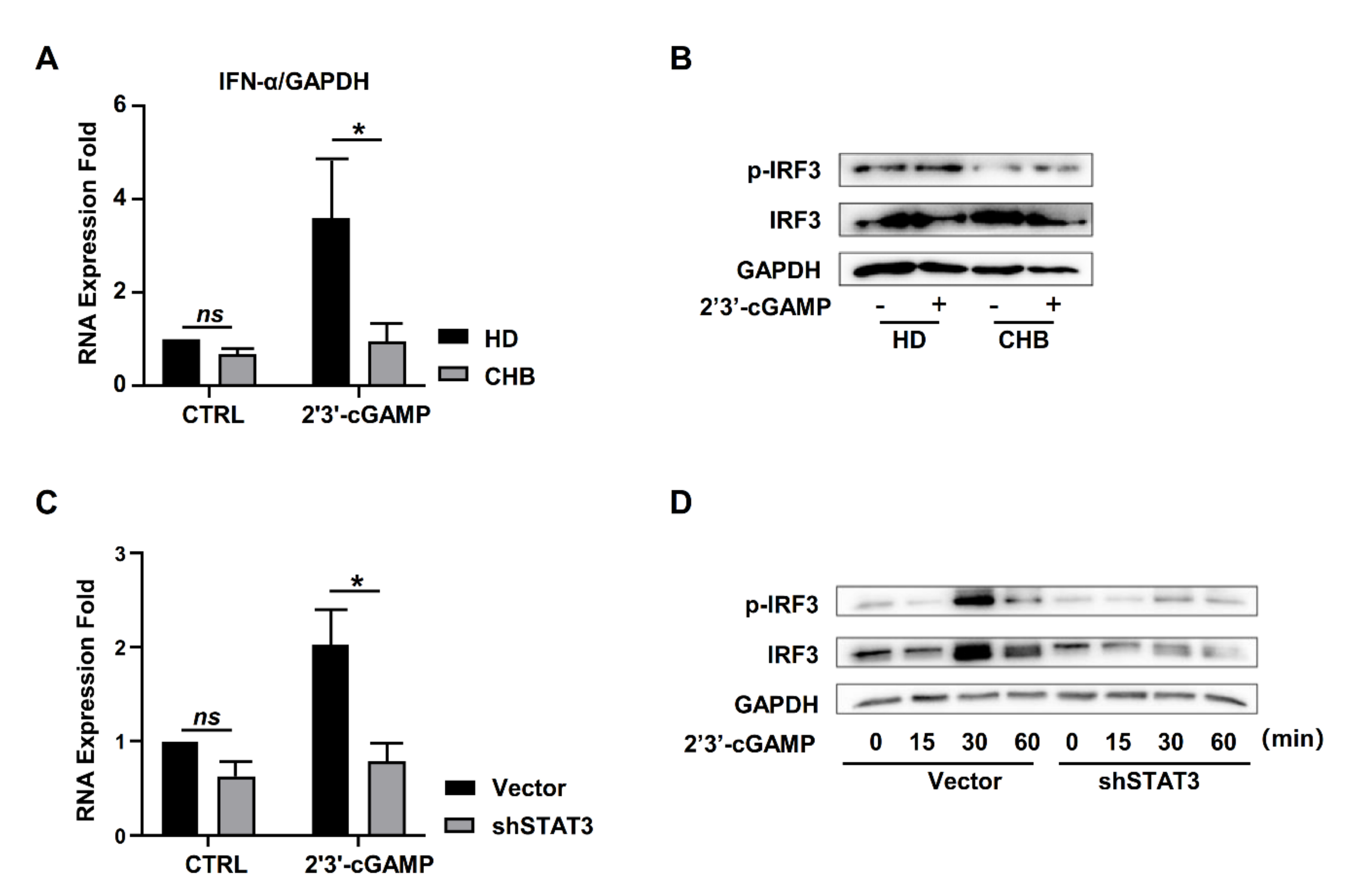
2.4. STAT3 Regulated STING Expression in NK Cells
2.5. STAT3 Directly Regulated STING Transcription in NK Cells
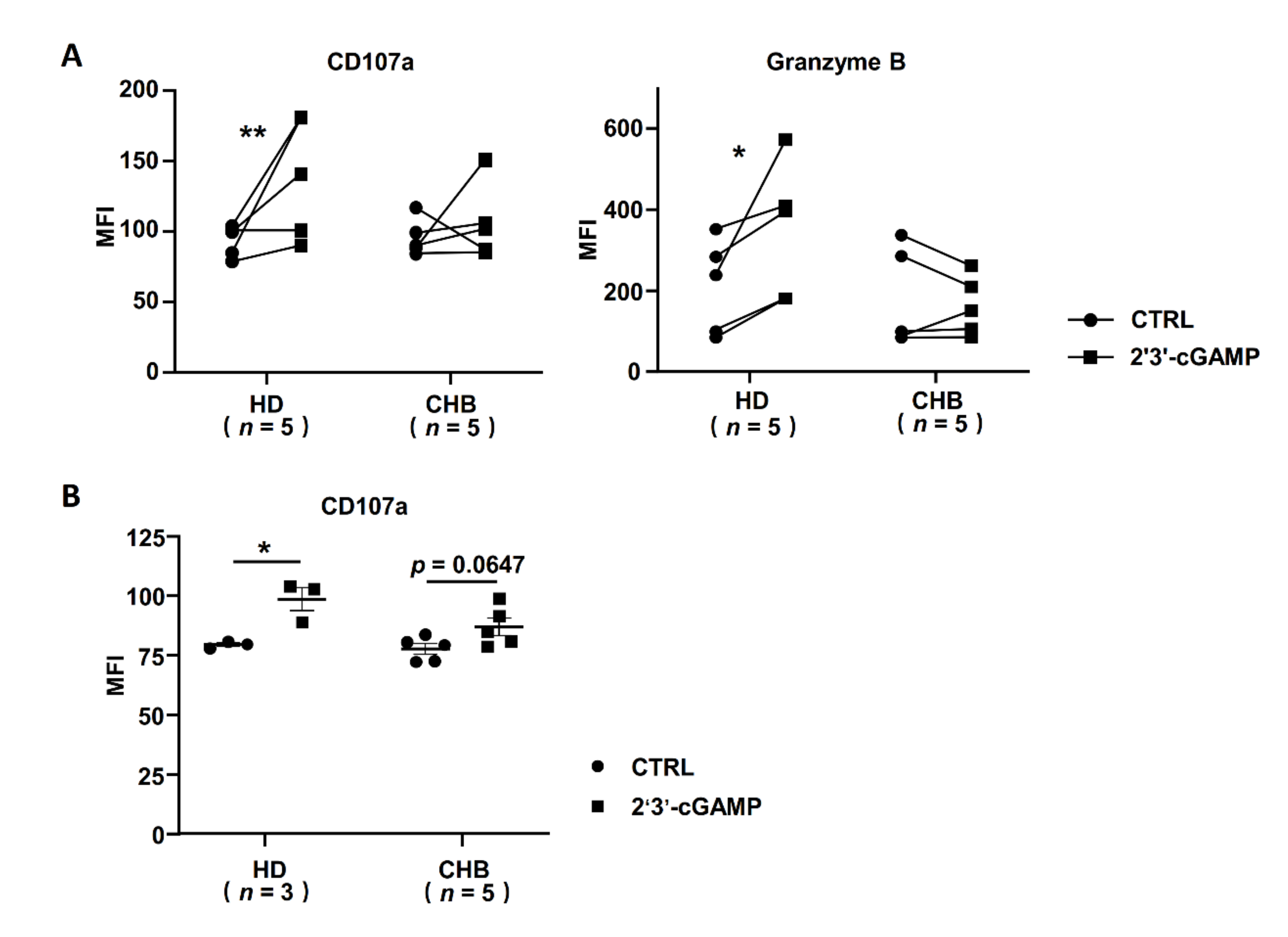
2.6. STING Associated IFNα Response Was Inhibited in NK Cells of HBeAg-Negative CHB Patients
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Cell Culture and Transfection
4.3. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
4.4. Western Blotting
4.5. Surface and Intracellular Immunostaining for FACS
4.6. Chromatin Immunoprecipitation
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Alexopoulou, A.; Vasilieva, L.; Karayiannis, P. New Approaches to the Treatment of Chronic Hepatitis B. J. Clin. Med. 2020, 9, 3187. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.D.; Yang, H.I.; Iloeje, U.H.; You, S.L.; Lu, S.N.; Wang, L.Y.; Su, J.; Sun, C.A.; Liaw, Y.F.; Chen, C.J. Carriers of inactive hepatitis B virus are still at risk for hepatocellular carcinoma and liver-related death. Gastroenterology 2010, 138, 1747–1754. [Google Scholar] [CrossRef]
- Ghosh, S.; Mondal, R.K.; Banerjee, P.; Nandi, M.; Sarkar, S.; Das, K.; Santra, A.; Banerjee, S.; Chowdhury, A.; Datta, S. Tracking the naturally occurring mutations across the full-length genome of hepatitis B virus of genotype D in different phases of chronic e-antigen-negative infection. Clin. Microbiol. Infect. 2012, 18, E412–E418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharm. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Guo, W.; Wei, J.; Zhong, X.; Zang, R.; Lian, H.; Hu, M.M.; Li, S.; Shu, H.B.; Yang, Q. SNX8 modulates the innate immune response to RNA viruses by regulating the aggregation of VISA. Cell Mol. Immunol. 2020, 17, 1126–1135. [Google Scholar] [CrossRef]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef]
- Real, C.I.; Lu, M.; Liu, J.; Huang, X.; Trippler, M.; Hossbach, M.; Deckert, J.; Jahn-Hofmann, K.; Ickenstein, L.M.; John, M.J.; et al. Hepatitis B virus genome replication triggers toll-like receptor 3-dependent interferon responses in the absence of hepatitis B surface antigen. Sci. Rep. 2016, 6, 24865. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Meng, Z.; Jiang, M.; Pei, R.; Trippler, M.; Broering, R.; Bucchi, A.; Sowa, J.P.; Dittmer, U.; Yang, D.; et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2009, 49, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 2010, 91, 2080–2090. [Google Scholar] [PubMed]
- Wang, X.; Li, Y.; Mao, A.; Li, C.; Li, Y.; Tien, P. Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-beta induction by disrupting the VISA-associated complex. Cell. Mol. Immunol. 2010, 7, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Op den Brouw, M.L.; Binda, R.S.; van Roosmalen, M.H.; Protzer, U.; Janssen, H.L.; van der Molen, R.G.; Woltman, A.M. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: A possible immune escape mechanism of hepatitis B virus. Immunology 2009, 126, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, Z.; Hu, C.; Qian, F.; Cheng, Y.; Wu, M.; Shi, B.; Chen, J.; Hu, Y.; Yuan, Z. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J. Immunol. 2013, 190, 5142–5151. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Imanishi, H.; Morisaki, H.; Liu, W.; Nakamura, H.; Morisaki, T.; Hada, T. Recombinant HBsAg inhibits LPS-induced COX-2 expression and IL-18 production by interfering with the NFkappaB pathway in a human monocytic cell line, THP-1. J. Hepatol. 2005, 43, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fu, B.; Gao, Y.; Liao, X.; Sun, R.; Tian, Z.; Wei, H. TGF-β1 down-regulation of NKG2D/DAP10 and 2B4/SAP expression on human NK cells contributes to HBV persistence. PLoS Pathog. 2012, 8, e1002594. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.J.; Shi, K.Q.; Chen, Y.P.; Ren, Y.F.; Song, Y.J.; Li, G.; Xue, Y.F.; Fang, Y.X.; Deng, Z.J.; et al. Hepatitis B surface antigen inhibits MICA and MICB expression via induction of cellular miRNAs in hepatocellular carcinoma cells. Carcinogenesis 2014, 35, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wei, H.; Wei, H.; Gao, Y.; Xu, L.; Yin, W.; Sun, R.; Tian, Z. Blocking the natural killer cell inhibitory receptor NKG2A increases activity of human natural killer cells and clears hepatitis B virus infection in mice. Gastroenterology 2013, 144, 392–401. [Google Scholar] [CrossRef]
- Noh, J.Y.; Yoon, S.R.; Kim, T.D.; Choi, I.; Jung, H. Toll-Like Receptors in Natural Killer Cells and Their Application for Immunotherapy. J. Immunol. Res. 2020, 2020, 2045860. [Google Scholar] [CrossRef]
- Yang, Y.; Han, Q.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol. Immunol. 2017, 14, 465–475. [Google Scholar] [CrossRef]
- Boni, C.; Vecchi, A.; Rossi, M.; Laccabue, D.; Giuberti, T.; Alfieri, A.; Lampertico, P.; Grossi, G.; Facchetti, F.; Brunetto, M.R.; et al. TLR7 Agonist Increases Responses of Hepatitis B Virus-Specific T Cells and Natural Killer Cells in Patients With Chronic Hepatitis B Treated With Nucleos(T)Ide Analogues. Gastroenterology 2018, 154, 1764–1777.e7. [Google Scholar] [CrossRef]
- Zhang, X.; Bai, X.C.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.R.; Bert, N.L.; Ho, S.S.; Shen, Y.J.; Tang, L.F.; Xiong, G.M.; Croxford, J.L.; Koo, C.X.; Ishii, K.J.; Akira, S.; et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014, 74, 2193–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bert, N.; Lam, A.R.; Ho, S.S.; Shen, Y.J.; Liu, M.M.; Gasser, S. STING-dependent cytosolic DNA sensor pathways regulate NKG2D ligand expression. Oncoimmunology 2014, 3, e29259. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Marcus, A.; Coscoy, L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol. Rev. 2017, 280, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.K.; Nandakumar, R.; Stadler, D.; Malo, A.; Valls, R.M.; Wang, F.; Reinert, L.S.; Dagnaes-Hansen, F.; Hollensen, A.K.; Mikkelsen, J.G.; et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 2016, 64, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Chen, Y.; Chen, L.; Yao, W.; Guan, J.; Liu, X.; Wei, X.; Lin, X. Impaired circulating CD56(dim) NK cells are associated with decompensation of HBV-related cirrhosis. Hum. Immunol. 2020, 81, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhai, N.; Wang, Z.; Song, H.; Yang, Y.; Cui, A.; Li, T.; Wang, G.; Niu, J.; Crispe, I.N.; et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 2018, 67, 2035–2044. [Google Scholar] [CrossRef]
- Xu, D.; Han, Q.; Hou, Z.; Zhang, C.; Zhang, J. miR-146a negatively regulates NK cell functions via STAT1 signaling. Cell Mol. Immunol. 2017, 14, 712–720. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Yang, Y.; Han, Q.; Yin, C.; Pan, Z.; Zhang, J. STAT3 directly regulates NKp46 transcription in NK cells of HBeAg-negative CHB patients. J. Leukoc. Biol. 2019, 106, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, Q.; Zhang, C.; Xiao, M.; Zhang, J. Hepatitis B virus antigens impair NK cell function. Int. Immunopharmacol. 2016, 38, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Lauterbach-Rivière, L.; Bergez, M.; Mönch, S.; Qu, B.; Riess, M.; Vondran, F.W.R.; Liese, J.; Hornung, V.; Urban, S.; König, R. Hepatitis B Virus DNA is a Substrate for the cGAS/STING Pathway but is not Sensed in Infected Hepatocytes. Viruses 2020, 12, 592. [Google Scholar] [CrossRef]
- Wang, L.; Wen, M.; Cao, X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science 2019, 365, eaav0758. [Google Scholar] [CrossRef]
- Yamashiro, L.H.; Wilson, S.C.; Morrison, H.M.; Karalis, V.; Chung, J.-Y.J.; Chen, K.J.; Bateup, H.S.; Szpara, M.L.; Lee, A.Y.; Cox, J.S.; et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat. Commun. 2020, 11, 3382. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Chen, M.; Zhang, R.; Zhang, W.; Wang, C.; Zhang, Y.; Wei, X.; Guan, Y.; Liu, J.; Feng, K.; et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020, 30, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, C.J.; Wolf, N.; Chang, I.C.; Kirn, G.; Marcus, A.; Ndubaku, C.O.; McWhirter, S.M.; Raulet, D.H. NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci. Immunol. 2020, 5, eaaz2738. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Deng, H.; Xu, Z. Targeting macrophage priming by polyphyllin VII triggers anti-tumor immunity via STING-governed cytotoxic T-cell infiltration in lung cancer. Sci. Rep. 2020, 10, 21360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Ye, S.B.; Ni, J.J.; Cai, T.T.; Liu, Y.N.; Huang, D.J.; Mai, H.Q.; Chen, Q.Y.; He, J.; Zhang, X.S.; et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. 2019, 26, 2314–2328. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Zhang, Y.; Luo, Q.; Zheng, W.; Li, W.; Zeng, X.; Li, Q.; Quan, J. STAT3 inhibition enhances CDN-induced STING signaling and antitumor immunity. Cancer Lett. 2019, 450, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, E.; Zabaleta, N.; Gil-Farina, I.; Gonzalez-Aparicio, M.; Echeverz, M.; Bähre, H.; Solano, C.; Lasa, I.; Gonzalez-Aseguinolaza, G.; Hommel, M. AdrA as a Potential Immunomodulatory Candidate for STING-Mediated Antiviral Therapy That Required Both Type I IFN and TNF-α Production. J. Immunol. 2021, 206, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Guo, R.; Ma, C.; Zeng, D.; Su, Z. Manganese Breaks the Immune Tolerance of HBs-Ag. Open Forum Infect. Dis. 2021, 8, ofab028. [Google Scholar] [CrossRef]
- Ito, H.; Kanbe, A.; Hara, A.; Ishikawa, T. Induction of humoral and cellular immune response to HBV vaccine can be up-regulated by STING ligand. Virology 2019, 531, 233–239. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HD | CHB | |
|---|---|---|
| Number | 90 | 107 |
| Gender (M/F) | 42/48 | 61/46 |
| Age (years) | 46.84 ± 11.51 | 41.14 ± 11.05 |
| HBsAg (Pos/Neg) | 0/90 | 107/107 |
| HBeAg (Pos/Neg) | 0/90 | 0/107 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, B.; Yu, Y.; Pan, Z.; Feng, Y.; Zhao, H.; Han, Q.; Zhang, J. HBsAg Dampened STING Associated Activation of NK Cells in HBeAg-Negative CHB Patients. Int. J. Mol. Sci. 2021, 22, 7643. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147643
Zheng B, Yu Y, Pan Z, Feng Y, Zhao H, Han Q, Zhang J. HBsAg Dampened STING Associated Activation of NK Cells in HBeAg-Negative CHB Patients. International Journal of Molecular Sciences. 2021; 22(14):7643. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147643
Chicago/Turabian StyleZheng, Bingqing, Yating Yu, Zhaoyi Pan, Yujie Feng, Huajun Zhao, Qiuju Han, and Jian Zhang. 2021. "HBsAg Dampened STING Associated Activation of NK Cells in HBeAg-Negative CHB Patients" International Journal of Molecular Sciences 22, no. 14: 7643. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147643