
Cigarette Smoke Specifically Affects Small Airway Epithelial Cell Populations and Triggers the Expansion of Inflammatory and Squamous Differentiation Associated Basal Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
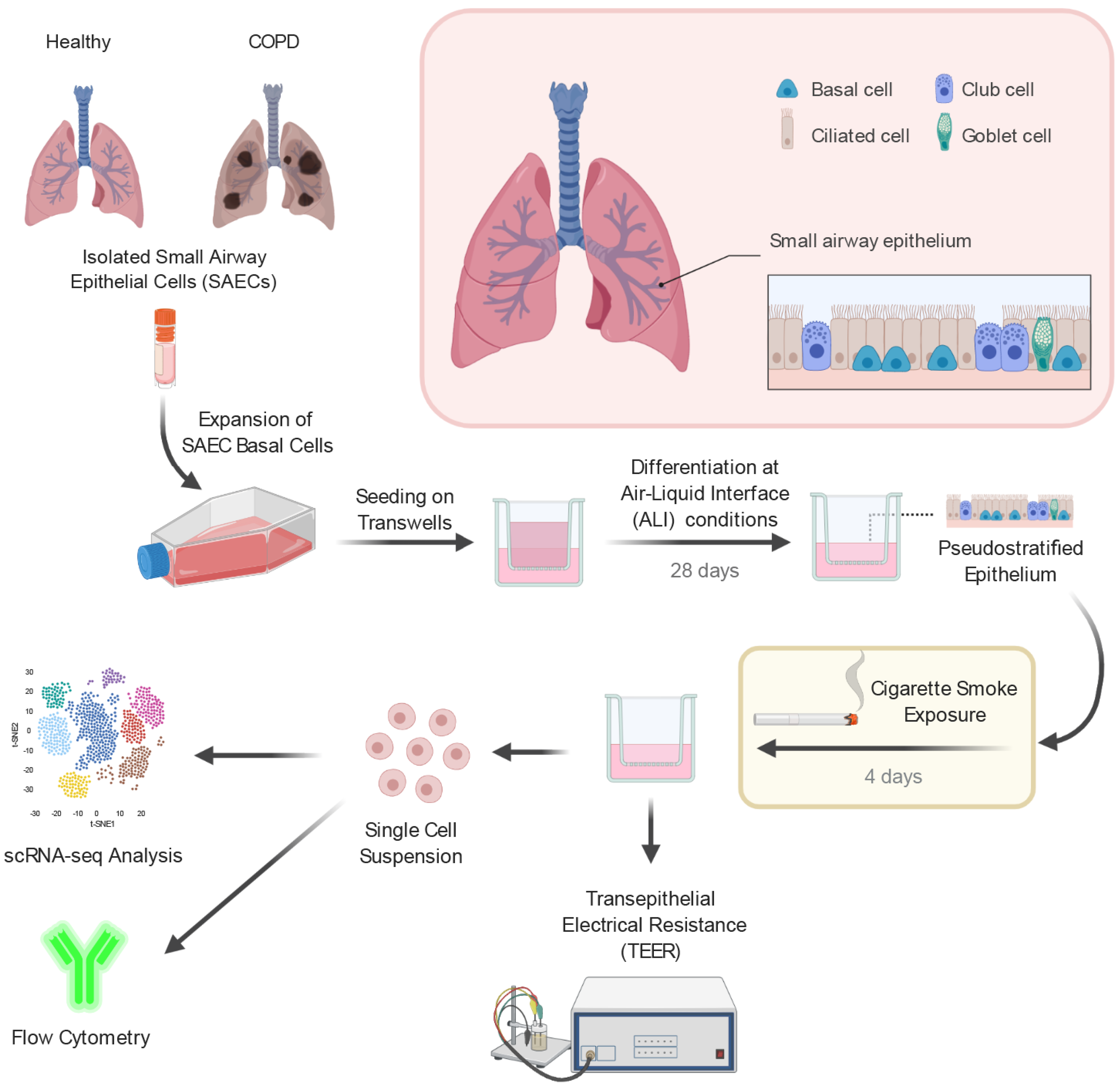
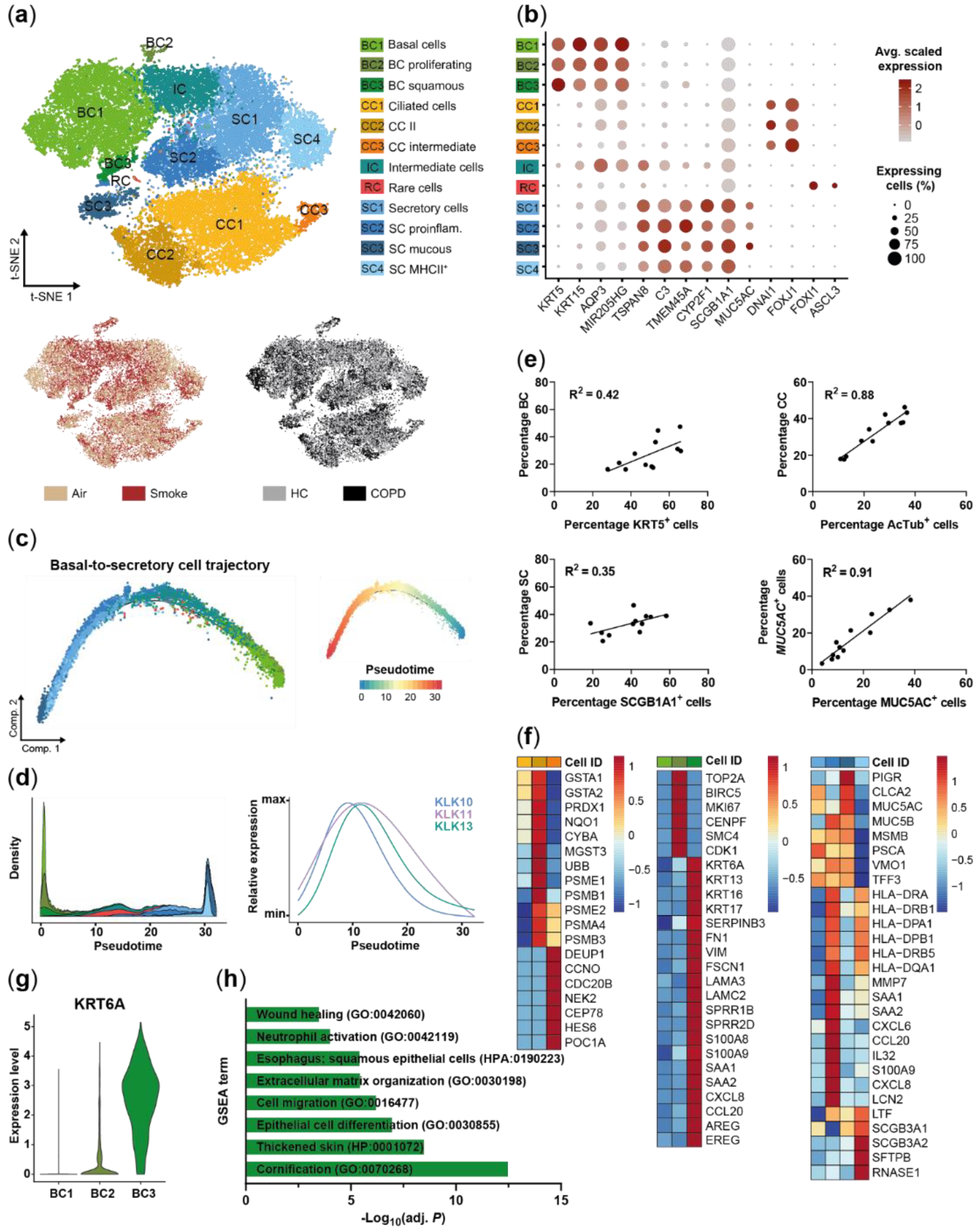
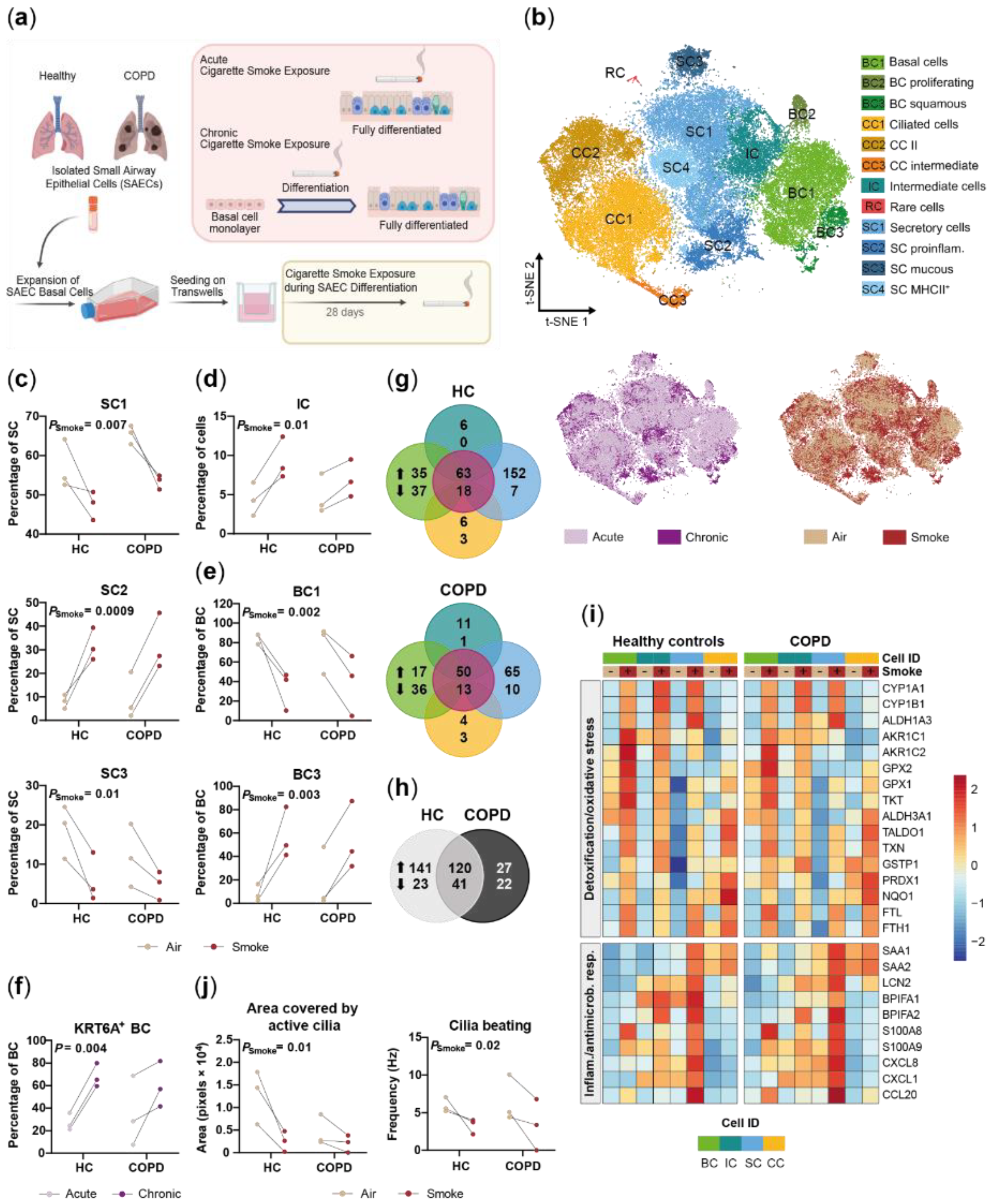
2.1. Small Airway Epithelial Cells—A Heterogeneous Cell Population
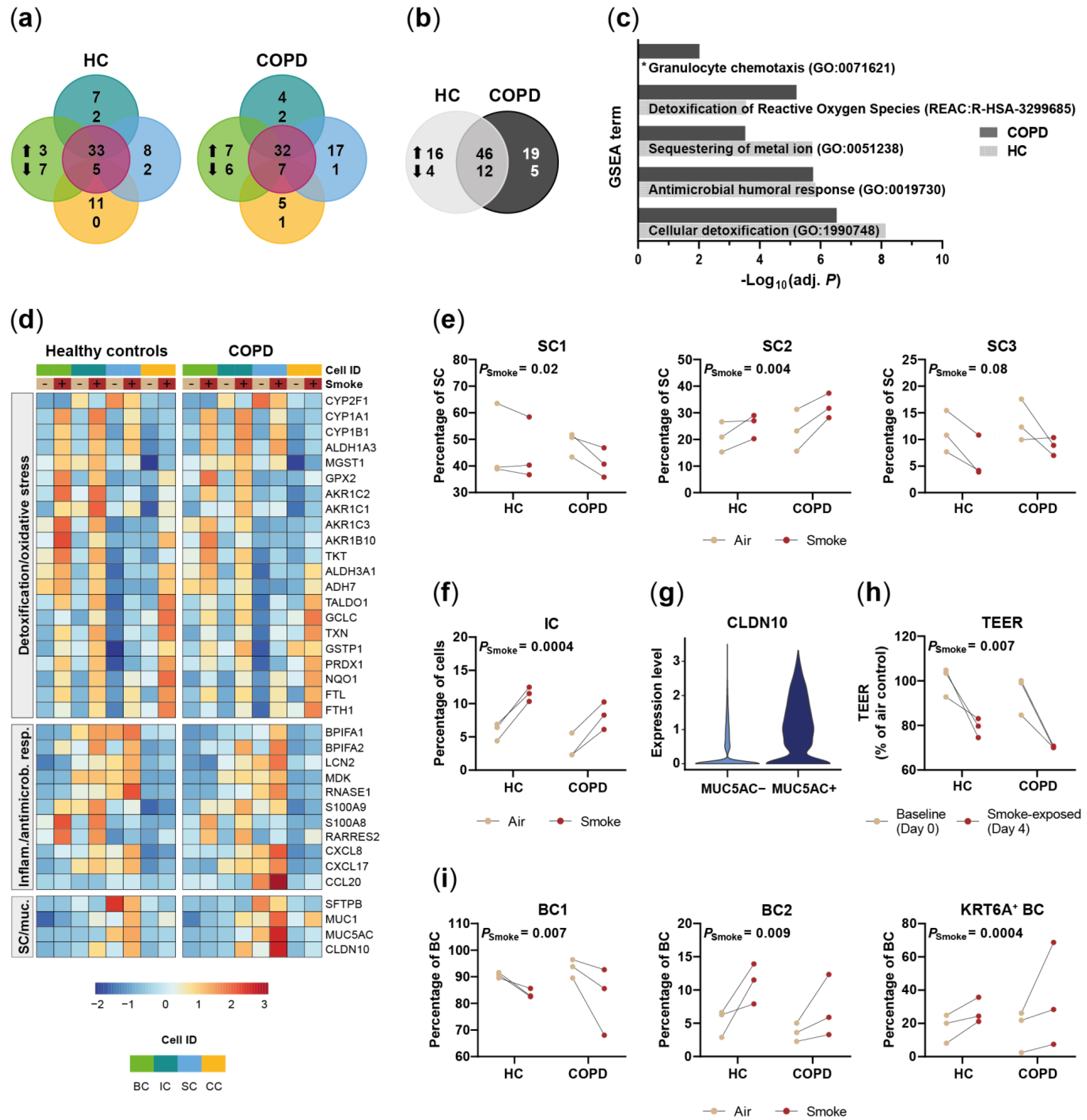
2.2. Disease-Relevant Characteristics in COPD-Derived ALI Cultures and Their Differences in Response to Cigarette Smoke Exposure
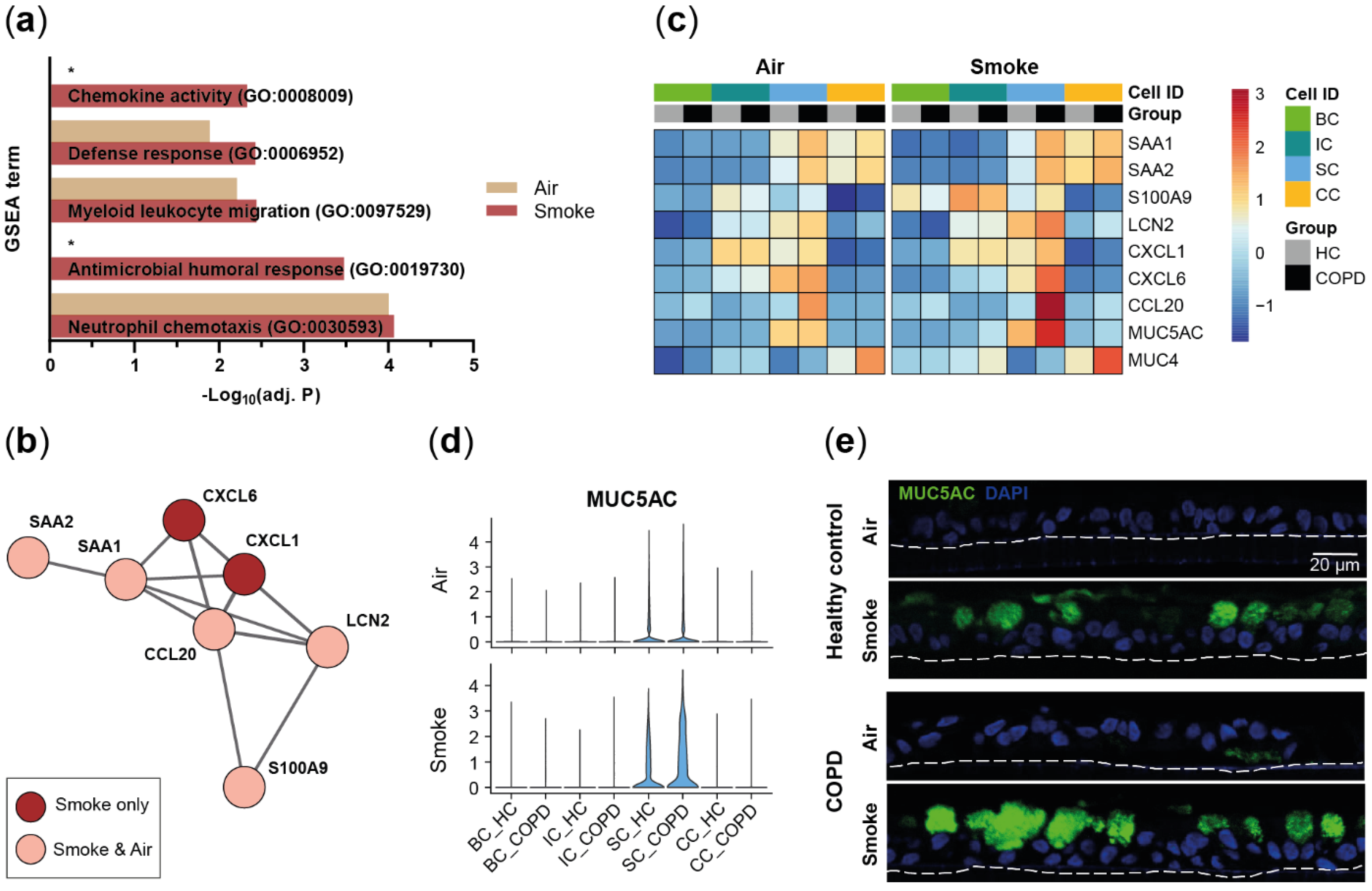
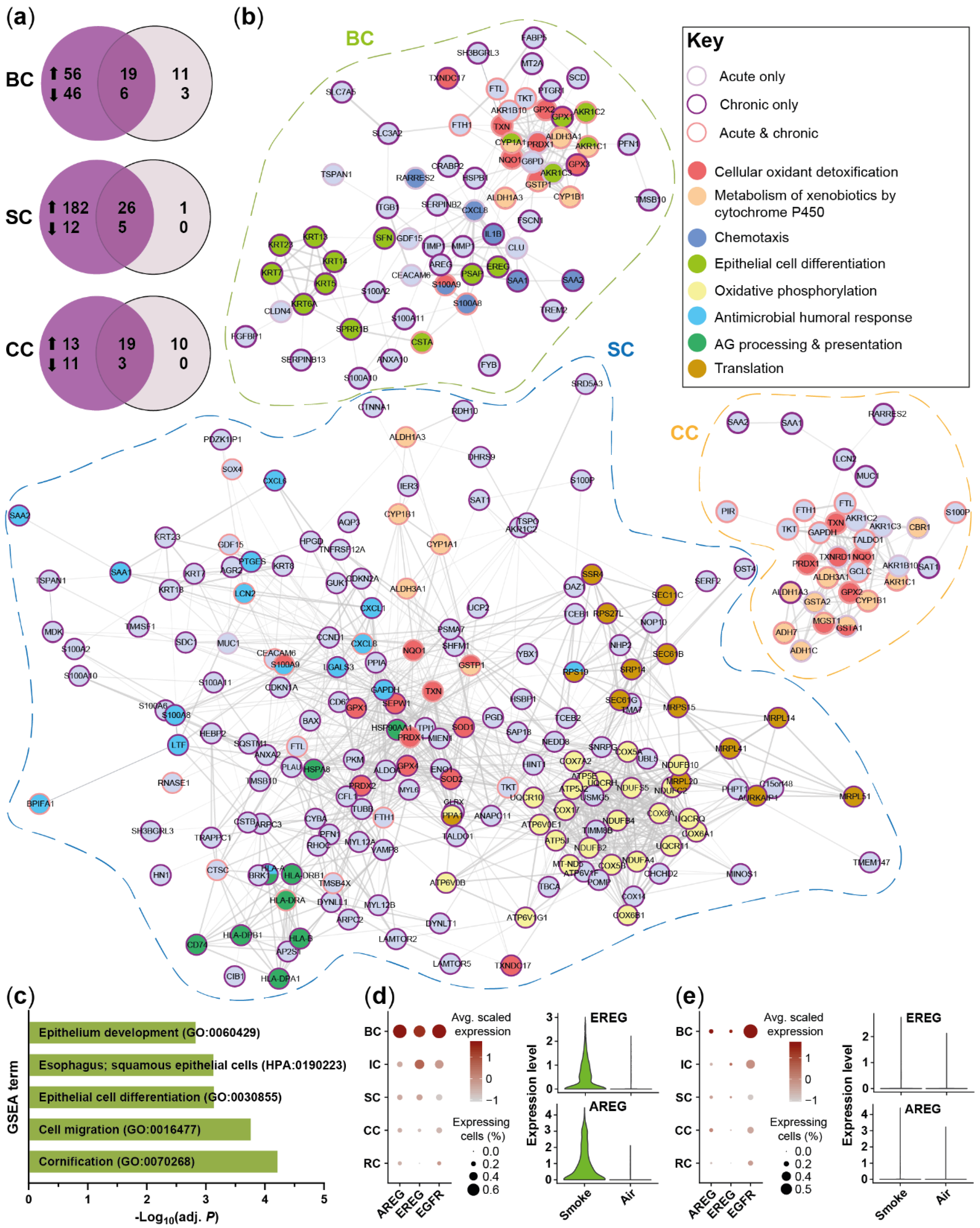
2.3. Acute Smoke Exposure Triggers Common and Cell Type-Specific Detoxification and Inflammatory Responses and Aberrant Basal Cell Activation and Differentiation
2.4. Chronic Smoke Exposure during Cell Differentiation Shifts Basal Cells towards the Squamous Differentiation Associated Phenotype
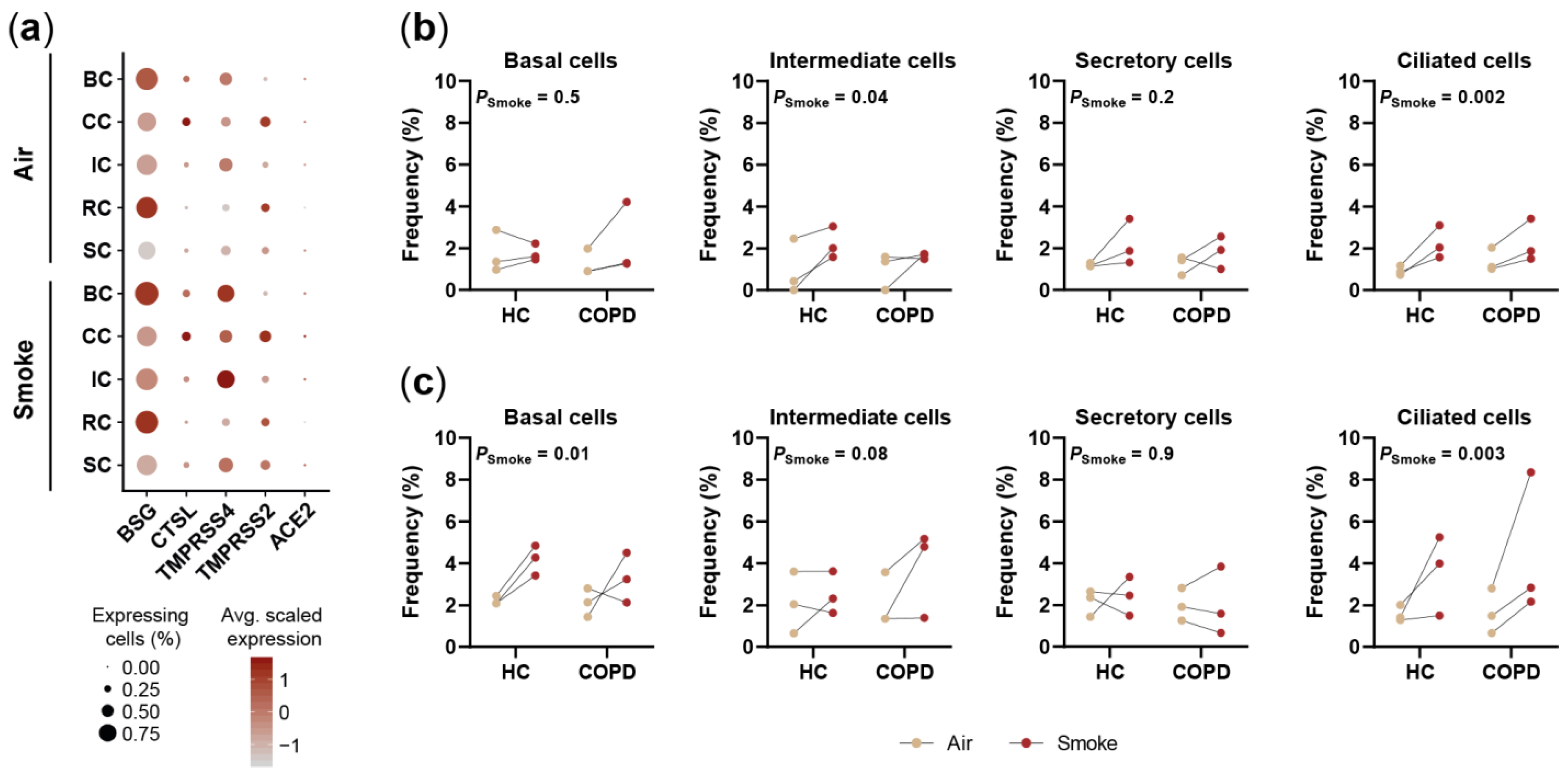
2.5. Smoking Affects the Expression of SARS-CoV-2 Entry Factors
3. Discussion
4. Materials and Methods
4.1. Small Airway Epithelial Cell Air–Liquid Interface Culture
4.2. Cigarette Smoke Exposure
4.3. Preparation of Single-Cell Suspensions
4.4. Single-Cell RNA-Sequencing
4.4.1. Library Preparation and Sequencing
4.4.2. Data Processing
4.4.3. Quality Control, Cluster Analysis and Visualization of scRNA-seq Data
4.4.4. Differential Expression Analysis
4.4.5. Gene Set Enrichment and Protein–Protein Interaction Network Analysis
4.4.6. Pseudotime Analysis
4.5. Transepithelial Electrical Resistance
4.6. Flow Cytometry
4.7. Histology
4.8. Statistical Analysis
4.9. Data Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Goldfarbmuren, K.C.; Jackson, N.D.; Sajuthi, S.P.; Dyjack, N.; Li, K.S.; Rios, C.L.; Plender, E.G.; Montgomery, M.T.; Everman, J.L.; Bratcher, P.E.; et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 2020, 11, 1–21. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Van Winkle, L.S.; Fanucchi, M.V.; Plopper, C.G. Cellular and molecular characteristics of basal cells in airway epithelium. Exp. Lung Res. 2001, 27, 401–415. [Google Scholar] [CrossRef]
- Rock, J.R.; Hogan, B.L. Epithelial Progenitor Cells in Lung Development, Maintenance, Repair, and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 493–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Hackett, N.R.; Shaykhiev, R.; Walters, M.S.; Wang, R.; Zwick, R.; Ferris, B.; Witover, B.; Salit, J.; Crystal, R.G. The Human Airway Epithelial Basal Cell Transcriptome. PLoS ONE 2011, 6, e18378. [Google Scholar] [CrossRef]
- Yoshida, T.; Tuder, R.M. Pathobiology of Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. Physiol. Rev. 2007, 87, 1047–1082. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborín, R. Smoking and Chronic Obstructive Pulmonary Disease (COPD). Parallel Epidemics of the 21st Century. Int. J. Environ. Res. Public Health 2009, 6, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Yanai, M.; Sekizawa, K.; Ohrui, T.; Sasaki, H.; Takishima, T. Site of airway obstruction in pulmonary disease: Direct measurement of intrabronchial pressure. J. Appl. Physiol. 1992, 72, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Macklem, P.T.; Thurlbeck, W.M. Site and Nature of Airway Obstruction in Chronic Obstructive Lung Disease. N. Engl. J. Med. 1968, 278, 1355–1360. [Google Scholar] [CrossRef]
- Lumsden, A.B.; McLean, A.; Lamb, D. Goblet and Clara cells of human distal airways: Evidence for smoking induced changes in their numbers. Thorax 1984, 39, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Saetta, M.; Turato, G.; Baraldo, S.; Zanin, A.; Braccioni, F.; Mapp, C.E.; Maestrelli, P.; Cavallesco, G.; Papi, A.; Fabbri, L. Goblet Cell Hyperplasia and Epithelial Inflammation in Peripheral Airways of Smokers with Both Symptoms of Chronic Bronchitis and Chronic Airflow Limitation. Am. J. Respir. Crit. Care Med. 2000, 161, 1016–1021. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Crystal, R.G. Early Events in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Smoking-induced Reprogramming of Airway Epithelial Basal Progenitor Cells. Ann. Am. Thorac. Soc. 2014, 11, S252–S258. [Google Scholar] [CrossRef] [Green Version]
- Auerbach, O.; Gere, J.B.; Forman, J.B.; Petrick, T.G.; Smolin, H.J.; Muehsam, G.E.; Kassouny, D.Y.; Stout, A.P. Changes in the Bronchial Epithelium in Relation to Smoking and Cancer of the Lung. N. Engl. J. Med. 1957, 256, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.M.; Vincent, T.L.; Salit, J.; Walters, M.S.; Agosto-Perez, F.; Shaykhiev, R.; Strulovici-Barel, Y.; Downey, R.J.; Buro-Auriemma, L.J.; Staudt, M.; et al. Smoking Dysregulates the Human Airway Basal Cell Transcriptome at COPD Risk Locus 19q13.2. PLoS ONE 2014, 9, e88051. [Google Scholar] [CrossRef] [Green Version]
- Crystal, R.G. Airway Basal Cells. The “Smoking Gun” of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2014, 190, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchelle, E.; Zahm, J.-M.; Tournier, J.-M.; Coraux, C. Airway Epithelial Repair, Regeneration, and Remodeling after Injury in Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Perotin, J.-M.; Adam, D.; Vella-Boucaud, J.; Delepine, G.; Sandu, S.; Jonvel, A.-C.; Prevost, A.; Berthiot, G.; Pison, C.; Lebargy, F.; et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir. Res. 2014, 15, 151. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Miller, Y.E.; Nakachi, I.; Kwon, J.B.; Barón, A.E.; Brantley, A.E.; Merrick, D.T.; Franklin, W.A.; Keith, R.L.; Vandivier, R.W. Exhaustion of Airway Basal Progenitor Cells in Early and Established Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2018, 197, 885–896. [Google Scholar] [CrossRef]
- Rigden, H.M.; Alias, A.; Havelock, T.; O’Donnell, R.; Djukanovic, R.; Davies, D.; Wilson, S.J. Squamous Metaplasia Is Increased in the Bronchial Epithelium of Smokers with Chronic Obstructive Pulmonary Disease. PLoS ONE 2016, 11, e0156009. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Cambier, S.; Markovics, J.A.; Wolters, P.; Jablons, D.; Hill, A.; Finkbeiner, W.; Jones, K.; Broaddus, V.C.; Sheppard, D.; et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J. Clin. Investig. 2007, 117, 3551–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polosukhin, V.V.; Cates, J.; Lawson, W.E.; Zaynagetdinov, R.; Milstone, A.P.; Massion, P.P.; Ocak, S.; Ware, L.B.; Lee, J.W.; Bowler, R.P.; et al. Bronchial Secretory Immunoglobulin A Deficiency Correlates With Airway Inflammation and Progression of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2011, 184, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Polosukhin, V.V.; Richmond, B.W.; Du, R.-H.; Cates, J.; Wu, P.; Nian, H.; Massion, P.P.; Ware, L.B.; Lee, J.W.; Kononov, A.; et al. Secretory IgA Deficiency in Individual Small Airways Is Associated with Persistent Inflammation and Remodeling. Am. J. Respir. Crit. Care Med. 2017, 195, 1010–1021. [Google Scholar] [CrossRef]
- Yaghi, A.; Dolovich, M.B. Airway Epithelial Cell Cilia and Obstructive Lung Disease. Cells 2016, 5, 40. [Google Scholar] [CrossRef]
- Rennard, S.I. Pathogenesis of COPD. Clin. Cornerstone 2003, 5. [Google Scholar] [CrossRef]
- Hessel, J.; Heldrich, J.; Fuller, J.; Staudt, M.; Radisch, S.; Hollmann, C.; Harvey, B.-G.; Kaner, R.J.; Salit, J.; Yee-Levin, J.; et al. Intraflagellar Transport Gene Expression Associated with Short Cilia in Smoking and COPD. PLoS ONE 2014, 9, e85453. [Google Scholar] [CrossRef] [Green Version]
- Yaghi, A.; Zaman, A.; Cox, G.; Dolovich, M.B. Ciliary beating is depressed in nasal cilia from chronic obstructive pulmonary disease subjects. Respir. Med. 2012, 106, 1139–1147. [Google Scholar] [CrossRef] [Green Version]
- Harvey, B.-G.; Heguy, A.; Leopold, P.L.; Carolan, B.J.; Ferris, B.; Crystal, R.G. Modification of gene expression of the small airway epithelium in response to cigarette smoking. J. Mol. Med. 2006, 85, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Hackett, N.R.; Butler, M.W.; Shaykhiev, R.; Salit, J.; Omberg, L.; Rodriguez-Flores, J.L.; Mezey, J.G.; Strulovici-Barel, Y.; Wang, G.; Didon, L.; et al. RNA-Seq quantification of the human small airway epithelium transcriptome. BMC Genom. 2012, 13, 82. [Google Scholar] [CrossRef] [Green Version]
- Duclos, G.E.; Teixeira, V.H.; Autissier, P.; Gesthalter, Y.B.; Reinders-Luinge, M.A.; Terrano, R.; Dumas, Y.M.; Liu, G.; Mazzilli, S.A.; Brandsma, C.-A.; et al. Characterizing smoking-induced transcriptional heterogeneity in the human bronchial epithelium at single-cell resolution. Sci. Adv. 2019, 5, eaaw3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.-L.; Rostami, M.R.; Shenoy, S.A.; LeBlanc, M.G.; Salit, J.; Strulovici-Barel, Y.; O’Beirne, S.L.; Kaner, R.J.; Leopold, P.L.; Mezey, J.G.; et al. Cell-specific expression of lung disease risk-related genes in the human small airway epithelium. Respir. Res. 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gindele, J.A.; Kiechle, T.; Benediktus, K.; Birk, G.; Brendel, M.; Heinemann, F.; Wohnhaas, C.T.; Leblanc, M.; Zhang, H.; Strulovici-Barel, Y.; et al. Intermittent exposure to whole cigarette smoke alters the differentiation of primary small airway epithelial cells in the air-liquid interface culture. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.-L.; Yang, J.; Gomi, K.; Chao, I.; Crystal, R.G.; Shaykhiev, R. EGF-Amphiregulin Interplay in Airway Stem/Progenitor Cells Links the Pathogenesis of Smoking-Induced Lesions in the Human Airway Epithelium. Stem Cells 2017, 35, 824–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.-L.; Yang, J.; Strulovici-Barel, Y.; Salit, J.; Rostami, M.; Mezey, J.G.; O’Beirne, S.; Kaner, R.J.; Crystal, R.G. Exaggerated BMP4 signalling alters human airway basal progenitor cell differentiation to cigarette smoking-related phenotypes. Eur. Respir. J. 2019, 53, 1702553. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Zhang, L.-J. Keratin 6, 16 and 17—Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, S.D.; Reynolds, P.R.; Pryhuber, G.S.; Finder, J.D.; Stripp, B.R. Secretoglobins SCGB3A1 and SCGB3A2 Define Secretory Cell Subsets in Mouse and Human Airways. Am. J. Respir. Crit. Care Med. 2002, 166, 1498–1509. [Google Scholar] [CrossRef]
- Yoneda, M.; Xu, L.; Kajiyama, H.; Kawabe, S.; Paiz, J.; Ward, J.M.; Kimura, S. Secretoglobin Superfamily Protein SCGB3A2 Alleviates House Dust Mite-Induced Allergic Airway Inflammation in Mice. Int. Arch. Allergy Immunol. 2016, 171, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Kurotani, R.; Kusakabe, T.; Miura, T.; Link, B.W.; Misawa, M.; Kimura, S. Uteroglobin-related Protein 1 Expression Suppresses Allergic Airway Inflammation in Mice. Am. J. Respir. Crit. Care Med. 2006, 173, 958–964. [Google Scholar] [CrossRef]
- Epaud, R.; Ikegami, M.; Whitsett, J.A.; Jobe, A.H.; Weaver, T.E.; Akinbi, H.T. Surfactant Protein B Inhibits Endotoxin-Induced Lung Inflammation. Am. J. Respir. Cell Mol. Biol. 2003, 28, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Bi, G.; Wu, L.; Huang, P.; Islam, S.; Heruth, D.P.; Zhang, L.Q.; Li, D.; Sampath, V.; Huang, W.; Simon, B.A.; et al. Up-regulation of SFTPB expression and attenuation of acute lung injury by pulmonary epithelial cell-specific NAMPT knockdown. FASEB J. 2018, 32, 3583–3596. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Blanco, J.A.; Fakih, D.; Arike, L.; Rodríguez-Piñeiro, A.M.; Abad, B.M.; Skansebo, E.; Jackson, S.; Root, J.; Singh, D.; McCrae, C.; et al. Attached stratified mucus separates bacteria from the epithelial cells in COPD lungs. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Wuyts, A.; Van Osselaer, N.; Haelens, A.; Samson, I.; Herdewijn, P.; Ben-Baruch, A.; Oppenheim, J.J.; Proost, A.P.; Van Damme, J. Characterization of Synthetic Human Granulocyte Chemotactic Protein 2: Usage of Chemokine Receptors CXCR1 and CXCR2 andin VivoInflammatory Properties. Biochemistry 1997, 36, 2716–2723. [Google Scholar] [CrossRef]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; Van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [Green Version]
- Dohlman, T.; Chauhan, S.K.; Kodati, S.; Hua, J.; Chen, Y.; Omoto, M.; Sadrai, Z.; Dana, R. The CCR6/CCL20 Axis Mediates Th17 Cell Migration to the Ocular Surface in Dry Eye Disease. Investig. Opthalmol. Vis. Sci. 2013, 54, 4081–4091. [Google Scholar] [CrossRef] [Green Version]
- Krzysiek, R.; Lefevre, E.A.; Bernard, J.; Foussat, A.; Galanaud, P.; Louache, F.; Richard, Y. Regulation of CCR6 chemokine receptor expression and responsiveness to macrophage inflammatory protein-3alpha/CCL20 in human B cells. Blood 2000, 96, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, M.; Etchart, N.; Goubier, A.; Lira, S.A.; Sirard, J.-C.; van Rooijen, N.; Caux, C.; Aït-Yahia, S.; Vicari, A.; Kaiserlian, D.; et al. Dendritic Cells Rapidly Recruited into Epithelial Tissues via CCR6/CCL20 Are Responsible for CD8+ T Cell Crosspriming In Vivo. Immunity 2006, 24, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Orino, K.; Lehman, L.; Tsuji, Y.; Ayaki, H.; Torti, S.V.; Torti, F.M. Ferritin and the response to oxidative stress. Biochem. J. 2001, 357, 241–247. [Google Scholar] [CrossRef]
- Koval, M. Claudin Heterogeneity and Control of Lung Tight Junctions. Annu. Rev. Physiol. 2013, 75, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Günzel, D.; Stuiver, M.; Kausalya, P.J.; Haisch, L.; Krug, S.; Rosenthal, R.; Meij, I.C.; Hunziker, W.; Fromm, M.; Müller, D. Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J. Cell Sci. 2009, 122, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Tesfaigzi, J.; Carlson, D.M. Expression, regulation, and function of the SPR family of proteins. Cell Biophys. 1999, 30, 243–265. [Google Scholar] [CrossRef]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev. Cell 2020, 53, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tao, Z.-W.; Wang, L.; Yuan, M.-L.; Liu, K.; Zhou, L.; Wei, P.-F.; Deng, Y.; Liu, J.; Liu, H.-G.; et al. Analysis of factors associated with disease outcomes in hospitalized patients with 2019 novel coronavirus disease. Chin. Med. J. 2020, 133, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.-Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.-X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Deprez, M.; Zaragosi, L.-E.; Truchi, M.; Becavin, C.; García, S.R.; Arguel, M.-J.; Plaisant, M.; Magnone, V.; Lebrigand, K.; Abelanet, S.; et al. A Single-Cell Atlas of the Human Healthy Airways. Am. J. Respir. Crit. Care Med. 2020, 202, 1636–1645. [Google Scholar] [CrossRef]
- Zuo, W.-L.; Rostami, M.R.; Leblanc, M.; Kaner, R.J.; O’Beirne, S.L.; Mezey, J.G.; Leopold, P.L.; Quast, K.; Visvanathan, S.; Fine, J.S.; et al. Dysregulation of club cell biology in idiopathic pulmonary fibrosis. PLoS ONE 2020, 15, e0237529. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef]
- Glanville, A.R.; Tazelaar, H.D.; Theodore, J.; Imoto, E.; Rouse, R.V.; Baldwin, J.C.; Robin, E.D. The Distribution of MHC Class I and II Antigens on Bronchial Epithelium. Am. Rev. Respir. Dis. 1989, 139, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Kalb, T.H.; Chuang, M.T.; Marom, Z.; Mayer, L. Evidence for Accessory Cell Function by Class II MHC Antigen-expressing Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 1991, 4, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Amatngalim, G.; Schrumpf, J.A.; Dishchekenian, F.; Mertens, T.C.; Ninaber, D.K.; Van Der Linden, A.C.; Pilette, C.; Taube, C.; Hiemstra, P.S.; van der Does, A. Aberrant epithelial differentiation by cigarette smoke dysregulates respiratory host defence. Eur. Respir. J. 2018, 51, 1701009. [Google Scholar] [CrossRef]
- Richmond, B.W.; Brucker, R.M.; Han, W.; Du, R.-H.; Zhang, Y.; Cheng, D.-S.; Gleaves, L.; Abdolrasulnia, R.; Polosukhina, D.; Clark, P.E.; et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat. Commun. 2016, 7, 11240. [Google Scholar] [CrossRef]
- Gohy, S.T.; Detry, B.R.; Lecocq, M.; Bouzin, C.; Weynand, B.A.; Amatngalim, G.; Sibille, Y.M.; Pilette, C. Polymeric Immunoglobulin Receptor Down-regulation in Chronic Obstructive Pulmonary Disease. Persistence in the Cultured Epithelium and Role of Transforming Growth Factor-β. Am. J. Respir. Crit. Care Med. 2014, 190, 509–521. [Google Scholar] [CrossRef]
- Rao, W.; Wang, S.; Duleba, M.; Niroula, S.; Goller, K.; Xie, J.; Mahalingam, R.; Neupane, R.; Liew, A.-A.; Vincent, M.; et al. Regenerative Metaplastic Clones in COPD Lung Drive Inflammation and Fibrosis. Cell 2020, 181, 848–864. [Google Scholar] [CrossRef]
- Wojcik, S.M.; Bundman, D.S.; Roop, D.R. Delayed Wound Healing in Keratin 6a Knockout Mice. Mol. Cell. Biol. 2000, 20, 5248–5255. [Google Scholar] [CrossRef] [Green Version]
- Coraux, C.; Roux, J.; Jolly, T.; Birembaut, P. Epithelial Cell-Extracellular Matrix Interactions and Stem Cells in Airway Epithelial Regeneration. Proc. Am. Thorac. Soc. 2008, 5, 689–694. [Google Scholar] [CrossRef]
- Riese, D.J.; Cullum, R.L. Epiregulin: Roles in normal physiology and cancer. Semin. Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demedts, I.K.; Bracke, K.R.; Van Pottelberge, G.; Testelmans, D.; Verleden, G.M.; Vermassen, F.E.; Joos, G.F.; Brusselle, G. Accumulation of Dendritic Cells and Increased CCL20 Levels in the Airways of Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2007, 175, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Treekitkarnmongkol, W.; Hassane, M.; Sinjab, A.; Chang, K.; Hara, K.; Rahal, Z.; Zhang, J.; Lu, W.; Sivakumar, S.; McDowell, T.L.; et al. Augmented Lipocalin-2 Is Associated with Chronic Obstructive Pulmonary Disease and Counteracts Lung Adenocarcinoma Development. Am. J. Respir. Crit. Care Med. 2021, 203, 90–101. [Google Scholar] [CrossRef] [PubMed]
- López-Campos, J.L.; Calero, C.; Rojano, B.; López-Porras, M.; Saenz, J.; Blanco, A.I.; Sánchez-López, V.; Tobar, D.; Montes-Worboys, A.; Arellano, E. C-Reactive Protein and Serum Amyloid A Overexpression in Lung Tissues of Chronic Obstructive Pulmonary Disease Patients: A Case-Control Study. Int. J. Med. Sci. 2013, 10, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.H.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wobbrock, J.O.; Findlater, L.; Gergle, D.; Higgins, J.J. The aligned rank transform for nonparametric factorial analyses using only anova procedures. In Proceedings of the 2011 Annual Conference on Human Factors in Computing Systems—CHI ’11, Vancouver, BC, Canada, 7–12 May 2011; ACM Press: New York, NY, USA, 2011; p. 143. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wohnhaas, C.T.; Gindele, J.A.; Kiechle, T.; Shen, Y.; Leparc, G.G.; Stierstorfer, B.; Stahl, H.; Gantner, F.; Viollet, C.; Schymeinsky, J.; et al. Cigarette Smoke Specifically Affects Small Airway Epithelial Cell Populations and Triggers the Expansion of Inflammatory and Squamous Differentiation Associated Basal Cells. Int. J. Mol. Sci. 2021, 22, 7646. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147646
Wohnhaas CT, Gindele JA, Kiechle T, Shen Y, Leparc GG, Stierstorfer B, Stahl H, Gantner F, Viollet C, Schymeinsky J, et al. Cigarette Smoke Specifically Affects Small Airway Epithelial Cell Populations and Triggers the Expansion of Inflammatory and Squamous Differentiation Associated Basal Cells. International Journal of Molecular Sciences. 2021; 22(14):7646. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147646
Chicago/Turabian StyleWohnhaas, Christian T., Julia A. Gindele, Tobias Kiechle, Yang Shen, Germán G. Leparc, Birgit Stierstorfer, Heiko Stahl, Florian Gantner, Coralie Viollet, Jürgen Schymeinsky, and et al. 2021. "Cigarette Smoke Specifically Affects Small Airway Epithelial Cell Populations and Triggers the Expansion of Inflammatory and Squamous Differentiation Associated Basal Cells" International Journal of Molecular Sciences 22, no. 14: 7646. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147646