
Adenosine and Inflammation: Here, There and Everywhere
by
 , and
, and
Silvia Pasquini
1 ,
,
Chiara Contri
1,
Pier Andrea Borea
2,
Fabrizio Vincenzi
1,* and
Katia Varani
1 1
Department of Translational Medicine, University of Ferrara, 44121 Ferrara, Italy
2
University of Ferrara, 44121 Ferrara, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(14), 7685; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147685
Submission received: 29 June 2021
/
Revised: 16 July 2021
/
Accepted: 16 July 2021
/
Published: 19 July 2021
(This article belongs to the Special Issue Pharmacological, Biochemical and Molecular Study of Adenosine Receptors)
Abstract
:Adenosine is a ubiquitous endogenous modulator with the main function of maintaining cellular and tissue homeostasis in pathological and stress conditions. It exerts its effect through the interaction with four G protein-coupled receptor (GPCR) subtypes referred as A1, A2A, A2B, and A3 adenosine receptors (ARs), each of which has a unique pharmacological profile and tissue distribution. Adenosine is a potent modulator of inflammation, and for this reason the adenosinergic system represents an excellent pharmacological target for the myriad of diseases in which inflammation represents a cause, a pathogenetic mechanism, a consequence, a manifestation, or a protective factor. The omnipresence of ARs in every cell of the immune system as well as in almost all cells in the body represents both an opportunity and an obstacle to the clinical use of AR ligands. This review offers an overview of the cardinal role of adenosine in the modulation of inflammation, showing how the stimulation or blocking of its receptors or agents capable of regulating its extracellular concentration can represent promising therapeutic strategies for the treatment of chronic inflammatory pathologies, neurodegenerative diseases, and cancer.
1. Introduction
Inflammation arises from a complex interplay between immune cells and many soluble mediators with the aim to protect the organism from harmful stimuli such as microorganism infections, damaged cells, or irritants as well as being a strong component of many pathological conditions like rheumatic diseases, neurological disorders, and cancer [1]. Adenosine, a pervasive autacoid, is considered a key mediator of the immune response. In physiological and unstressed conditions, the extracellular concentrations of adenosine are maintained at a low level as a result of the rapid metabolism and uptake [2]. Nevertheless, its levels rise considerably during conditions involving increased metabolic demand, hypoxia, tissue injury, and inflammation regulating the immune response.
Adenosine exerts its functions through the interaction with four adenosine receptors (ARs), all of them are transmembrane G protein-coupled receptors (GPCRs) named A1, A2A, A2B, and A3ARs. Interestingly, the A1 and A3 subtypes are coupled to Gi proteins and have an inhibitory effect on adenylyl cyclase (AC) activity; while A2A and A2BARs stimulate it, through the coupling to Gs proteins, with a consequent modulation of cyclic AMP (cAMP) levels [3]. Adenosine production occurs through different mechanisms, the principal is by the dephosphorylation of the adenine nucleotides (ATP, ADP, and AMP) to adenosine. Especially ATP serves as the reservoir for the production of adenosine because it is the most abundant molecule in the cell [4]. Under resting conditions, some ATP is dephosphorylated to adenosine, but stressful stimuli can increase rates of intracellular conversion of ATP to adenosine. More commonly, these stimuli trigger the release of adenine nucleotides into the extracellular space where they are dephosphorylated to adenosine mainly by the combined action of two hydrolyzing enzymes termed ectonucleoside triphosphate diphosphohydrolase (CD39) and ecto-5′-nucleotidase (CD73) [3]. Once formed or released into the extracellular space, adenosine can be deaminated to inosine through adenosine deaminase (ADA) or taken up directly by cells by specific nucleoside transporters (ENT1 and ENT2) and re-phosphorylated to ATP [5,6] (Figure 1).
Although there are instances in which adenosine exerts detrimental effects in various pathological conditions, it is generally considered a protective and homeostatic mediator against tissue damages and stress conditions [4,5]. In particular, adenosine presents immune-regulatory effects, mostly anti-inflammatory, which strengthen its general tissue-protective functions. In some cases, however, the effect of adenosine on the immune system is deleterious, as prolonged adenosine signaling may impede antitumor and antibacterial immunity, thereby promoting the development and progression of cancer and sepsis, respectively [1,7].
The focal point of this review is to describe the current knowledge about the role of ARs in inflammation, starting from the regulation of immune cells by ARs and then discussing their role in different pathological conditions where inflammation is predominant such as rheumatic diseases, lung and intestinal disorders, neuroinflammation, and cancer.
2. Regulation of Immune Cells
Adenosine is considered a crucial mediator of the immune response. ARs are expressed in all kinds of immune cells where they participate in the regulation of immune and inflammatory responses, usually with anti-inflammatory effects supporting the protective role of adenosine (Figure 2) [1]. ARs are present on both monocytes and macrophages, with different expression levels depending on maturation advancement from monocytes to macrophages. In particular, in quiescent monocytes, a modest expression of A1, A2A, and A3ARs has been reported which rises over differentiation into macrophages [8]. Many studies also investigated the role of proinflammatory cytokines on AR expression reporting that interleukin (IL)-1 and tumor necrosis factor α (TNF-α) enhance both A2AAR expression and functionality, averting receptor desensitization, in human monocytes [9,10]. The anti-inflammatory effect of ARs in macrophages is supported both by A2A, A2B, and A3AR activation and by extracellular adenosine. The Ars’ activation blocks the release of proinflammatory mediators like TNF-α, IL-6, IL-12, nitric oxide, and macrophage inflammatory protein (MIP)-1α [11,12,13] while adenosine, acting on A2A and A2BArs, endorses the release of IL-10, an anti-inflammatory cytokine [14,15,16]; while the A3AR stimulation promotes macrophages chemotaxis towards apoptotic cells [17]. Adenosine is also involved in the regulation of dendritic cells, the antigen-presenting cells able to trigger adaptive immune response [1,18]. A1 and A3ARs are mainly present in immature dendritic cells where they regulate chemotaxis through intracellular calcium rise. Differently, in mature dendritic cells mostly A2AARs are expressed leading to a decrease in proinflammatory cytokines [19]. On the contrary, a proinflammatory effect of A2BARs in dendritic cells has been reported. In fact, they have the capability to shift bone marrow cell differentiation to a specific subpopulation of dendritic cells which activate Th17 cell response [20]. Th17 cells play a role in host defense against extracellular pathogens, particularly at the mucosal and epithelial barriers, but aberrant activation has been linked to the pathogenesis of various autoimmune diseases [21]. Moreover, A2BARs in association with ADA form a complex that interacts with CD26 on T cell surface leading to TNF-α and interferon γ (IFN-γ) release [22]. Other evidence, supporting the proinflammatory role of this receptor subtype, reports that the activation of A2BARs moves dendritic cell differentiation to a proangiogenic and proinflammatory phenotype. In particular, in hypoxic conditions A2BARs stimulation provokes the release of proangiogenic mediators such as IL-6, IL-8, IL-10, transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF), indoleamine 2,3 dioxygenase (IDO-1), and cyclooxygenase 2 (COX-2) [23].
Even though the role of ARs in monocyte, macrophages, and dendritic cells has been extensively studied, their role in mast cells remains poorly understood. Mast cells are an important component of the immune system and are present throughout the body playing crucial roles in the maintenance of many physiological functions, such as vasodilation, angiogenesis, bacterial, and parasite elimination, as well as in the pathophysiology of diseases. Moreover, mast cells regulate the functions of several cell types, including dendritic cells, macrophages, T and B cells, fibroblasts, eosinophils, endothelial cells, and epithelial cells [24,25]. Studies have reported that ARs are expressed in human skin mast cells; conversely, studies carried out in the human mast cell line LAD2 and HMC-1 cells demonstrated that both cell lines express A2A, A2B, and A3, but not A1ARs [26,27]. In murine mast cells, A2B and A3AR activation prompts degranulation leading to histamine, serotonin, chemokine, and protease release [26]. Other studies in human mast cells confirmed that A2BAR activation is principally implicated in mast cell degranulation, while A3AR stimulation seems to mediate anti-inflammatory effects [28].
Adenosine also regulates the activation of neutrophils, which are pivotal modulators of both inflammation and immune responses. Neutrophils are the most copious leukocytes of the immune system with a great phenotypic heterogeneity and functional versatility [29]. Neutrophils also represent a large source of adenosine in particular in inflammatory conditions when they release ATP that is rapidly converted into adenosine through CD39 and CD73 expressed on neutrophils surface [30,31]. Notably, inflammation reduces adenosine metabolism through ADA deactivation, and equilibrative transporters decrease [30]. Studies have demonstrated that all four AR subtypes are expressed in neutrophils [25]. In particular, A1ARs promote neutrophil chemotaxis, whereas A2A and A2BARs inhibit neutrophil activation [32]. In fact, treatment with the A2AAR agonist, ATL313, inhibits critical steps in integrin-dependent neutrophil adhesion in vitro and in vivo [33]. A subsequent study, investigating the signaling under the inhibitory function of A2AARs stimulation in neutrophils, reported that following treatment with the A2AAR agonist CGS 21680 the phosphorylation of p38 MAPK, Erk-1/2, PI3K/Akt, HCK, and SYK protein kinases was decreased [34]. Furthermore, A2AARs dampen IL-8 release, impairing neutrophil degranulation [35]. The activation of A1 and A2AARs is also involved in neutrophils phagocytosis with an opposite role: A1ARs increase phagocytosis while A2AARs reduce it. Even the A3AR agonist 2-Cl-IB-MECA promoted the formation and rapid extension of projections that improve bacterial phagocytosis and chemotaxis [36]. Moreover, adenosine has a different effect on reactive oxygen species (ROS) generation based on the receptor subtype activated [31]. In particular, the stimulation of A1ARs induces ROS production from activated neutrophils, whereas the activation of A2AARs down-regulates ROS generation [35]. A2B or A3AR agonists suppressed stimulus-induced superoxide production in wild-type but not in A2B or A3AR deficient neutrophils [37].
The cells responsible for cell-mediated immune response are T lymphocytes. These cells are triggered by antigen-presenting cells such as dendritic cells or macrophages evoking T cell differentiation, cytokine production, and cytotoxic activity [38]. ARs can forge many lymphocyte functions. A2AAR stimulation blocks IL-4 and IFN-γ production in both naive CD4+ T cells and Th1 and Th2 cells [1]. Recently, a crucial role of A2AARs in maintaining T follicular help cell/T follicular regulatory cell ratios including the overall ratio between T to B cells into the germinal centers has been reported [39]. Regulatory T cells (Treg) are a subpopulation of T lymphocytes with the task to control and suppress autoreactive T cells preventing autoimmunity [40]. Tregs produce adenosine, which stimulates A2AARs thus reducing proinflammatory cytokine release via nuclear factor-κB activation [41]. The adenosine generated by Tregs acting on A2AARs activates Tregs cell expansion leading to additional immunosuppression with a self-reinforcing loop [42]. Recently, a critical role of the adenosine system in the modulation of B cell functions has been pointed out. ARs are expressed in both murine and human B cells, as well as the complex network of ectoenzymes (nucleotidases, deaminases, kinases) and nucleoside transporters [43]. Adenosine seems to be involved in regulating the development, implantation, and maintenance of the plasma cell population in bone marrow for the primary immune response but also in managing immunoglobulin class switching [1]. In particular, in inactivated B cells there is a higher extracellular concentration of adenosine, while once activated their ATP release increases. This mechanism seems to defend activated B cells from the adenosine-induced inhibitory effect and exerts a proinflammatory role [44]. Interestingly, as observed with Treg cells, adenosine can regulate the function of Breg cells, a subset of immunosuppressive cells that support immunological tolerance. In particular, Bregs were able to regulate both their own function and T cell activity via an adenosine signaling originating from the enzymatic degradation of ATP, released in the extracellular space from activated immune cells [45].
3. Rheumatic Diseases and Osteoarthritis
Rheumatic diseases are characterized by inflammation of the joints, ligaments, tendons, bones, or muscles and which in some cases can also involve other organs; several studies have shown that both the innate and adaptive immune systems can contribute to the inflammation seen in diseases such as rheumatoid arthritis (RA) and psoriatic arthritis [46]. Inflammation of the synovial membrane is characterized by the infiltration of leukocytes [47] leading to changes in endothelial permeability and increasing the adhesion of immune cells, releasing ATP [48]. In response to the increase in ATP concentrations, there is an increase in the activity of ectonucleotidases (enzymes that metabolize nucleotides into nucleosides) to control the inflammatory response and promote the formation of adenosine [49]. Adenosine production has emerged as an important cellular mechanism for regulating inflammation in chronic rheumatic diseases [50].
Methotrexate has been used in the treatment of RA for decades and is often the first-line medication for RA treatment. It is well known that methotrexate increases adenosine levels, and its induction of adenosine signaling is probably the most widely accepted explanation for the methotrexate mechanism in RA [46,51]. ARs are expressed in a large number of cells, including those involved in the pathology of RA such as lymphocytes, neutrophils, macrophages, and synovial cells, where they have predominantly anti-inflammatory effects [52]. Several studies demonstrated greater expression of A2AAR and A3AR receptors on lymphocytes and neutrophils isolated from RA patients compared to healthy subjects. No changes were observed in A1 or A2BAR [53,54,55]. The main anti-inflammatory effects of adenosine occur through the stimulation of A2AAR and A3AR, the expression of which is dynamically regulated by proinflammatory cytokines [56]. Up-regulation and stimulation of A2AAR and A3AR in peripheral leukocytes of RA patients inhibits the NF-κB pathway by decreasing IL-1β, IL-6, and TNF-α [54]. However, the expression of A2AAR and A3AR is normalized when anti-TNF agents are administered, demonstrating that their expression varies with inflammatory levels during RA [53]. Treatment with A2AAR agonists has been shown to increase serum levels of IL-10, an anti-inflammatory cytokine, and reduce the characteristic signs and symptoms of the disease in two different animal models of arthritis [57,58]. In the model of collagen-induced arthritis, stimulation of A2AARs by a CD73-dependent prodrug markedly reduced joint inflammation [59]. In support of the importance of CD73 as a source of adenosine in arthritis, CD73-deficient mice were found to be significantly more susceptible to collagen-induced arthritis than wild-type mice [60].
CD4 germinal center (GC)-follicular helper T (Tfh) cells have an important role in the pathogenesis of autoimmune diseases [61,62]. In an interesting recent study using a mouse model of autoimmune arthritis, A2AAR stimulation diverts autoreactive CD4 T cell differentiation away from the GC-Tfh cell lineage, thus reducing the induction of autoreactive B cells that promote arthritis [63].
In different animal models of arthritis, the A3AR agonist CF101 exerted significant antirheumatic effects reducing the clinical and histological features of arthritis mainly by a decrease of TNF-α production [64]. Similar effects were obtained with the A3AR positive allosteric modulator LUF6000 through deregulation of NF-κB [65]. Another A3AR agonist, CF502, induced an inhibitory effect on the proliferation of fibroblast-like synoviocytes from RA patients and in adjuvant-induced arthritis rats and suppressed the clinical and pathological manifestations of arthritis in the rat model [66].
Despite promising results with CF101 in animal models, the results of early clinical trials have not demonstrated with certainty its ability to improve the course of RA in human patients [67]. Later, in two phase II clinical studies where CF101 was administered to RA patients as a stand-alone drug, a significant antirheumatic effect was observed. Furthermore, a direct significant correlation between receptor expression at baseline and patients’ response to the drug was found, suggesting that A3AR may be used as a predictive biomarker [68].
A2BAR’s role is not entirely clear and current research findings seem ambiguous. While some studies indicate that Th17 differentiation is stimulated by increased production of IL-6 in dendritic cells [69], others argue that A2BAR, when stimulated, promotes T cell differentiation into Treg [67]. The effects of A2BAR binding on osteoclast differentiation and bone resorption are also controversial. Researchers observed that the A2BAR agonist inhibits RANKL-induced osteoclast formation in mouse cells [70]. On the other hand, one study found that adenosine via A2BAR abolished the osteoclast inhibition induced by methotrexate [71]. A1AR stimulation, in turn, is essential for the bone remodeling process, through the differentiation of giant cells into osteoclasts during RA [72].
ADA levels increased in plasma and synovial fluid of RA patients [73,74,75]. It is conceivable that the increase in ADA is a pathogenic factor, as the increased adenosine deamination will result in reduced bioavailability and decreased AR-mediated inflammation suppression [76]. Furthermore, ADA activity can serve as a useful marker for monitoring the effects of methotrexate as this drug acts in the metabolism of adenosine [77].
The most common form of arthritis is osteoarthritis (OA), which is the most important cause of disability in the elderly. Adenosine signaling plays a critical role in maintaining joint cartilage and can serve as a new therapy for OA. Mice lacking A2AAR or CD73 develop spontaneous OA by 16 weeks of age [78]. Exogenous adenosine treatment by intra-articular injection of liposomal suspensions containing adenosine prevents the development of OA in rats. As an approach to extend its half-life, adenosine conjugated to biodegradable nanoparticles prevented the development of OA in a rat post-traumatic model [79]. Recently, in the human TC28a2 cell line, A2AAR stimulation increased activation and nuclear localization of FoxO1 and FoxO3, promoted an increase in autophagic flux, improved metabolic function, and reduced apoptosis [80]. All these findings offer evidence that A2AARs may be a useful pharmacological target for OA by promoting chondrocyte and cartilage homeostasis.
The NF-κB signaling pathway is an important factor involved in the pathogenesis of OA. It has been reported that NF-κB is deregulated by the presence of the A3AR agonist CF101 resulting in the reduction of TNF-α in a rat model of OA induced by monosodium iodoacetate. Furthermore, CF101 induces apoptosis of inflammatory cells and acts as a protective agent of cartilage, suggesting that it may be a suitable candidate for the treatment of OA [81]. The safety and efficacy of IB-MECA were also evaluated in a phase II clinical study with patients with knee OA [82].
In vitro studies on joint cells have suggested that pulsed electromagnetic field (PEMF) exposure mediates significant protection against the catabolic effect of proinflammatory cytokines and anabolic action that increases matrix synthesis and cell proliferation [83]. Mechanistically, PEMFs were able to mediate the up-regulation of A2AAR and A3AR in bovine chondrocytes and synoviocytes [84,85]. Thereafter, in human chondrocytes and osteoblasts, stimulation with PEMFs has been observed to potentiate the AR-mediated reduction of prostaglandin E2 (PGE2), IL-6, and IL-8, suggesting their potential in the treatment of inflammatory disorders of the bone and joints [86]. Therefore, PEMFs could be an innovative physiological alternative to the use of AR agonists as they can mediate the effects of tissue-specific agonists without any desensitization and down-regulation [50].
4. Chronic Lung Diseases and Pulmonary Inflammation
Chronic lung diseases such as asthma and chronic obstructive pulmonary disease (COPD) are characterized by persistent inflammation and tissue remodeling processes leading to a progressive loss of pulmonary functions [87]. Although the pathogenesis of chronic lung diseases is multifactorial, a common feature among these pathologies is excessive recruitment and aberrant activation of effector cells. These include mast cells, eosinophils, macrophages, neutrophils, lymphocytes, and dendritic cells as well as airway epithelial cells and fibroblasts. Dysregulation of these cells leads to the release of a plethora of mediators that contribute to pulmonary inflammation and remodeling [88,89]. ARs are expressed in all these cell types and various studies indicate that their activation by increased levels of adenosine participates in the pathogenesis of chronic lung diseases [90]. Indeed, elevated adenosine levels were initially reported in bronchoalveolar lavage fluid (BAL) from asthmatics [91,92] and then confirmed in COPD patients [93,94]. As for other chronic lung diseases, there is little evidence of elevated adenosine levels in patients with interstitial lung disease or idiopathic pulmonary fibrosis, although increased concentrations have been found in animal models of these diseases [95,96]. These chronically elevated adenosine levels seem to induce the release of inflammatory mediators leading to tissue injury and fibrosis [90]. The first evidence derived from animal models of airway disease suggested a clinical benefit to the use of A1, A2B, or A3AR antagonists in the treatment of asthma and COPD [97,98]. Subsequent studies have identified the A2BAR as the most promising candidate, among AR subtypes, as a target for the treatment of chronic lung diseases. The activation of A2BARs on various pulmonary and immune cell types has in fact been correlated with aberrant cell differentiation and elevated levels of proinflammatory and pro-fibrotic mediators including IL-4, IL-6, IL-8, fibronectin, and TGF-β [99,100,101]. In mice exposed to bleomycin, the most common animal model of pulmonary fibrosis, direct A2BAR activation on vascular cells promoted IL-6 and endothelin-1 release. In the same study, genetic removal of the A2BAR or treatment with the A2BAR antagonist GS-6201 attenuated vascular remodeling and hypertension [100]. In a subsequent study using the bleomycin model, the conditional knockout mice lacking A2BAR on myeloid cells presented with attenuated fibrosis, improved lung function, and showed no evidence of pulmonary hypertension compared with control mice exposed to bleomycin. Furthermore, the authors found a reduction of IL-6 and hyaluronan release and a reduced expression of CD206 and arginase-1, markers of alternatively activated macrophages [102]. The implication of adenosine in chronic lung diseases is corroborated by the fact that ADA-deficient mice develop pulmonary inflammation and injury reminiscent of that seen in asthma [103]. ADA-deficient mice exhibited extensive lung mast cell degranulation [103], increased number of alveolar macrophages, and elevated monocyte chemoattractant protein-3 in the bronchial epithelium [104]. Different studies were performed with the aim to elucidate the role of the specific AR subtypes in ADA-deficient mice-induced pulmonary inflammation. When ADA-deficient mice were treated with the antagonist CVT-6883, they exhibited less pulmonary inflammation, fibrosis, and alveolar airspace enlargement with a significant reduction of proinflammatory cytokines and chemokines as well as mediators of fibrosis and airway destruction [105]. The A3AR was found to be expressed in eosinophils and mucus-producing cells in the airways of ADA-deficient mice. Genetic deletion A3AR or their blockade with the selective antagonist MRS 1523 prevented airway eosinophilia and mucus production [106]. On the contrary, genetic removal of the A1AR from ADA-deficient mice resulted in increased pulmonary inflammation, mucus metaplasia, and alveolar destruction along with exaggerated expression of IL-4, IL-13, chemokines, and matrix metalloproteinases [107]. These findings suggested that the A1AR plays an anti-inflammatory and/or protective role in the pulmonary phenotype seen in ADA-deficient mice. Similar results were obtained in ADA/A2AAR double knockout mice where the authors observed enhanced inflammation comprised largely of macrophages and neutrophils, mucin production in the bronchial airways, and elevated levels of chemoattractant protein-1 and chemokine (C-X-C motif) ligand (CXCL1) in comparison with ADA-deficient mice with the A2AAR. This suggested a protective role of A2AARs in pulmonary inflammation [108].
Alteration of ARs has been found in chronic lung diseases strengthening the hypothesis of a potential role played by adenosine and its receptors in the pathogenesis of these diseases. In lung parenchyma obtained from subjects with COPD compared with control smokers, binding experiments revealed an increased density of A1, A2A, and A3ARs, while a decrease of A2BAR expression. Furthermore, a significant correlation was found between the affinity and density of the ARs and the FEV1/FVC ratio [109]. In a subsequent study, the authors found a significant decrease of A2BAR density in BAL macrophages from patients with COPD when compared with healthy smokers [110].
As opposed to its detrimental role in the chronic setting, elevated levels of extracellular adenosine promote tissue-protective responses in acute pulmonary inflammation. Most of the studies conducted have identified the A2A and A2BARs as the major factors responsible for the protective action of adenosine in acute pulmonary inflammation [111,112,113,114]. In a model of acute lung injury, aerosolized BAY 60-6583 treatment, an A2BAR agonist, was associated with attenuated pulmonary edema, improved histologic lung injury, and dampened lung inflammation. In this model, tissue-specific deletion of A2BARs suggested that alveolar epithelial A2BAR signaling contributes to lung protection during acute pulmonary inflammation [115]. In a murine model of lipopolysaccharide (LPS)-induced pulmonary inflammation, A2BAR on hematopoietic cells was found to be crucial for the anti-inflammatory effect obtained with the inhibition of SDF-1 receptors CXCR4 and CXCR7 [116]. In another study based on LPS-induced acute lung injury, treatment with adenosine or NECA, an AR non-selective agonist, recovered lung vascular barrier and reduced inflammation [117]. The activation of heme oxygenase 1 by hemin significantly decreased leukocyte migration and chemokine levels into the lung after LPS inhalation, an effect abolished in A2A and A2BAR knockout mice [118]. In a mouse model of carrageenan-induced pleurisy, administration of the A2AAR agonist CGS 21680 caused a significant reduction of neutrophil infiltration, nitric oxide and cytokine production, NF-κB expression, and PARP activation [119].
Apart from A2A and A2BARs, few studies have investigated the role of the other AR subtypes. In a murine model of LPS-induced lung injury, treatment with the A1AR agonist 2’-Me-CCPA attenuated polymorphonuclear cells accumulation in the interstitium and alveolar space, microvascular permeability, and reduced TNF-α, IL-6, and CXCL2/3 levels in the BAL [120]. Activation of A3AR by the selective agonist Cl-IB-MECA dampened lung dysfunction, inflammation, and neutrophil infiltration after ischemia-reperfusion in wild-type but not A3AR knockout mice [121]. In mice exposed to bleomycin, genetic deletion of A3ARs resulted in enhanced inflammatory responses associated with an increase of eosinophils [122].
It is worth mentioning that recently, a COVID-19 patient with unresponsive respiratory failure was treated with adenosine for compassionate use, showing a rapid improvement of clinical conditions [123].
5. Intestinal Inflammation
Inflammatory bowel diseases (IBDs), including Crohn’s disease and ulcerative colitis, are chronic, progressive inflammatory conditions presenting an overactive intestinal immune system [124]. IBDs are characterized by an overproduction of proinflammatory mediators associated with morpho-functional alterations of the enteric nervous system, and intestinal dysfunctions [125]. Over the years, several studies have emphasized the crucial role of adenosine signaling in the pathophysiology of IBDs [126]. Consistent with the recognized anti-inflammatory role of A2AARs, most studies have identified A2AAR stimulation as the most promising approach for the potential treatment of IBDs compared to the pharmacological modulation of the other AR subtypes. In the acute model of rabbit immune colitis, treatment with the A2AAR agonist ATL-146e reduced inflammation in the intestinal mucosa by inhibiting proinflammatory cytokines such as TNF-α, INF-γ, and IL-4, and decreasing leukocyte infiltration into the colonic mucosa [127]. In a rat model of experimental colitis, Antonioli and colleagues further elucidated the role of A2AAR on colonic motility. In this model, the A2AAR antagonist ZM 241385 increased transmural electrical stimulation-induced contractions with a more noticeable effect inflamed respect to control animals, while the A2AAR agonist CGS 21680 induced a concentration-dependent reduction of contractile responses [128]. Furthermore, overexpression of A2AAR was found in colonic tissues isolated from inflamed animals. The results of a subsequent study by the same group indicated that, while in normal colon, both A1 and A2AARs contribute to the inhibitory control of motor functions at the neuronal level, under bowel inflammation, only A2AARs preserved an inhibitory control of colonic neuromotility [129]. More recently, in a rat model of oxazolone-induced colitis, the A2AAR agonist PSB-0777 improved body weight, and reduced inflammatory parameters and colonic myeloperoxidase levels [130].
Using adoptive T cell transfer studies, Naganuma et al. suggested that A2AARs play a critical role in the T cell-mediated regulation of colitis by suppressing the expression of proinflammatory cytokines while sparing anti-inflammatory activity mediated by IL-10 and TGF-β [131]. In the 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced chronic model of experimental colitis, the involvement of A2AARs was investigated with the antagonist SCH-442416, suggesting a protective effect of their activation [132]. In mice with toxin A-induced enteritis ATL 313, an A2AAR agonist, reduced tissue injury and inflammation by reducing toxin A-induced secretion and edema, mucosal disruption, and neutrophil infiltration [133].
More recently, the stimulation of A2AARs with polydeoxyribonucleotide ameliorated the clinical symptoms and weight loss associated with two rat models of experimental colitis represented by intra-colonic instillation of dinitrobenzenesulfonic acid (DNBS) and dextran sulfate sodium (DSS) in drinking water [134]. Polydeoxyribonucleotide promoted the histological repair of damaged tissues, reduced the production of inflammatory cytokines, and decreased myeloperoxidase activity and malondialdehyde.
It was also found that the anti-inflammatory effect of STW 5 (Iberogast®), an established herbal combination showing clinical efficacy in functional dyspepsia and irritable bowel syndrome, was dependent upon A2AAR activation in a rat-intestinal inflammatory model induced by intraluminal instillation of TNBS [135].
The role of A2BARs in intestinal inflammation is more controversial than that of A2AARs. The proinflammatory role of A2BAR activation was suggested by the administration of A2BAR antagonist ATL-801 in DSS-treated mice. Compared to mice treated with DSS alone, animals receiving ATL-801 showed a significantly lower extent and severity of colitis with a reduction of IL-6 levels, histological scores, and proliferative indices [136]. The detrimental role of this receptor subtype was further corroborated by the fact that A2BAR genetic deletion attenuated colonic inflammation induced by DSS or TNBS [137]. Moreover, a recent study demonstrated that the inhibition of A2BAR with the antagonist PSB1115 attenuated intestinal injury in a neonatal rat model of necrotizing enterocolitis [138]. By contrast, an opposite effect was reported in the DSS colitis model, where both the administration of PSB1115 and the genetic deletion of A2BAR resulted in an increase in the severity of colitis [139].
Various preclinical studies have indicated the activation of A3ARs as a promising pharmacological approach for attenuating bowel inflammation [125,140]. In particular, using various experimental models of colitis, A3AR agonists were shown to attenuate inflammatory cell infiltration and to reduce proinflammatory mediator levels resulting in an improvement of the intestinal injury [141,142]. In murine DSS colitis, the activation of A3ARs attenuated NF-κB activation leading to the reduced expression of TNF-α and IL-1β in colonic epithelia [143]. A more recent study highlighted a down-regulated A3AR expression in the colonic mucosa of patients with ulcerative colitis. In cultured colonic mucosal tissue from these patients, the stimulation of A3AR with the agonist 2-Cl-IB-MECA significantly decreased TNF-α and IL-1β production and attenuated the NF-κB p65 activation [144].
As shown in other inflammatory conditions, the increase of endogenous adenosine levels obtained by blocking catabolic enzymes like ADA or adenosine kinase, or inhibiting nucleoside transporters, also resulted in an improvement in experimental settings of bowel inflammation [77,145,146]. Antonioli et al. showed that inhibiting ADA with EHNA in different mouse models of intestinal inflammation produced beneficial effects characterized by a reduction of proinflammatory cytokines, inflammatory cell infiltration, and a general reduction of colonic damage [147,148,149].
6. Neuroinflammation
The central nervous system (CNS), being separated from the periphery by the blood-brain barrier, is characterized by unique features of immune cell distribution and inflammatory responses. Neuroinflammation is mediated by several proinflammatory mediators produced by activated CNS-resident cells such as microglia and astrocytes, which are innate immune cells without direct counterparts in the periphery [150]. In addition, endothelial cells, perivascular macrophages, and other CNS-infiltrating immune cells are also important in interpreting and propagating these inflammatory signals within the CNS [151]. The duration and intensity of the inflammatory signals often determine whether this response is harmful or beneficial for the CNS. Low and transient neuroinflammatory responses can in fact represent important signals for the maintenance of cerebral homeostasis, for the processes of memory and learning [152], and tissue repair and remodeling [153]. By contrast, chronic, uncontrolled inflammation in the CNS has been recognized as a pathological factor for neurodegenerative diseases [154,155,156] as well as for neuropsychiatric disorders [157,158]. Pathological neuroinflammation is associated with activation of glial cells with proinflammatory cytokine and chemokine production, increased blood-brain barrier permeability, infiltration of peripheral immune cells, neuronal damage and death, and neuronal atrophy over time [151].
In the CNS, adenosine acts as a neuromodulator. Following neuronal stress and damage, extracellular brain adenosine concentration is dramatically increased resulting in the promotion or attenuation of neuroinflammation. Many of the immunomodulatory effects of adenosine in the CNS are mediated by A1 and A2AAR subtypes [159,160], although recent evidence also points to the involvement of A2B and A3ARs [161,162]. The expression of all four ARs has been detected in astrocytes, oligodendrocytes, and microglia [163]. During neuroinflammation, A2AAR stimulates activated microglia to assume their characteristic amoeboid morphology [164]. A composite and regional-specific effect of A2AAR antagonist SCH-58261 pretreatment on glial cell activation was observed in a rat model of striatal excitotoxicity obtained by unilateral intrastriatal injection of quinolinic acid [165]. In microglia, A2AAR blockage has been suggested to directly potentiated the neuroprotective cannabinoid CB2 receptor signaling, likely due to conformational changes within the A2AAR-CB2 receptor heteromer [166]. In two neonatal rat models of neuroinflammation and microglial activation, the gestational low protein diet model, and postnatal ibotenate intracerebral injections, A2AARs and CD73 were increased and the A2AAR antagonist SCH-58261 reduced M1 markers [167]. SCH-58261 also prevented the LPS-induced recruitment of activated microglial cells and the increase in inflammatory cytokines in the hippocampus [168]. In contrast, the stimulation of A1ARs inhibited the ATP-induced activation of microglia, likely suppressing the Ca2+ influx induced by ATP treatment [169]. In a recent study performed on N13 microglial cells, selective A1AR agonists or A2AAR antagonists prevented the inflammatory effect induced by a cytokine cocktail. Interestingly, the combined effect of A1AR agonists and A2AAR antagonists showed a synergistic effect [170].
A1AR knockout mice exhibited increased microglial response in an experimental brain traumatic injury model in mice. The attenuated responses induced by A1AR stimulation were confirmed in vitro using BV-2 microglial cells [171]. A reduction of microglia activation was also observed stimulating A3ARs with the agonist IB-MECA, an effect likely underlining their antinociceptive action [172]. The selective A3AR agonist 2-Cl-IB-MECA was effective in controlling microglia reactivity induced by elevated hydrostatic pressure, with potential positive repercussions in glaucoma [173]. In a model of subarachnoid hemorrhage, 2-CI-IB-MECA markedly directed microglia towards the M2, or more precisely M(IL-4) phenotype, reduced inflammation, and improved neurological dysfunction [174]. Marked attenuation of neuroinflammation was also observed with the A3AR agonist MRS5980 in a mouse model of traumatic brain injury [162]. Among ARs, the A2BAR subtype seems the least involved in the process of neuroinflammation. Nevertheless, A2BAR activation was shown to increase IL-6 production and cell proliferation in murine primary microglial cells, an effect involving PLC, PKC-ε, PKC-δ, and p38 pathways, thus suggesting their contribution to microglial activation and neuroinflammation [175].
The modulation of neuroinflammation by ARs makes them an attractive pharmacological target for neurodegenerative diseases that share chronic brain inflammation as a ubiquitous common feature, including Alzheimer’s disease [176,177], Parkinson’s disease [178,179], multiple sclerosis [180,181], and Huntington’s disease [182]. Remarkably, an alteration of A2AARs was found in the brain and peripheral blood cells of patients affected by such neurodegenerative diseases [183,184,185,186,187,188].
Experimental autoimmune encephalomyelitis (EAE) is the most commonly used experimental model for the inflammatory demyelinating disease, multiple sclerosis [189]. Compared with wild-type mice, A1AR knockout mice developed a more severe form of EAE characterized by worsened demyelination, axonal injury, and enhanced microglial activation [190]. More recently, a similar exacerbation of EAE was found in mice lacking A2AARs [191]. A subsequent work using A2AAR knockout mice suggested a dual role for A2AARs in EAE: while providing protection at early stages of the disease by exerting anti-inflammatory effects on T cells, A2AARs seem to be detrimental during later stages contributing to tissue damage within the inflamed CNS [192]. Using a different approach, mice lacking CD73 were resistant to EAE. According to the authors, the protection was not caused by a deficiency in T cell responsiveness and CD73 must be expressed either on T cells or in the CNS for disease induction [193]. The treatment with the A2AAR antagonist SCH-58261 protected wild-type mice from EAE by inhibiting the entry of lymphocytes into the CNS. In a subsequent work by the same authors, results obtained with bone marrow chimeric mice revealed that A2AAR expression on nonimmune cells is required for efficient EAE development, while A2AAR expressed on lymphocytes is essential for limiting the severity of the inflammatory response [194]. The confounding role of A2AARs in EAE seems to be related to the identification of the best therapeutic window: depending on the treatment period, both A2AAR agonist and antagonist have been proved to protect against EAE development [195,196,197].
7. Immunity, Inflammation, and Cancer
Cancer is a complex disease, caused by multiple cellular dysregulations [198]. The immune system is involved in all stages of cancer progression. In physiological conditions, the immune system is able to remove abnormal cells, but when cancer cells escape the immune surveillance and start growing, immune cells infiltrate the tumor [199]. The cancer immune-escaping mechanism uses different factors. The involvement of adenosine in controlling inflammation and preventing exaggerated immune response is well established. Some kinds of tumors are infiltrated by different immune cells and can use adenosine as an immunosuppressive agent to block immune response versus cancer cells [198]. Indeed, in cancer cells there is commonly an augmented level of adenosine accompanied by overexpression of CD39 and CD73 thus limiting the immune cell activity in the tumor microenvironment [200,201,202,203,204]. Many studies demonstrated that the intratumoral overexpression of ectonucleotidases could be principally due to hypoxia and inflammation [205,206]. The activation of HIF-1α, as well as the signaling pathways triggered by proinflammatory mediators (IL-1β, IL-6, TNF-α, TGF-β), may boost CD39 and CD73 levels [207,208,209,210,211,212]. It is now understood that T cells are a key player in tumor control; the elevated levels of adenosine in the tumor can potently impair T cells mediated antitumor response by inducing accumulation of intracellular cAMP [199,213].
In the tumor microenvironment, there are two types of infiltrating macrophages, called tumor-associated macrophages: classically activated macrophages (M1) and alternatively activated macrophages (M2) [214]. Studies so far conducted reported that M1 predominately expresses A2AARs, while A2BARs are responsible for the alternative macrophages’ activation into the M2 phenotype [215]. Adenosine exerts many suppressive effects on antitumoral M1 macrophages mostly through A2AAR activation, which decreases proinflammatory cytokine release while increasing the secretion of IL-10, an anti-inflammatory mediator [198]. M2 macrophages exhibit protumoral functions; in particular, a subtype of M2, called M2d, is activated by A2AARs [216]. As a result, they release proangiogenic factors such as VEGF and IL-10. In addition, IL-10 endorses the polarization of Th17 and Treg leading to inflammatory response inhibition. Evidence collected so far suggests that M2 macrophages affect tissue repair and matrix remodeling hence promoting tumor growth, angiogenesis, and metastasis development [217,218].
Similar to macrophages, neutrophils are a crucial player in the interaction between the immune system and cancer cells. When infiltrated in the tumor, they fine-tune adaptive cells enrollment through cytokine and chemokine production [219]. Neutrophils can be subjected to two different polarizations, similarly to macrophages, achieving an antitumor phenotype (N1) or a protumor (N2) phenotype. The major difference between these two cell types is their capacity for the production of pro- or anti-inflammatory cytokines and that N2 cells are proangiogenic and prometastatic [220]. Several studies on tumor infiltrated neutrophils reported that adenosine hinders the migration of these cells from the bloodstream to the inflammatory site, blocking their adhesion to endothelial cells. Moreover, the activation of A2AARs on neutrophils inhibits proinflammatory cytokine and chemokine production such as TNF-α, CCL3, CCL4, and others, causing an inefficient enrollment of immune cells [221]. Other studies showed that, besides A2AArs, A3AR activation is also involved in the inhibition of neutrophil degranulation thus reducing their proinflammatory potential [222].
The adenosine in the tumor microenvironment also acts on natural killer cells. A2AARs stimulation interrupts the maturation process of these cells, which can result in natural killer cell death [198]. Indeed, in A2AAR knockout mice a greater amount of mature natural killer cells has been found associated with a reduced number of immature cells. Furthermore, the mature natural killer cells showed an increased ability to control tumor initiation and growth [223]. A recent paper reports that infiltrated natural killer cells are modulated by cancer cells through CD137 engagement on the cell surface, inducing the translocation of vesicles containing CD73 on the natural killer cell membrane, thus promoting the secretion of IL-10 and TGF-β [224]. IL-10 inhibits the proliferation of CD4+ T cells and its IFN-γ production, thus blocking adaptive antitumor response, and CD73 on natural killer cells increases adenosine concentration in the tumor microenvironment leading to immunosuppression [224].
In recent studies, the role of mast cells in the tumor microenvironment has been investigated. Apart from their role in allergic reactions and inflammation, they are also involved in wound healing and angiogenesis; thus, they can result in protumoral cells. Indeed, mast cells can be triggered by direct contact with cancer cells when they infiltrate the tumor [225,226]. This activation results in IL-8, IL-6, and VEGF secretion [221]. These mediators promote new blood vessels angiogenesis supporting tumor growth and metastasis. Both mechanisms involve CD73 activation on mast cells and autocrine signaling of adenosine through A3ARs stimulation [225,226].
Adenosine affects the normal function of dendritic cells. They are antigen-presenting cells responsible for the activation of T cells and induction of their differentiation [198]. Dendritic cells derive from bone marrow monocyte-dendritic cell progenitor, which differentiate into common the dendritic cell’s precursor. During inflammatory reactions caused by cancer or infections, it was observed, both in vivo and in vitro, that monocytes can give rise to a subset of dendritic cells called monocyte-derived dendritic cells [227]. Mature dendritic cells express A2AARs, thus their stimulation with adenosine or agonists resulted in increased levels of intracellular cAMP and inhibited the production of IL-12. In addition, dendritic cells maturated in the presence of adenosine produce IL-10 and have a reduced capacity of induction of Th1 [227]. Studies performed on differentiation of human monocytes, mouse peritoneal macrophages, and hematopoietic progenitor cells into myeloid dendritic cells, in the presence of increased levels of adenosine, revealed an impaired activation and function of resulting dendritic cells, as they fail to activate naive T cells and they produce anti-inflammatory and angiogenic factors [23]. This adenosine-mediated effect was due to the activation of A2BARs [23].
Given the harmful role of A2A and A2BAR stimulation on tumor-infiltrating immune cells which suppress their antitumor effect, many studies have been conducted using antagonists, in order to restore antitumor immunity and enhance the efficacy of cancer immunotherapies [228]. Interestingly, A2AAR antagonists ZM241385 and SCH58261 were able to restrain primary tumor growth, prevent negative T cells signaling, and inhibit angiogenesis in a mice model of non-small cell lung cancer [229]. Three A2AAR antagonists are currently undergoing phase I/II clinical trials as single agents for the treatment of solid tumors: CPI-444 (NCT02655822), AZD4635 (NCT02740985), and NIR178 (NCT02403193, NCT03207867), respectively. A2BAR signaling in tumor cells themselves further promotes their survival and metastasis [228]. Notably, administration of A2BAR antagonists decreases tumor growth in mice tumor model and metastasis [230,231]. In vivo, the block of A2BARs increases CD8+ T cells, natural killer cells, and the production of TNF-α and IFN-γ in the tumor microenvironment, accompanied by reduced levels of IL-10, VEGF, and angiogenesis [232,233,234]. From these encouraging preclinical results, a dose-escalation phase I clinical trial (NCT03274479) administering PBF-1129, a selective A2BAR antagonist, in patients with advanced non-small cell lung cancer has been started [212].
8. Conclusions
Adenosine, through interaction with its four receptor subtypes, is a ubiquitous and powerful modulator of inflammatory processes affecting almost all physiological and pathophysiological functions. All immune cells express ARs and respond in a different manner to adenosine, the concentration of which varies enormously in stressful situations including inflammation. Considering that inflammation is a hallmark of many chronic diseases, both in the periphery and in the CNS, the modulation of ARs has a huge potential as a therapeutic strategy. However, the wide distribution of ARs and the multiple functions of adenosine in the body was supposed to be a limiting factor for the drug development acting on this system. Indeed, once in the clinic, AR ligands have often shown problems of low efficacy and/or manifestation of adverse effects. Nevertheless, the recent approval of the A2AARs antagonist istradefylline for Parkinson’s disease and the encouraging results obtained so far on cancer immunotherapy has again turned the spotlight on the adenosine system as a drug target. Great efforts are being made to identify new and more selective AR ligands as well as adenosine-regulating agents capable of modulating the concentration of endogenous adenosine. As a matter of fact, numerous clinical trials are currently being performed on AR ligands (Table 1).
In conclusion, the adenosinergic system has considerable therapeutic potential in all pathological states and in particular in those diseases where regulation of inflammation and modulation of the immune system is required.
Author Contributions
Conceptualization, F.V. and S.P.; writing—original draft preparation, F.V., S.P. and C.C.; writing—review and editing, P.A.B. and K.V.; visualization, F.V. and S.P.; supervision, P.A.B. and K.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AC | Adenylate cyclase |
| ADA | Adenosine deaminase |
| ADP | Adenosine diphosphate |
| AMP | Adenosine monophosphate |
| ARs | Adenosine receptors |
| ATP | Adenosine triphosphate |
| BAL | Bronchoalveolar lavage fluid |
| cAMP | Cyclic adenosine monophosphate |
| CD39 | Ectonucleoside triphosphate diphosphohydrolase |
| CD73 | Ecto-5′-nucleotidase |
| CNS | Central nervous system |
| COPD | Chronic obstructive pulmonary disease |
| COX-2 | Cyclooxygenase 2 |
| CXCL | Chemokine (C-X-C motif) ligand |
| DNBS | Dinitrobenzenesulfonic acid |
| DSS | Dextran sulfate sodium |
| EAE | Experimental autoimmune encephalomyelitis |
| ENT | Equilibrative nucleoside transporter |
| GC | Germinal center |
| GPCR | G protein-coupled receptor |
| IBDs | Inflammatory bowel diseases |
| IDO-1 | Indoleamine 2,3 dioxygenase |
| IFN-γ | Interferon γ |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MIP | Macrophage inflammatory protein |
| NF-kB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| OA | Osteoarthritis |
| PEMFs | Pulsed electromagnetic fields |
| PGE2 | Prostaglandin E2 |
| RA | Rheumatoid arthritis |
| ROS | Reactive oxygen species |
| TGF-β | Tissue growth factor β |
| TNBS | 2,4,6-trinitrobenzene sulphonic acid |
| TNF-α | Tumor necrosis factor α |
| VEGF | Vascular endothelial growth factor |
References
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Haskó, G. Adenosine Signaling and the Immune System: When a Lot Could Be Too Much. Immunol. Lett. 2019, 205, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine Receptors: Therapeutic Aspects for Inflammatory and Immune Diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological Overproduction: The Bad Side of Adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does It Exert Its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Fredholm, B.B. Adenosine--a Physiological or Pathophysiological Agent? J. Mol. Med. Berl. Ger. 2014, 92, 201–206. [Google Scholar] [CrossRef]
- Antonioli, L.; Csóka, B.; Fornai, M.; Colucci, R.; Kókai, E.; Blandizzi, C.; Haskó, G. Adenosine and Inflammation: What’s New on the Horizon? Drug Discov. Today 2014, 19, 1051–1068. [Google Scholar] [CrossRef]
- Thiele, A.; Kronstein, R.; Wetzel, A.; Gerth, A.; Nieber, K.; Hauschildt, S. Regulation of Adenosine Receptor Subtypes during Cultivation of Human Monocytes: Role of Receptors in Preventing Lipopolysaccharide-Triggered Respiratory Burst. Infect. Immun. 2004, 72, 1349–1357. [Google Scholar] [CrossRef] [Green Version]
- Khoa, N.D.; Postow, M.; Danielsson, J.; Cronstein, B.N. Tumor Necrosis Factor-α Prevents Desensitization of Gαs-Coupled Receptors by Regulating GRK2 Association with the Plasma Membrane. Mol. Pharmacol. 2006, 69, 1311–1319. [Google Scholar] [CrossRef]
- Khoa, N.D.; Montesinos, M.C.; Reiss, A.B.; Delano, D.; Awadallah, N.; Cronstein, B.N. Inflammatory Cytokines Regulate Function and Expression of Adenosine A2A Receptors in Human Monocytic THP-1 Cells. J. Immunol. 2001, 167, 4026–4032. [Google Scholar] [CrossRef] [Green Version]
- Haskó, G.; Pacher, P.; Deitch, E.A.; Vizi, E.S. Shaping of Monocyte and Macrophage Function by Adenosine Receptors. Pharmacol. Ther. 2007, 113, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-S.; Chung, H.-J.; Lee, H.W.; Jeong, L.S.; Lee, S.K. Suppression of Inflammation Response by a Novel A3 Adenosine Receptor Agonist Thio-Cl-IB-MECA through Inhibition of Akt and NF-ΚB Signaling. Immunobiology 2011, 216, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection from Tissue Damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Haskó, G.; Csóka, B.; Németh, Z.H.; Vizi, E.S.; Pacher, P. A2B Adenosine Receptors in Immunity and Inflammation. Trends Immunol. 2009, 30, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Koscsó, B.; Csóka, B.; Kókai, E.; Németh, Z.H.; Pacher, P.; Virág, L.; Leibovich, S.J.; Haskó, G. Adenosine Augments IL-10-Induced STAT3 Signaling in M2c Macrophages. J. Leukoc. Biol. 2013, 94, 1309–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Németh, Z.H.; Lutz, C.S.; Csóka, B.; Deitch, E.A.; Leibovich, S.J.; Gause, W.C.; Tone, M.; Pacher, P.; Vizi, E.S.; Haskó, G. Adenosine Augments IL-10 Production by Macrophages through an A2B Receptor-Mediated Posttranscriptional Mechanism. J. Immunol. 2005, 175, 8260–8270. [Google Scholar] [CrossRef] [Green Version]
- Joós, G.; Jákim, J.; Kiss, B.; Szamosi, R.; Papp, T.; Felszeghy, S.; Sághy, T.; Nagy, G.; Szondy, Z. Involvement of Adenosine A3 Receptors in the Chemotactic Navigation of Macrophages towards Apoptotic Cells. Immunol. Lett. 2017, 183, 62–72. [Google Scholar] [CrossRef]
- Ghislat, G.; Lawrence, T. Autophagy in Dendritic Cells. Cell. Mol. Immunol. 2018, 15, 944–952. [Google Scholar] [CrossRef]
- Schnurr, M.; Toy, T.; Shin, A.; Hartmann, G.; Rothenfusser, S.; Soellner, J.; Davis, I.D.; Cebon, J.; Maraskovsky, E. Role of Adenosine Receptors in Regulating Chemotaxis and Cytokine Production of Plasmacytoid Dendritic Cells. Blood 2004, 103, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Zuo, A.; Shao, H.; Chen, M.; Kaplan, H.J.; Sun, D. A2B Adenosine Receptor Activation Switches Differentiation of Bone Marrow Cells to a CD11c+Gr-1+ Dendritic Cell Subset That Promotes the Th17 Response. Immun. Inflamm. Dis. 2015, 3, 360–373. [Google Scholar] [CrossRef]
- Awasthi, A.; Kuchroo, V.K. Th17 Cells: From Precursors to Players in Inflammation and Infection. Int. Immunol. 2009, 21, 489–498. [Google Scholar] [CrossRef]
- Pacheco, R.; Martinez-Navio, J.M.; Lejeune, M.; Climent, N.; Oliva, H.; Gatell, J.M.; Gallart, T.; Mallol, J.; Lluis, C.; Franco, R. CD26, Adenosine Deaminase, and Adenosine Receptors Mediate Costimulatory Signals in the Immunological Synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 9583–9588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine Receptors in Regulation of Dendritic Cell Differentiation and Function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef]
- Garcia-Garcia, L.; Olle, L.; Martin, M.; Roca-Ferrer, J.; Muñoz-Cano, R. Adenosine Signaling in Mast Cells and Allergic Diseases. Int. J. Mol. Sci. 2021, 22, 5203. [Google Scholar] [CrossRef]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.-G.; Jacobson, K.A. Purinergic Signaling in Mast Cell Degranulation and Asthma. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudich, N.; Dekel, O.; Sagi-Eisenberg, R. Down-Regulation of the A3 Adenosine Receptor in Human Mast Cells Upregulates Mediators of Angiogenesis and Remodeling. Mol. Immunol. 2015, 65, 25–33. [Google Scholar] [CrossRef]
- Rudich, N.; Ravid, K.; Sagi-Eisenberg, R. Mast Cell Adenosine Receptors Function: A Focus on the A3 Adenosine Receptor and Inflammation. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Barletta, K.E.; Ley, K.; Mehrad, B. Regulation of Neutrophil Function by Adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 856–864. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, D. Purinergic Regulation of Neutrophil Function. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riff, R.; Naamani, O.; Mazar, J.; Haviv, Y.S.; Chaimovitz, C.; Douvdevani, A. A1 and A2A Adenosine Receptors Play a Protective Role to Reduce Prevalence of Autoimmunity Following Tissue Damage. Clin. Exp. Immunol. 2021. [Google Scholar] [CrossRef]
- Yago, T.; Tsukamoto, H.; Liu, Z.; Wang, Y.; Thompson, L.F.; McEver, R.P. Multi-Inhibitory Effects of A2A Adenosine Receptor Signaling on Neutrophil Adhesion Under Flow. J. Immunol. 2015, 195, 3880–3889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giambelluca, M.S.; Pouliot, M. Early Tyrosine Phosphorylation Events Following Adenosine A2A Receptor in Human Neutrophils: Identification of Regulated Pathways. J. Leukoc. Biol. 2017, 102, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Frasson, A.P.; Menezes, C.B.; Goelzer, G.K.; Gnoatto, S.C.B.; Garcia, S.C.; Tasca, T. Adenosine Reduces Reactive Oxygen Species and Interleukin-8 Production by Trichomonas Vaginalis-Stimulated Neutrophils. Purinergic Signal. 2017, 13, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Corriden, R.; Self, T.; Akong-Moore, K.; Nizet, V.; Kellam, B.; Briddon, S.J.; Hill, S.J. Adenosine-A3 Receptors in Neutrophil Microdomains Promote the Formation of Bacteria-Tethering Cytonemes. EMBO Rep. 2013, 14, 726–732. [Google Scholar] [CrossRef]
- Van der Hoeven, D.; Wan, T.C.; Gizewski, E.T.; Kreckler, L.M.; Maas, J.E.; Van Orman, J.; Ravid, K.; Auchampach, J.A. A Role for the Low-Affinity A2B Adenosine Receptor in Regulating Superoxide Generation by Murine Neutrophils. J. Pharmacol. Exp. Ther. 2011, 338, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, J.; Cekic, C. Regulation of Lymphocyte Function by Adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2097–2103. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.K.; Silva, M.; Labuda, J.; Thayer, M.; Cain, D.W.; Philbrook, P.; Sethumadhavan, S.; Hatfield, S.; Ohta, A.; Sitkovsky, M. The GS Protein-Coupled A2a Adenosine Receptor Controls T Cell Help in the Germinal Center. J. Biol. Chem. 2017, 292, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Kasheta, M.; Painter, C.A.; Moore, F.E.; Lobbardi, R.; Bryll, A.; Freiman, E.; Stachura, D.; Rogers, A.B.; Houvras, Y.; Langenau, D.M.; et al. Identification and Characterization of T Reg–like Cells in Zebrafish. J. Exp. Med. 2017, 214, 3519–3530. [Google Scholar] [CrossRef]
- Romio, M.; Reinbeck, B.; Bongardt, S.; Hüls, S.; Burghoff, S.; Schrader, J. Extracellular Purine Metabolism and Signaling of CD73-Derived Adenosine in Murine Treg and Teff Cells. Am. J. Physiol. Cell Physiol. 2011, 301, C530–C539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The Development and Immunosuppressive Functions of CD4+ CD25+ FoxP3+ Regulatory T Cells Are under Influence of the Adenosine-A2A Adenosine Receptor Pathway. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakowicz-Burkiewicz, M.; Kocbuch, K.; Grden, M.; Maciejewska, I.; Szutowicz, A.; Pawelczyk, T. Impact of adenosine receptors on immunoglobulin production by human peripheral blood b lymphocytes. J. Physiol. Pharmacol. 2012, 63, 661–668. [Google Scholar]
- Przybyła, T.; Sakowicz-Burkiewicz, M.; Pawełczyk, T. Purinergic Signalling in B Cells. Acta Biochim. Pol. 2018, 65, 1–7. [Google Scholar] [CrossRef]
- Saze, Z.; Schuler, P.J.; Hong, C.-S.; Cheng, D.; Jackson, E.K.; Whiteside, T.L. Adenosine Production by Human B Cells and B Cell–Mediated Suppression of Activated T Cells. Blood 2013, 122, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Cronstein, B.N.; Sitkovsky, M. Adenosine and Adenosine Receptors in the Pathogenesis and Treatment of Rheumatic Diseases. Nat. Rev. Rheumatol. 2017, 13, 41–51. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Sattar, N.; McCarey, D.W.; Capell, H.; McInnes, I.B. Explaining How “High-Grade” Systemic Inflammation Accelerates Vascular Risk in Rheumatoid Arthritis. Circulation 2003, 108, 2957–2963. [Google Scholar] [CrossRef]
- Varani, K.; Padovan, M.; Govoni, M.; Vincenzi, F.; Trotta, F.; Borea, P.A. The Role of Adenosine Receptors in Rheumatoid Arthritis. Autoimmun. Rev. 2010, 10, 61–64. [Google Scholar] [CrossRef]
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 Adenosine Receptor: History and Perspectives. Pharmacol. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef] [Green Version]
- Friedman, B.; Cronstein, B. Methotrexate Mechanism in Treatment of Rheumatoid Arthritis. Joint Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef]
- Padovan, M.; Vincenzi, F.; Govoni, M.; Bortoluzzi, A.; Borea, P.A.; Varani, K. Adenosine and Adenosine Receptors in Rheumatoid Arthritis. Int. J. Clin. Rheumatol. 2013, 8, 13. [Google Scholar] [CrossRef]
- Varani, K.; Massara, A.; Vincenzi, F.; Tosi, A.; Padovan, M.; Trotta, F.; Borea, P.A. Normalization of A2A and A3 Adenosine Receptor Up-Regulation in Rheumatoid Arthritis Patients by Treatment with Anti-Tumor Necrosis Factor Alpha but Not Methotrexate. Arthritis Rheum. 2009, 60, 2880–2891. [Google Scholar] [CrossRef]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and Function of A2A and A₃ Adenosine Receptors in Patients with Ankylosing Spondylitis, Psoriatic Arthritis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Padovan, M.; Vincenzi, F.; Targa, M.; Trotta, F.; Govoni, M.; Borea, P.A. A2A and A3 Adenosine Receptor Expression in Rheumatoid Arthritis: Upregulation, Inverse Correlation with Disease Activity Score and Suppression of Inflammatory Cytokine and Metalloproteinase Release. Arthritis Res. Ther. 2011, 13, R197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, J.L.G.; Passos, D.F.; Bernardes, V.M.; Leal, D.B.R. ATP and Adenosine: Role in the Immunopathogenesis of Rheumatoid Arthritis. Immunol. Lett. 2019, 214, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Padovan, M.; Targa, M.; Corciulo, C.; Giacuzzo, S.; Merighi, S.; Gessi, S.; Govoni, M.; Borea, P.A.; Varani, K. A(2A) Adenosine Receptors Are Differentially Modulated by Pharmacological Treatments in Rheumatoid Arthritis Patients and Their Stimulation Ameliorates Adjuvant-Induced Arthritis in Rats. PLoS ONE 2013, 8, e54195. [Google Scholar] [CrossRef] [Green Version]
- Mazzon, E.; Esposito, E.; Impellizzeri, D.; DI Paola, R.; Melani, A.; Bramanti, P.; Pedata, F.; Cuzzocrea, S. CGS 21680, an Agonist of the Adenosine (A2A) Receptor, Reduces Progression of Murine Type II Collagen-Induced Arthritis. J. Rheumatol. 2011, 38, 2119–2129. [Google Scholar] [CrossRef]
- Flögel, U.; Burghoff, S.; van Lent, P.L.E.M.; Temme, S.; Galbarz, L.; Ding, Z.; El-Tayeb, A.; Huels, S.; Bönner, F.; Borg, N.; et al. Selective Activation of Adenosine A2A Receptors on Immune Cells by a CD73-Dependent Prodrug Suppresses Joint Inflammation in Experimental Rheumatoid Arthritis. Sci. Transl. Med. 2012, 4, 146ra108. [Google Scholar] [CrossRef]
- Chrobak, P.; Charlebois, R.; Rejtar, P.; El Bikai, R.; Allard, B.; Stagg, J. CD73 Plays a Protective Role in Collagen-Induced Arthritis. J. Immunol. 2015, 194, 2487–2492. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shan, Y.; Jiang, Z.; Feng, J.; Li, C.; Ma, L.; Jiang, Y. High Frequencies of Activated B Cells and T Follicular Helper Cells Are Correlated with Disease Activity in Patients with New-Onset Rheumatoid Arthritis. Clin. Exp. Immunol. 2013, 174, 212–220. [Google Scholar] [CrossRef]
- Ma, J.; Zhu, C.; Ma, B.; Tian, J.; Baidoo, S.E.; Mao, C.; Wu, W.; Chen, J.; Tong, J.; Yang, M.; et al. Increased Frequency of Circulating Follicular Helper T Cells in Patients with Rheumatoid Arthritis. Clin. Dev. Immunol. 2012, 2012, 827480. [Google Scholar] [CrossRef] [Green Version]
- Schmiel, S.E.; Kalekar, L.A.; Zhang, N.; Blankespoor, T.W.; Robinson, L.J.; Mueller, D.L. Adenosine 2a Receptor Signals Block Autoimmune Arthritis by Inhibiting Pathogenic Germinal Center T Follicular Helper Cells. Arthritis Rheumatol. 2019, 71, 773–783. [Google Scholar] [CrossRef]
- Baharav, E.; Bar-Yehuda, S.; Madi, L.; Silberman, D.; Rath-Wolfson, L.; Halpren, M.; Ochaion, A.; Weinberger, A.; Fishman, P. Antiinflammatory Effect of A3 Adenosine Receptor Agonists in Murine Autoimmune Arthritis Models. J. Rheumatol. 2005, 32, 469–476. [Google Scholar] [PubMed]
- Cohen, S.; Barer, F.; Bar-Yehuda, S.; IJzerman, A.P.; Jacobson, K.A.; Fishman, P. A₃ Adenosine Receptor Allosteric Modulator Induces an Anti-Inflammatory Effect: In Vivo Studies and Molecular Mechanism of Action. Mediat. Inflamm. 2014, 2014, 708746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Amital, H.; Jacobson, K.A.; Joshi, B.V.; Gao, Z.G.; Barer, F.; Patoka, R.; Del Valle, L.; et al. The A3 Adenosine Receptor Agonist CF502 Inhibits the PI3K, PKB/Akt and NF-KappaB Signaling Pathway in Synoviocytes from Rheumatoid Arthritis Patients and in Adjuvant-Induced Arthritis Rats. Biochem. Pharmacol. 2008, 76, 482–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, M.H.; Strand, V.; Markovits, D.; Nahir, M.; Reitblat, T.; Molad, Y.; Rosner, I.; Rozenbaum, M.; Mader, R.; Adawi, M.; et al. Clinical Evidence for Utilization of the A3 Adenosine Receptor as a Target to Treat Rheumatoid Arthritis: Data from a Phase II Clinical Trial. J. Rheumatol. 2008, 35, 41–48. [Google Scholar]
- Fishman, P.; Cohen, S. The A3 Adenosine Receptor (A3AR): Therapeutic Target and Predictive Biological Marker in Rheumatoid Arthritis. Clin. Rheumatol. 2016, 35, 2359–2362. [Google Scholar] [CrossRef]
- Passos, D.F.; Bernardes, V.M.; da Silva, J.L.G.; Schetinger, M.R.C.; Leal, D.B.R. Adenosine Signaling and Adenosine Deaminase Regulation of Immune Responses: Impact on the Immunopathogenesis of HIV Infection. Purinergic Signal. 2018, 14, 309–320. [Google Scholar] [CrossRef]
- He, W.; Cronstein, B.N. Adenosine A1 Receptor Regulates Osteoclast Formation by Altering TRAF6/TAK1 Signaling. Purinergic Signal. 2012, 8, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Teramachi, J.; Kukita, A.; Li, Y.-J.; Ushijima, Y.; Ohkuma, H.; Wada, N.; Watanabe, T.; Nakamura, S.; Kukita, T. Adenosine Abolishes MTX-Induced Suppression of Osteoclastogenesis and Inflammatory Bone Destruction in Adjuvant-Induced Arthritis. Lab. Investig. J. Tech. Methods Pathol. 2011, 91, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Kara, F.M.; Chitu, V.; Sloane, J.; Axelrod, M.; Fredholm, B.B.; Stanley, E.R.; Cronstein, B.N. Adenosine A1 Receptors (A1Rs) Play a Critical Role in Osteoclast Formation and Function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2325–2333. [Google Scholar] [CrossRef] [Green Version]
- Hameed, A.K.; El-Said, T.O.; Askar, H.Y.; ElKady, B.A. Performance of Serum Adenosine Deaminase in Measuring Disease Activity in Rheumatoid Arthritis Patients. Egypt. Rheumatol. 2019, 41, 81–85. [Google Scholar] [CrossRef]
- Vinapamula, K.S.; Pemmaraju, S.V.L.N.; Bhattaram, S.K.; Bitla, A.R.; Manohar, S.M. Serum Adenosine Deaminase as Inflammatory Marker in Rheumatoid Arthritis. J. Clin. Diagn. Res. JCDR 2015, 9, BC08–BC10. [Google Scholar] [CrossRef]
- Valadbeigi, S.; Saghiri, R.; Ebrahimi-Rad, M.; Khatami, S.; Akhbari, H. Adenosine Deaminase Activity and HLA-DRB as Diagnostic Markers for Rheumatoid Arthritis. Curr. Rheumatol. Rev. 2019, 15, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases. Pharmacol. Rev. 2019, 71, 345–382. [Google Scholar] [CrossRef]
- Antonioli, L.; Colucci, R.; La Motta, C.; Tuccori, M.; Awwad, O.; Da Settimo, F.; Blandizzi, C.; Fornai, M. Adenosine Deaminase in the Modulation of Immune System and Its Potential as a Novel Target for Treatment of Inflammatory Disorders. Curr. Drug Targets 2012, 13, 842–862. [Google Scholar] [CrossRef]
- Corciulo, C.; Lendhey, M.; Wilder, T.; Schoen, H.; Cornelissen, A.S.; Chang, G.; Kennedy, O.D.; Cronstein, B.N. Endogenous Adenosine Maintains Cartilage Homeostasis and Exogenous Adenosine Inhibits Osteoarthritis Progression. Nat. Commun. 2017, 8, 15019. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Corciulo, C.; Arabagian, S.; Ulman, A.; Cronstein, B.N. Adenosine-Functionalized Biodegradable PLA-b-PEG Nanoparticles Ameliorate Osteoarthritis in Rats. Sci. Rep. 2019, 9, 7430. [Google Scholar] [CrossRef]
- Friedman, B.; Corciulo, C.; Castro, C.M.; Cronstein, B.N. Adenosine A2A Receptor Signaling Promotes FoxO Associated Autophagy in Chondrocytes. Sci. Rep. 2021, 11, 968. [Google Scholar] [CrossRef]
- Bar-Yehuda, S.; Rath-Wolfson, L.; Del Valle, L.; Ochaion, A.; Cohen, S.; Patoka, R.; Zozulya, G.; Barer, F.; Atar, E.; Piña-Oviedo, S.; et al. Induction of an Antiinflammatory Effect and Prevention of Cartilage Damage in Rat Knee Osteoarthritis by CF101 Treatment. Arthritis Rheum. 2009, 60, 3061–3071. [Google Scholar] [CrossRef]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and Therapeutic Effects of A3 Adenosine Receptor Agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Fini, M.; Pagani, S.; Giavaresi, G.; De Mattei, M.; Ongaro, A.; Varani, K.; Vincenzi, F.; Massari, L.; Cadossi, M. Functional Tissue Engineering in Articular Cartilage Repair: Is There a Role for Electromagnetic Biophysical Stimulation? Tissue Eng. Part B Rev. 2013, 19, 353–367. [Google Scholar] [CrossRef]
- Varani, K.; De Mattei, M.; Vincenzi, F.; Gessi, S.; Merighi, S.; Pellati, A.; Ongaro, A.; Caruso, A.; Cadossi, R.; Borea, P.A. Characterization of Adenosine Receptors in Bovine Chondrocytes and Fibroblast-like Synoviocytes Exposed to Low Frequency Low Energy Pulsed Electromagnetic Fields. Osteoarth. Cartil. 2008, 16, 292–304. [Google Scholar] [CrossRef] [Green Version]
- De Mattei, M.; Varani, K.; Masieri, F.F.; Pellati, A.; Ongaro, A.; Fini, M.; Cadossi, R.; Vincenzi, F.; Borea, P.A.; Caruso, A. Adenosine Analogs and Electromagnetic Fields Inhibit Prostaglandin E2 Release in Bovine Synovial Fibroblasts. Osteoarth. Cartil. 2009, 17, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Vincenzi, F.; Targa, M.; Corciulo, C.; Gessi, S.; Merighi, S.; Setti, S.; Cadossi, R.; Goldring, M.B.; Borea, P.A.; Varani, K. Pulsed Electromagnetic Fields Increased the Anti-Inflammatory Effect of A₂A and A₃ Adenosine Receptors in Human T/C-28a2 Chondrocytes and HFOB 1.19 Osteoblasts. PLoS ONE 2013, 8, e65561. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Schneider, D.J.; Blackburn, M.R. Adenosine Signaling and the Regulation of Chronic Lung Disease. Pharmacol. Ther. 2009, 123, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Inflammatory Mechanisms in Patients with Chronic Obstructive Pulmonary Disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Cellular and Molecular Mechanisms of Asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef] [Green Version]
- Le, T.-T.T.; Berg, N.K.; Harting, M.T.; Li, X.; Eltzschig, H.K.; Yuan, X. Purinergic Signaling in Pulmonary Inflammation. Front. Immunol. 2019, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Driver, A.G.; Kukoly, C.A.; Ali, S.; Mustafa, S.J. Adenosine in Bronchoalveolar Lavage Fluid in Asthma. Am. Rev. Respir. Dis. 1993, 148, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Huszár, E.; Vass, G.; Vizi, E.; Csoma, Z.; Barát, E.; Molnár Világos, G.; Herjavecz, I.; Horváth, I. Adenosine in Exhaled Breath Condensate in Healthy Volunteers and in Patients with Asthma. Eur. Respir. J. 2002, 20, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Esther, C.R.; Lazaar, A.L.; Bordonali, E.; Qaqish, B.; Boucher, R.C. Elevated Airway Purines in COPD. Chest 2011, 140, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh Patidar, B.; Meena, A.; Kumar, M.; Menon, B.; Rohil, V.; Kumar Bansal, S. Adenosine Metabolism in COPD: A Study on Adenosine Levels, 5’-Nucleotidase, Adenosine Deaminase and Its Isoenzymes Activity in Serum, Lymphocytes and Erythrocytes. COPD 2018, 15, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M.R. Too Much of a Good Thing: Adenosine Overload in Adenosine-Deaminase-Deficient Mice. Trends Pharmacol. Sci. 2003, 24, 66–70. [Google Scholar] [CrossRef]
- Ma, B.; Blackburn, M.R.; Lee, C.G.; Homer, R.J.; Liu, W.; Flavell, R.A.; Boyden, L.; Lifton, R.P.; Sun, C.-X.; Young, H.W.; et al. Adenosine Metabolism and Murine Strain-Specific IL-4-Induced Inflammation, Emphysema, and Fibrosis. J. Clin. Investig. 2006, 116, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Polosa, R.; Blackburn, M.R. Adenosine Receptors as Targets for Therapeutic Intervention in Asthma and Chronic Obstructive Pulmonary Disease. Trends Pharmacol. Sci. 2009, 30, 528–535. [Google Scholar] [CrossRef] [Green Version]
- Polosa, R. Adenosine-Receptor Subtypes: Their Relevance to Adenosine-Mediated Responses in Asthma and Chronic Obstructive Pulmonary Disease. Eur. Respir. J. 2002, 20, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J.; Lindsay, J.C.; Zhou, Y.; Molina, J.G.; Blackburn, M.R. Adenosine and Osteopontin Contribute to the Development of Chronic Obstructive Pulmonary Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Karmouty-Quintana, H.; Zhong, H.; Acero, L.; Weng, T.; Melicoff, E.; West, J.D.; Hemnes, A.; Grenz, A.; Eltzschig, H.K.; Blackwell, T.S.; et al. The A2B Adenosine Receptor Modulates Pulmonary Hypertension Associated with Interstitial Lung Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 2546–2557. [Google Scholar] [CrossRef] [Green Version]
- Zaynagetdinov, R.; Ryzhov, S.; Goldstein, A.E.; Yin, H.; Novitskiy, S.V.; Goleniewska, K.; Polosukhin, V.V.; Newcomb, D.C.; Mitchell, D.; Morschl, E.; et al. Attenuation of Chronic Pulmonary Inflammation in A2B Adenosine Receptor Knockout Mice. Am. J. Respir. Cell Mol. Biol. 2010, 42, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Karmouty-Quintana, H.; Philip, K.; Acero, L.F.; Chen, N.-Y.; Weng, T.; Molina, J.G.; Luo, F.; Davies, J.; Le, N.-B.; Bunge, I.; et al. Deletion of ADORA2B from Myeloid Cells Dampens Lung Fibrosis and Pulmonary Hypertension. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chunn, J.L.; Volmer, J.B.; Fozard, J.R.; Blackburn, M.R. Adenosine-Mediated Mast Cell Degranulation in Adenosine Deaminase-Deficient Mice. J. Pharmacol. Exp. Ther. 2001, 298, 433–440. [Google Scholar]
- Chunn, J.L.; Young, H.W.; Banerjee, S.K.; Colasurdo, G.N.; Blackburn, M.R. Adenosine-Dependent Airway Inflammation and Hyperresponsiveness in Partially Adenosine Deaminase-Deficient Mice. J. Immunol. 2001, 167, 4676–4685. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-X.; Zhong, H.; Mohsenin, A.; Morschl, E.; Chunn, J.L.; Molina, J.G.; Belardinelli, L.; Zeng, D.; Blackburn, M.R. Role of A2B Adenosine Receptor Signaling in Adenosine-Dependent Pulmonary Inflammation and Injury. J. Clin. Investig. 2006, 116, 2173–2182. [Google Scholar] [CrossRef] [Green Version]
- Young, H.W.J.; Molina, J.G.; Dimina, D.; Zhong, H.; Jacobson, M.; Chan, L.-N.L.; Chan, T.-S.; Lee, J.J.; Blackburn, M.R. A3 Adenosine Receptor Signaling Contributes to Airway Inflammation and Mucus Production in Adenosine Deaminase-Deficient Mice. J. Immunol. 2004, 173, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-X.; Young, H.W.; Molina, J.G.; Volmer, J.B.; Schnermann, J.; Blackburn, M.R. A Protective Role for the A1 Adenosine Receptor in Adenosine-Dependent Pulmonary Injury. J. Clin. Investig. 2005, 115, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsenin, A.; Mi, T.; Xia, Y.; Kellems, R.E.; Chen, J.-F.; Blackburn, M.R. Genetic Removal of the A2A Adenosine Receptor Enhances Pulmonary Inflammation, Mucin Production, and Angiogenesis in Adenosine Deaminase-Deficient Mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L753–L761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, K.; Caramori, G.; Vincenzi, F.; Adcock, I.; Casolari, P.; Leung, E.; MacLennan, S.; Gessi, S.; Morello, S.; Barnes, P.J.; et al. Alteration of Adenosine Receptors in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2006, 173, 398–406. [Google Scholar] [CrossRef]
- Varani, K.; Caramori, G.; Vincenzi, F.; Tosi, A.; Barczyk, A.; Contoli, M.; Casolari, P.; Triggiani, M.; Hansel, T.; Leung, E.; et al. Oxidative/Nitrosative Stress Selectively Altered A(2B) Adenosine Receptors in Chronic Obstructive Pulmonary Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 1192–1204. [Google Scholar] [CrossRef]
- Eckle, T.; Grenz, A.; Laucher, S.; Eltzschig, H.K. A2B Adenosine Receptor Signaling Attenuates Acute Lung Injury by Enhancing Alveolar Fluid Clearance in Mice. J. Clin. Investig. 2008, 118, 3301–3315. [Google Scholar] [CrossRef] [Green Version]
- Eckle, T.; Hughes, K.; Ehrentraut, H.; Brodsky, K.S.; Rosenberger, P.; Choi, D.-S.; Ravid, K.; Weng, T.; Xia, Y.; Blackburn, M.R.; et al. Crosstalk between the Equilibrative Nucleoside Transporter ENT2 and Alveolar Adora2b Adenosine Receptors Dampens Acute Lung Injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 3078–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, F.M.; Neudeck, G.; Vollmer, I.; Ngamsri, K.C.; Thiel, M.; Reutershan, J. Protective Effects of Pentoxifylline in Pulmonary Inflammation Are Adenosine Receptor A2A Dependent. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 3524–3535. [Google Scholar] [CrossRef]
- Folkesson, H.G.; Kuzenko, S.R.; Lipson, D.A.; Matthay, M.A.; Simmons, M.A. The Adenosine 2A Receptor Agonist GW328267C Improves Lung Function after Acute Lung Injury in Rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L259–L271. [Google Scholar] [CrossRef] [PubMed]
- Hoegl, S.; Brodsky, K.S.; Blackburn, M.R.; Karmouty-Quintana, H.; Zwissler, B.; Eltzschig, H.K. Alveolar Epithelial A2B Adenosine Receptors in Pulmonary Protection during Acute Lung Injury. J. Immunol. 2015, 195, 1815–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, F.M.; Meichssner, N.; Bury, A.; Ngamsri, K.-C.; Reutershan, J. Inhibition of SDF-1 Receptors CXCR4 and CXCR7 Attenuates Acute Pulmonary Inflammation via the Adenosine A2B-Receptor on Blood Cells. Cell Death Dis. 2017, 8, e2832. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, J.N.; Gorshkov, B.; Varn, M.N.; Zemskova, M.A.; Zemskov, E.A.; Sridhar, S.; Lucas, R.; Verin, A.D. Protective Effect of Adenosine Receptors against Lipopolysaccharide-Induced Acute Lung Injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L497–L507. [Google Scholar] [CrossRef] [Green Version]
- Konrad, F.M.; Zwergel, C.; Ngamsri, K.-C.; Reutershan, J. Anti-Inflammatory Effects of Heme Oxygenase-1 Depend on Adenosine A2A- and A2B-Receptor Signaling in Acute Pulmonary Inflammation. Front. Immunol. 2017, 8, 1874. [Google Scholar] [CrossRef] [Green Version]
- Impellizzeri, D.; Di Paola, R.; Esposito, E.; Mazzon, E.; Paterniti, I.; Melani, A.; Bramanti, P.; Pedata, F.; Cuzzocrea, S. CGS 21680, an Agonist of the Adenosine (A2A) Receptor, Decreases Acute Lung Inflammation. Eur. J. Pharmacol. 2011, 668, 305–316. [Google Scholar] [CrossRef]
- Ngamsri, K.-C.; Wagner, R.; Vollmer, I.; Stark, S.; Reutershan, J. Adenosine Receptor A1 Regulates Polymorphonuclear Cell Trafficking and Microvascular Permeability in Lipopolysaccharide-Induced Lung Injury. J. Immunol. 2010, 185, 4374–4384. [Google Scholar] [CrossRef] [Green Version]
- Mulloy, D.P.; Sharma, A.K.; Fernandez, L.G.; Zhao, Y.; Lau, C.L.; Kron, I.L.; Laubach, V.E. Adenosine A3 Receptor Activation Attenuates Lung Ischemia-Reperfusion Injury. Ann. Thorac. Surg. 2013, 95, 1762–1767. [Google Scholar] [CrossRef] [Green Version]
- Morschl, E.; Molina, J.G.; Volmer, J.B.; Mohsenin, A.; Pero, R.S.; Hong, J.-S.; Kheradmand, F.; Lee, J.J.; Blackburn, M.R. A3 Adenosine Receptor Signaling Influences Pulmonary Inflammation and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, C.; Caracciolo, M.; Correale, P.; Macheda, S.; Vadalà, E.G.; La Scala, S.; Tescione, M.; Danieli, R.; Ferrarelli, A.; Tarsitano, M.G.; et al. Can Adenosine Fight COVID-19 Acute Respiratory Distress Syndrome? J. Clin. Med. 2020, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, D.; Antonioli, L.; Lambertucci, C.; Fornai, M.; Blandizzi, C.; Volpini, R. Purinergic Ligands as Potential Therapeutic Tools for the Treatment of Inflammation-Related Intestinal Diseases. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Moss, A.; Jiang, Z.G.; Robson, S.C. Purinergic Signaling during Intestinal Inflammation. J. Mol. Med. Berl. Ger. 2017, 95, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Odashima, M.; Bamias, G.; Rivera-Nieves, J.; Linden, J.; Nast, C.C.; Moskaluk, C.A.; Marini, M.; Sugawara, K.; Kozaiwa, K.; Otaka, M.; et al. Activation of A2A Adenosine Receptor Attenuates Intestinal Inflammation in Animal Models of Inflammatory Bowel Disease. Gastroenterology 2005, 129, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Fornai, M.; Colucci, R.; Ghisu, N.; Blandizzi, C.; Del Tacca, M. A2a Receptors Mediate Inhibitory Effects of Adenosine on Colonic Motility in the Presence of Experimental Colitis. Inflamm. Bowel Dis. 2006, 12, 117–122. [Google Scholar] [CrossRef]
- Antonioli, L.; Fornai, M.; Colucci, R.; Awwad, O.; Ghisu, N.; Tuccori, M.; Del Tacca, M.; Blandizzi, C. Differential Recruitment of High Affinity A1 and A2A Adenosine Receptors in the Control of Colonic Neuromuscular Function in Experimental Colitis. Eur. J. Pharmacol. 2011, 650, 639–649. [Google Scholar] [CrossRef]
- Antonioli, L.; El-Tayeb, A.; Pellegrini, C.; Fornai, M.; Awwad, O.; Giustarini, G.; Natale, G.; Ryskalin, L.; Németh, Z.H.; Müller, C.E.; et al. Anti-Inflammatory Effect of a Novel Locally Acting A2A Receptor Agonist in a Rat Model of Oxazolone-Induced Colitis. Purinergic Signal. 2018, 14, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Naganuma, M.; Wiznerowicz, E.B.; Lappas, C.M.; Linden, J.; Worthington, M.T.; Ernst, P.B. Cutting Edge: Critical Role for A2A Adenosine Receptors in the T Cell-Mediated Regulation of Colitis. J. Immunol. 2006, 177, 2765–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimian, R.; Fakhfouri, G.; Daneshmand, A.; Mohammadi, H.; Bahremand, A.; Rasouli, M.R.; Mousavizadeh, K.; Dehpour, A.R. Adenosine A2A Receptors and Uric Acid Mediate Protective Effects of Inosine against TNBS-Induced Colitis in Rats. Eur. J. Pharmacol. 2010, 649, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, I.C.; Castro, M.V.; Barreto, A.R.F.; Sullivan, G.W.; Vale, M.; Almeida, P.R.C.; Linden, J.; Rieger, J.M.; Cunha, F.Q.; Guerrant, R.L.; et al. Effect of Novel A2A Adenosine Receptor Agonist ATL 313 on Clostridium Difficile Toxin A-Induced Murine Ileal Enteritis. Infect. Immun. 2006, 74, 2606–2612. [Google Scholar] [CrossRef] [Green Version]
- Pallio, G.; Bitto, A.; Pizzino, G.; Galfo, F.; Irrera, N.; Squadrito, F.; Squadrito, G.; Pallio, S.; Anastasi, G.P.; Cutroneo, G.; et al. Adenosine Receptor Stimulation by Polydeoxyribonucleotide Improves Tissue Repair and Symptomology in Experimental Colitis. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Michael, S.; Abdel-Aziz, H.; Weiser, D.; Müller, C.E.; Kelber, O.; Nieber, K. Adenosine A2A Receptor Contributes to the Anti-Inflammatory Effect of the Fixed Herbal Combination STW 5 (Iberogast®) in Rat Small Intestinal Preparations. Naunyn. Schmiedebergs Arch. Pharmacol. 2012, 385, 411–421. [Google Scholar] [CrossRef]
- Kolachala, V.; Ruble, B.; Vijay-Kumar, M.; Wang, L.; Mwangi, S.; Figler, H.; Figler, R.; Srinivasan, S.; Gewirtz, A.; Linden, J.; et al. Blockade of Adenosine A2B Receptors Ameliorates Murine Colitis. Br. J. Pharmacol. 2008, 155, 127–137. [Google Scholar] [CrossRef]
- Kolachala, V.L.; Vijay–Kumar, M.; Dalmasso, G.; Yang, D.; Linden, J.; Wang, L.; Gewirtz, A.; Ravid, K.; Merlin, D.; Sitaraman, S.V. A2B Adenosine Receptor Gene Deletion Attenuates Murine Colitis. Gastroenterology 2008, 135, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Fan, J.; Chen, Y.-X.; Wang, J.-H. Inhibition of A2B Adenosine Receptor Attenuates Intestinal Injury in a Rat Model of Necrotizing Enterocolitis. Mediators Inflamm. 2020, 2020, 1562973. [Google Scholar] [CrossRef]
- Frick, J.-S.; MacManus, C.F.; Scully, M.; Glover, L.E.; Eltzschig, H.K.; Colgan, S.P. Contribution of Adenosine A2B Receptors to Inflammatory Parameters of Experimental Colitis. J. Immunol. 2009, 182, 4957–4964. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Fornai, M.; Colucci, R.; Ghisu, N.; Tuccori, M.; Del Tacca, M.; Blandizzi, C. Pharmacological Modulation of Adenosine System: Novel Options for Treatment of Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2008, 14, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Mabley, J.; Soriano, F.; Pacher, P.; Haskó, G.; Marton, A.; Wallace, R.; Salzman, A.; Szabó, C. The Adenosine A3 Receptor Agonist, N6-(3-Iodobenzyl)-Adenosine-5′-N-Methyluronamide, Is Protective in Two Murine Models of Colitis. Eur. J. Pharmacol. 2003, 466, 323–329. [Google Scholar] [CrossRef]
- Guzman, J.; Yu, J.G.; Suntres, Z.; Bozarov, A.; Cooke, H.; Javed, N.; Auer, H.; Palatini, J.; Hassanain, H.H.; Cardounel, A.J.; et al. ADOA3R as a Therapeutic Target in Experimental Colitis: Proof by Validated High-Density Oligonucleotide Microarray Analysis. Inflamm. Bowel Dis. 2006, 12, 766–789. [Google Scholar] [CrossRef]
- Ren, T.; Tian, T.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Qiu, Y.; Yu, C.; He, Y.; Zeng, J.; et al. An Adenosine A3 Receptor Agonist Inhibits DSS-Induced Colitis in Mice through Modulation of the NF-ΚB Signaling Pathway. Sci. Rep. 2015, 5, 9047. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.H.; Lv, M.M.; An, X.M.; Leung, W.K.; Seto, W.-K. Activation of Adenosine A3 Receptor Inhibits Inflammatory Cytokine Production in Colonic Mucosa of Patients with Ulcerative Colitis by Down-Regulating the Nuclear Factor-Kappa B Signaling. J. Dig. Dis. 2020, 21, 38–45. [Google Scholar] [CrossRef]
- Brown, J.B.; Lee, G.; Grimm, G.R.; Barrett, T.A. Therapeutic Benefit of Pentostatin in Severe IL-10−/− Colitis. Inflamm. Bowel Dis. 2008, 14, 880–887. [Google Scholar] [CrossRef]
- Siegmund, B.; Rieder, F.; Albrich, S.; Firestein, G.S.; Boyle, D.; Hartmann, G.; Endres, S.; Eigler, A. Adenosine Kinase Inhibitor GP515 Improves Experimental Colitis in Mice by Inhibition of TH1 Cytokine Synthesis. Gastroenterology 2000, 118, A578. [Google Scholar] [CrossRef]
- Antonioli, L.; Fornai, M.; Colucci, R.; Ghisu, N.; Da Settimo, F.; Natale, G.; Kastsiuchenka, O.; Duranti, E.; Virdis, A.; Vassalle, C.; et al. Inhibition of Adenosine Deaminase Attenuates Inflammation in Experimental Colitis. J. Pharmacol. Exp. Ther. 2007, 322, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Fornai, M.; Colucci, R.; Awwad, O.; Ghisu, N.; Tuccori, M.; Da Settimo, F.; La Motta, C.; Natale, G.; Duranti, E.; et al. The Blockade of Adenosine Deaminase Ameliorates Chronic Experimental Colitis through the Recruitment of Adenosine A2A and A3 Receptors. J. Pharmacol. Exp. Ther. 2010, 335, 434–442. [Google Scholar] [CrossRef] [Green Version]
- La Motta, C.; Sartini, S.; Mugnaini, L.; Salerno, S.; Simorini, F.; Taliani, S.; Marini, A.M.; Da Settimo, F.; Lavecchia, A.; Novellino, E.; et al. Exploiting the Pyrazolo[3,4-d]Pyrimidin-4-One Ring System as a Useful Template To Obtain Potent Adenosine Deaminase Inhibitors. J. Med. Chem. 2009, 52, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate Immunity in the Central Nervous System. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Derecki, N.C.; Cardani, A.N.; Yang, C.H.; Quinnies, K.M.; Crihfield, A.; Lynch, K.R.; Kipnis, J. Regulation of Learning and Memory by Meningeal Immunity: A Key Role for IL-4. J. Exp. Med. 2010, 207, 1067–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, D.P.; Stevens, B. Phagocytic Glial Cells: Sculpting Synaptic Circuits in the Developing Nervous System. Curr. Opin. Neurobiol. 2013, 23, 1034–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in Neurodegenerative Disorders: The Roles of Microglia and Astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. An Introduction to the Roles of Purinergic Signalling in Neurodegeneration, Neuroprotection and Neuroregeneration. Neuropharmacology 2016, 104, 4–17. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and Depression: A Review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and Psychiatric Illness. J. Neuroinflamm. 2013, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Martí Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine Receptors as Neuroinflammation Modulators: Role of A1 Agonists and A2A Antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Pedata, F.; Pugliese, A.M.; Coppi, E.; Dettori, I.; Maraula, G.; Cellai, L.; Melani, A. Adenosine A 2A Receptors Modulate Acute Injury and Neuroinflammation in Brain Ischemia. Mediators Inflamm. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Lana, D.; Giovannini, M.G.; Pedata, F.; Pugliese, A.M. A2B Adenosine Receptors: When Outsiders May Become an Attractive Target to Treat Brain Ischemia or Demyelination. Int. J. Mol. Sci. 2020, 21, 9697. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.A.; Cuzzocrea, S.; Esposito, E.; Campolo, M.; Niehoff, M.L.; Doyle, T.M.; Salvemini, D. Adenosine A3 Receptor as a Novel Therapeutic Target to Reduce Secondary Events and Improve Neurocognitive Functions Following Traumatic Brain Injury. J. Neuroinflamm. 2020, 17, 339. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Chen, J.-F.; Fredholm, B.B. Adenosine Signalling and Function in Glial Cells. Cell Death Differ. 2010, 17, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, A.G.; Orr, A.L.; Li, X.-J.; Gross, R.E.; Traynelis, S.F. Adenosine A(2A) Receptor Mediates Microglial Process Retraction. Nat. Neurosci. 2009, 12, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L.; Greco, A.; Potenza, R.L.; Pezzola, A.; Blum, D.; Bantubungi, K.; Popoli, P. Effects of the Adenosine A2A Receptor Antagonist SCH 58621 on Cyclooxygenase-2 Expression, Glial Activation, and Brain-Derived Neurotrophic Factor Availability in a Rat Model of Striatal Neurodegeneration. J. Neuropathol. Exp. Neurol. 2007, 66, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jiménez, J.; Raïch, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; Sánchez de Medina, V.; et al. Potentiation of Cannabinoid Signaling in Microglia by Adenosine A2A Receptor Antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef] [PubMed]
- Colella, M.; Zinni, M.; Pansiot, J.; Cassanello, M.; Mairesse, J.; Ramenghi, L.; Baud, O. Modulation of Microglial Activation by Adenosine A2a Receptor in Animal Models of Perinatal Brain Injury. Front. Neurol. 2018, 9, 605. [Google Scholar] [CrossRef]
- Rebola, N.; Simões, A.P.; Canas, P.M.; Tomé, A.R.; Andrade, G.M.; Barry, C.E.; Agostinho, P.M.; Lynch, M.A.; Cunha, R.A. Adenosine A2A Receptors Control Neuroinflammation and Consequent Hippocampal Neuronal Dysfunction. J. Neurochem. 2011, 117, 100–111. [Google Scholar] [CrossRef]
- Luongo, L.; Guida, F.; Imperatore, R.; Napolitano, F.; Gatta, L.; Cristino, L.; Giordano, C.; Siniscalco, D.; Di Marzo, V.; Bellini, G.; et al. The A1 Adenosine Receptor as a New Player in Microglia Physiology. Glia 2014, 62, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Ben, D.D.; Lambertucci, C.; Navia, A.M.; Spinaci, A.; Volpini, R.; Buccioni, M. Combined Therapy of A1AR Agonists and A2AAR Antagonists in Neuroinflammation. Molecules 2021, 26, 1188. [Google Scholar] [CrossRef]
- Haselkorn, M.L.; Shellington, D.K.; Jackson, E.K.; Vagni, V.A.; Janesko-Feldman, K.; Dubey, R.K.; Gillespie, D.G.; Cheng, D.; Bell, M.J.; Jenkins, L.W.; et al. Adenosine A1 Receptor Activation as a Brake on the Microglial Response after Experimental Traumatic Brain Injury in Mice. J. Neurotrauma 2010, 27, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Terayama, R.; Tabata, M.; Maruhama, K.; Iida, S. A3 Adenosine Receptor Agonist Attenuates Neuropathic Pain by Suppressing Activation of Microglia and Convergence of Nociceptive Inputs in the Spinal Dorsal Horn. Exp. Brain Res. 2018, 236, 3203–3213. [Google Scholar] [CrossRef]
- Ferreira-Silva, J.; Aires, I.D.; Boia, R.; Ambrósio, A.F.; Santiago, A.R. Activation of Adenosine A3 Receptor Inhibits Microglia Reactivity Elicited by Elevated Pressure. Int. J. Mol. Sci. 2020, 21, 7218. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, X.; Deng, P.; Wang, D.; Bai, X.; Li, Y.; Luo, C.; Belguise, K.; Wang, X.; Wei, X.; et al. Activation of Adenosine A3 Receptor Reduces Early Brain Injury by Alleviating Neuroinflammation after Subarachnoid Hemorrhage in Elderly Rats. Aging 2020, 13, 694–713. [Google Scholar] [CrossRef]
- Merighi, S.; Bencivenni, S.; Vincenzi, F.; Varani, K.; Borea, P.A.; Gessi, S. A2B Adenosine Receptors Stimulate IL-6 Production in Primary Murine Microglia through P38 MAPK Kinase Pathway. Pharmacol. Res. 2017, 117, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Poloni, T.E.; Terrazzan, A.; Moretti, E.; Gessi, S.; Ferrari, D. Alzheimer and Purinergic Signaling: Just a Matter of Inflammation? Cells 2021, 10, 1267. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Poloni, T.E.; Pelloni, L.; Pasquini, S.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. An Open Question: Is the A2A Adenosine Receptor a Novel Target for Alzheimer’s Disease Treatment? Front. Pharmacol. 2021, 12, 652455. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.M.; Fathalla, A.M.; Moustafa, A.A. Adenosine Role in Brain Functions: Pathophysiological Influence on Parkinson’s Disease and Other Brain Disorders. Pharmacol. Rep. 2018, 70, 661–667. [Google Scholar] [CrossRef]
- Nazario, L.R.; da Silva, R.S.; Bonan, C.D. Targeting Adenosine Signaling in Parkinson’s Disease: From Pharmacological to Non-Pharmacological Approaches. Front. Neurosci. 2017, 11, 658. [Google Scholar] [CrossRef] [Green Version]
- Safarzadeh, E.; Jadidi-Niaragh, F.; Motallebnezhad, M.; Yousefi, M. The Role of Adenosine and Adenosine Receptors in the Immunopathogenesis of Multiple Sclerosis. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. Al 2016, 65, 511–520. [Google Scholar] [CrossRef]
- Cieślak, M.; Kukulski, F.; Komoszyński, M. Emerging Role of Extracellular Nucleotides and Adenosine in Multiple Sclerosis. Purinergic Signal. 2011, 7, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, D.; Chern, Y.; Domenici, M.R.; Buée, L.; Lin, C.-Y.; Rea, W.; Ferré, S.; Popoli, P. The Role of Adenosine Tone and Adenosine Receptors in Huntington’s Disease. J. Caffeine Adenosine Res. 2018, 8, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Corciulo, C.; Targa, M.; Merighi, S.; Gessi, S.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Varani, K. Multiple Sclerosis Lymphocytes Upregulate A2A Adenosine Receptors That Are Antiinflammatory When Stimulated. Eur. J. Immunol. 2013, 43, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors Is Reflected in Platelets of Patients with Alzheimer’s Disease. J. Alzheimers Dis. JAD 2021, 80, 1105–1117. [Google Scholar] [CrossRef]
- Casetta, I.; Vincenzi, F.; Bencivelli, D.; Corciulo, C.; Gentile, M.; Granieri, E.; Borea, P.A.; Varani, K. A(2A) Adenosine Receptors and Parkinson’s Disease Severity. Acta Neurol. Scand. 2014, 129, 276–281. [Google Scholar] [CrossRef]
- Varani, K.; Vincenzi, F.; Tosi, A.; Gessi, S.; Casetta, I.; Granieri, G.; Fazio, P.; Leung, E.; MacLennan, S.; Granieri, E.; et al. A2A Adenosine Receptor Overexpression and Functionality, as Well as TNF-Alpha Levels, Correlate with Motor Symptoms in Parkinson’s Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 587–598. [Google Scholar] [CrossRef]
- Varani, K.; Bachoud-Lévi, A.-C.; Mariotti, C.; Tarditi, A.; Abbracchio, M.P.; Gasperi, V.; Borea, P.A.; Dolbeau, G.; Gellera, C.; Solari, A.; et al. Biological Abnormalities of Peripheral A(2A) Receptors in a Large Representation of Polyglutamine Disorders and Huntington’s Disease Stages. Neurobiol. Dis. 2007, 27, 36–43. [Google Scholar] [CrossRef]
- Varani, K.; Abbracchio, M.P.; Cannella, M.; Cislaghi, G.; Giallonardo, P.; Mariotti, C.; Cattabriga, E.; Cattabeni, F.; Borea, P.A.; Squitieri, F.; et al. Aberrant A2A Receptor Function in Peripheral Blood Cells in Huntington’s Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 2148–2150. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, S.; Schnermann, J.; Noorbakhsh, F.; Henry, S.; Yong, V.W.; Winston, B.W.; Warren, K.; Power, C. A1 Adenosine Receptor Upregulation and Activation Attenuates Neuroinflammation and Demyelination in a Model of Multiple Sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 1521–1529. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.-Q.; Li, Z.-Z.; Huang, Q.-Y.; Li, F.; Wang, Z.-W.; Augusto, E.; He, J.-C.; Wang, X.-T.; Chen, J.-F.; Zheng, R.-Y. Genetic Inactivation of the Adenosine A(2A) Receptor Exacerbates Brain Damage in Mice with Experimental Autoimmune Encephalomyelitis. J. Neurochem. 2012, 123, 100–112. [Google Scholar] [CrossRef]
- Ingwersen, J.; Wingerath, B.; Graf, J.; Lepka, K.; Hofrichter, M.; Schröter, F.; Wedekind, F.; Bauer, A.; Schrader, J.; Hartung, H.-P.; et al. Dual Roles of the Adenosine A2a Receptor in Autoimmune Neuroinflammation. J. Neuroinflamm. 2016, 13, 48. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.H.; Thompson, L.F.; Mueller, C.; Waickman, A.T.; Jalkanen, S.; Niemela, J.; Airas, L.; Bynoe, M.S. CD73 Is Required for Efficient Entry of Lymphocytes into the Central Nervous System during Experimental Autoimmune Encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 9325–9330. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.H.; Kim, D.-G.; Krenz, A.; Chen, J.-F.; Bynoe, M.S. A2A Adenosine Receptor Signaling in Lymphocytes and the Central Nervous System Regulates Inflammation during Experimental Autoimmune Encephalomyelitis. J. Immunol. 2012, 188, 5713–5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, Z.-X.; Zheng, L.-P.; Wang, L.; Liu, Y.-F.; Yin, W.-Y.; Chen, Y.-Y.; Wang, X.-S.; Hou, S.-T.; Chen, J.-F.; et al. The Adenosine A2A Receptor Antagonist SCH58261 Reduces Macrophage/Microglia Activation and Protects against Experimental Autoimmune Encephalomyelitis in Mice. Neurochem. Int. 2019, 129, 104490. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Alahiri, M.; Ulloa, B.; Xie, B.; Sadiq, S.A. Adenosine A2A Receptor Agonist Ameliorates EAE and Correlates with Th1 Cytokine-Induced Blood Brain Barrier Dysfunction via Suppression of MLCK Signaling Pathway. Immun. Inflamm. Dis. 2018, 6, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Loram, L.C.; Strand, K.A.; Taylor, F.R.; Sloane, E.; Van Dam, A.-M.; Rieger, J.; Maier, S.F.; Watkins, L.R. Adenosine 2A Receptor Agonism: A Single Intrathecal Administration Attenuates Motor Paralysis in Experimental Autoimmune Encephalopathy in Rats. Brain. Behav. Immun. 2015, 46, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strakhova, R.; Cadassou, O.; Cros-Perrial, E.; Jordheim, L.P. Regulation of Tumor Infiltrated Innate Immune Cells by Adenosine. Purinergic Signal. 2020, 16, 289–295. [Google Scholar] [CrossRef]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The Prognostic Value of Infiltrating Immune Cells in Cancer. Br. J. Cancer 2017, 117, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Adenosine as an Endogenous Immunoregulator in Cancer Pathogenesis: Where to Go? Purinergic Signal. 2013, 9, 145–165. [Google Scholar] [CrossRef] [Green Version]
- Aliagas, E.; Vidal, A.; Texidó, L.; Ponce, J.; Condom, E.; Martín-Satué, M. High Expression of Ecto-Nucleotidases CD39 and CD73 in Human Endometrial Tumors. Mediators Inflamm. 2014, 2014, 509027. [Google Scholar] [CrossRef] [Green Version]
- Bastid, J.; Regairaz, A.; Bonnefoy, N.; Déjou, C.; Giustiniani, J.; Laheurte, C.; Cochaud, S.; Laprevotte, E.; Funck-Brentano, E.; Hemon, P.; et al. Inhibition of CD39 Enzymatic Function at the Surface of Tumor Cells Alleviates Their Immunosuppressive Activity. Cancer Immunol. Res. 2015, 3, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.-Y.; Ni, X.-C.; Yi, Y.; He, H.-W.; Wang, J.-X.; Fu, Y.-P.; Sun, J.; Zhou, J.; Cheng, Y.-F.; Jin, J.-J.; et al. Overexpression of CD39 in Hepatocellular Carcinoma Is an Independent Indicator of Poor Outcome after Radical Resection. Medicine 2016, 95, e4989. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, I.; Vigano, S.; Faouzi, M.; Treilleux, I.; Michielin, O.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; de Leval, L. CD73 Expression and Clinical Significance in Human Metastatic Melanoma. Oncotarget 2018, 9, 26659–26669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional Regulation by Hypoxia Inducible Factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Koizume, S.; Miyagi, Y. Diverse Mechanisms of Sp1-Dependent Transcriptional Regulation Potentially Involved in the Adaptive Response of Cancer Cells to Oxygen-Deficient Conditions. Cancers 2015, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Tak, E.; Jung, D.-H.; Kim, S.-H.; Park, G.-C.; Jun, D.Y.; Lee, J.; Jung, B.; Kirchner, V.A.; Hwang, S.; Song, G.-W.; et al. Protective Role of Hypoxia-Inducible Factor-1α-Dependent CD39 and CD73 in Fulminant Acute Liver Failure. Toxicol. Appl. Pharmacol. 2017, 314, 72–81. [Google Scholar] [CrossRef]
- Poth, J.M.; Brodsky, K.; Ehrentraut, H.; Grenz, A.; Eltzschig, H.K. Transcriptional Control of Adenosine Signaling by Hypoxia-Inducible Transcription Factors during Ischemic or Inflammatory Disease. J. Mol. Med. 2013, 91, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Canale, F.P.; Ramello, M.C.; Núñez, N.; Furlan, C.L.A.; Bossio, S.N.; Serrán, M.G.; Boari, J.T.; del Castillo, A.; Ledesma, M.; Sedlik, C.; et al. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8+ T Cells. Cancer Res. 2018, 78, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Pagnotta, S.M.; Laudanna, C.; Pancione, M.; Sabatino, L.; Votino, C.; Remo, A.; Cerulo, L.; Zoppoli, P.; Manfrin, E.; Colantuoni, V.; et al. Ensemble of Gene Signatures Identifies Novel Biomarkers in Colorectal Cancer Activated through PPARγ and TNFα Signaling. PLoS ONE 2013, 8, e72638. [Google Scholar] [CrossRef] [Green Version]
- Ryzhov, S.V.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Zhang, Q.; Owens, P.; Feoktistov, I.; Moses, H.L.; Novitskiy, S.V. Role of TGFβ Signaling in Generation of CD39+CD73+ Myeloid Cells in Tumors. J. Immunol. 2014, 193, 3155–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T Lymphocyte Interplay in Controlling Tumor Growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskó, G.; Pacher, P. Regulation of Macrophage Function by Adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 865–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The Adenosine-Dependent Angiogenic Switch of Macrophages to an M2-like Phenotype Is Independent of Interleukin-4 Receptor Alpha (IL4Rα) Signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Velot, E.; Haas, B.; Léonard, F.; Ernens, I.; Rolland-Turner, M.; Schwartz, C.; Longrois, D.; Devaux, Y.; Wagner, D.R. Activation of the Adenosine-A3 Receptor Stimulates Matrix Metalloproteinase-9 Secretion by Macrophages. Cardiovasc. Res. 2008, 80, 246–254. [Google Scholar] [CrossRef]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine Promotes Alternative Macrophage Activation via A2A and A2B Receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef] [Green Version]
- Rubenich, D.S.; de Souza, P.O.; Omizzollo, N.; Lenz, G.S.; Sevigny, J.; Braganhol, E. Neutrophils: Fast and Furious—The Nucleotide Pathway. Purinergic Signal. 2021. [Google Scholar] [CrossRef] [PubMed]
- McColl, S.R.; St-Onge, M.; Dussault, A.-A.; Laflamme, C.; Bouchard, L.; Boulanger, J.; Pouliot, M. Immunomodulatory Impact of the A2A Adenosine Receptor on the Profile of Chemokines Produced by Neutrophils. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 187–189. [Google Scholar] [CrossRef] [Green Version]
- Gessi, S.; Varani, K.; Merighi, S.; Cattabriga, E.; Iannotta, V.; Leung, E.; Baraldi, P.G.; Borea, P.A. A3 Adenosine Receptors in Human Neutrophils and Promyelocytic HL60 Cells: A Pharmacological and Biochemical Study. Mol. Pharmacol. 2002, 61, 415–424. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Neo, S.Y.; Yang, Y.; Record, J.; Ma, R.; Chen, X.; Chen, Z.; Tobin, N.P.; Blake, E.; Seitz, C.; Thomas, R.; et al. CD73 Immune Checkpoint Defines Regulatory NK Cells within the Tumor Microenvironment. J. Clin. Investig. 2020, 130, 1185–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorzalczany, Y.; Akiva, E.; Klein, O.; Merimsky, O.; Sagi-Eisenberg, R. Mast Cells Are Directly Activated by Contact with Cancer Cells by a Mechanism Involving Autocrine Formation of Adenosine and Autocrine/Paracrine Signaling of the Adenosine A3 Receptor. Cancer Lett. 2017, 397, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gorzalczany, Y.; Merimsky, O.; Sagi-Eisenberg, R. Mast Cells Are Directly Activated by Cancer Cell–Derived Extracellular Vesicles by a CD73- and Adenosine-Dependent Mechanism. Transl. Oncol. 2019, 12, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Coillard, A.; Segura, E. In Vivo Differentiation of Human Monocytes. Front. Immunol. 2019, 10, 1907. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The Adenosine Pathway in Immuno-Oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Luddy, K.; Noyes, D.; Khalil, F.K.; Neuger, A.M.; Soliman, H.; Antonia, S.J. Antagonism of Adenosine A2A Receptor Expressed by Lung Adenocarcinoma Tumor Cells and Cancer Associated Fibroblasts Inhibits Their Growth. Cancer Biol. Ther. 2013, 14, 860–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A Receptors Potently Suppresses the Metastasis of CD73+ Tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [Green Version]
- Mittal, D.; Sinha, D.; Barkauskas, D.; Young, A.; Kalimutho, M.; Stannard, K.; Caramia, F.; Haibe-Kains, B.; Stagg, J.; Khanna, K.K.; et al. Adenosine 2B Receptor Expression on Cancer Cells Promotes Metastasis. Cancer Res. 2016, 76, 4372–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Blockade of A2b Adenosine Receptor Reduces Tumor Growth and Immune Suppression Mediated by Myeloid-Derived Suppressor Cells in a Mouse Model of Melanoma. Neoplasia 2013, 15, 1400–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekic, C.; Sag, D.; Li, Y.; Theodorescu, D.; Strieter, R.M.; Linden, J. Adenosine A2B Receptor Blockade Slows Growth of Bladder and Breast Tumors. J. Immunol. 2012, 188, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-Derived Suppressor Cells Contribute to A2B Adenosine Receptor-Induced VEGF Production and Angiogenesis in a Mouse Melanoma Model. Oncotarget 2015, 6, 27478–27489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Adenosine metabolism and transport in the extracellular and intracellular milieu. Abbreviations: ATP: adenosine triphosphate; ADP: adenosine diphosphate; AMP: adenosine monophosphate; ADA: adenosine deaminase; AK: adenosine kinase; ENTs: equilibrative nucleoside transporters; ARs: adenosine receptors.
Figure 1.
Adenosine metabolism and transport in the extracellular and intracellular milieu. Abbreviations: ATP: adenosine triphosphate; ADP: adenosine diphosphate; AMP: adenosine monophosphate; ADA: adenosine deaminase; AK: adenosine kinase; ENTs: equilibrative nucleoside transporters; ARs: adenosine receptors.

Figure 2.
Schematic representation of main actions mediated by adenosine receptors (ARs) in human immune cells. Abbreviations: TNF-α: tumor necrosis factor α; IL: interleukin; MIP: macrophage inflammatory protein; IFN-γ: interferon γ; TGF-β: tissue growth factor β; VEGF: vascular endothelial growth factor; IDO-1: indoleamine 2:3 dioxygenase; COX-2: cyclooxygenase 2; ROS: reactive oxygen species; NF-κB: nuclear factor κ-light-chain-enhancer of activated B cells; ↑: increase; ↓: decrease.
Figure 2.
Schematic representation of main actions mediated by adenosine receptors (ARs) in human immune cells. Abbreviations: TNF-α: tumor necrosis factor α; IL: interleukin; MIP: macrophage inflammatory protein; IFN-γ: interferon γ; TGF-β: tissue growth factor β; VEGF: vascular endothelial growth factor; IDO-1: indoleamine 2:3 dioxygenase; COX-2: cyclooxygenase 2; ROS: reactive oxygen species; NF-κB: nuclear factor κ-light-chain-enhancer of activated B cells; ↑: increase; ↓: decrease.


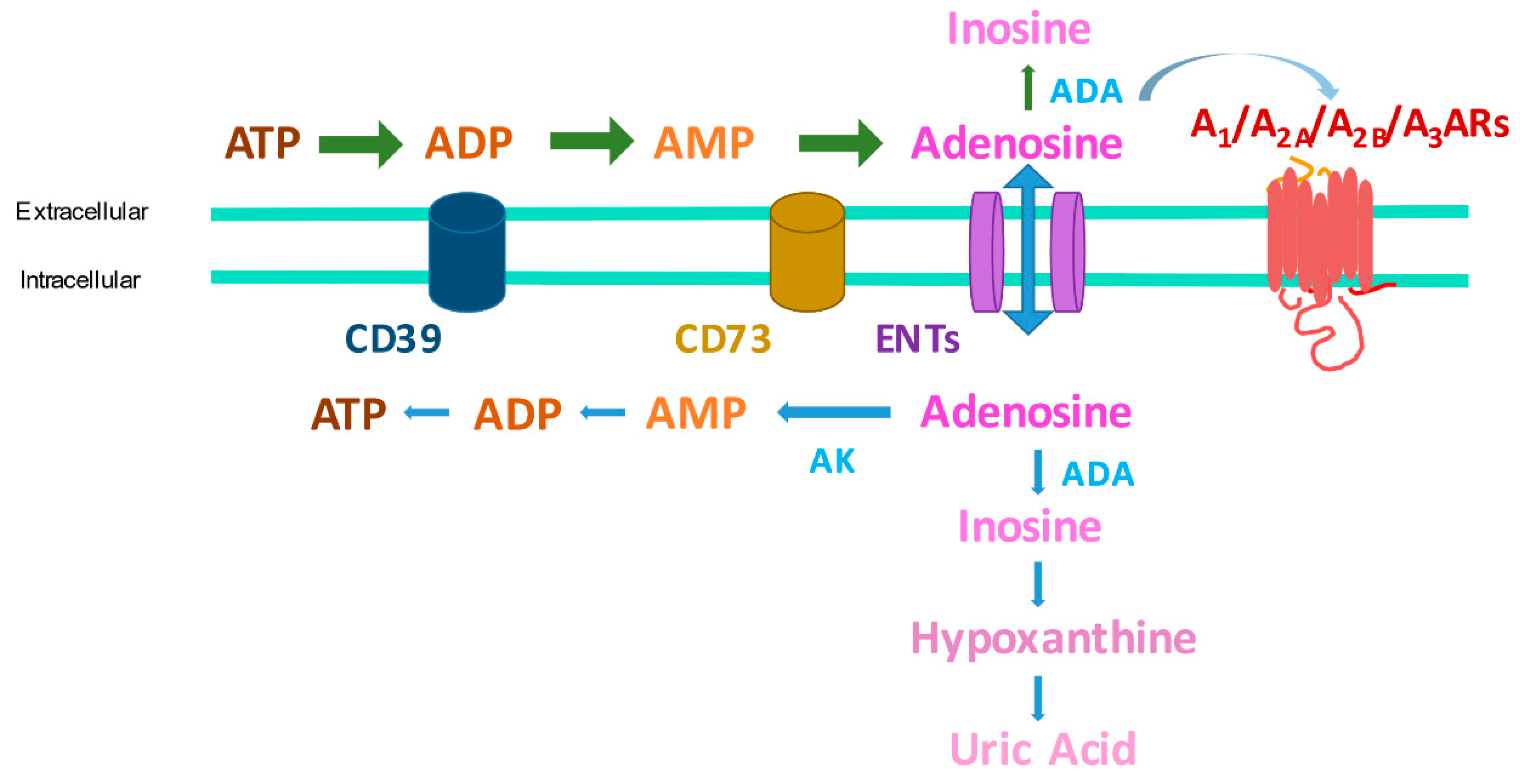{kind=link}
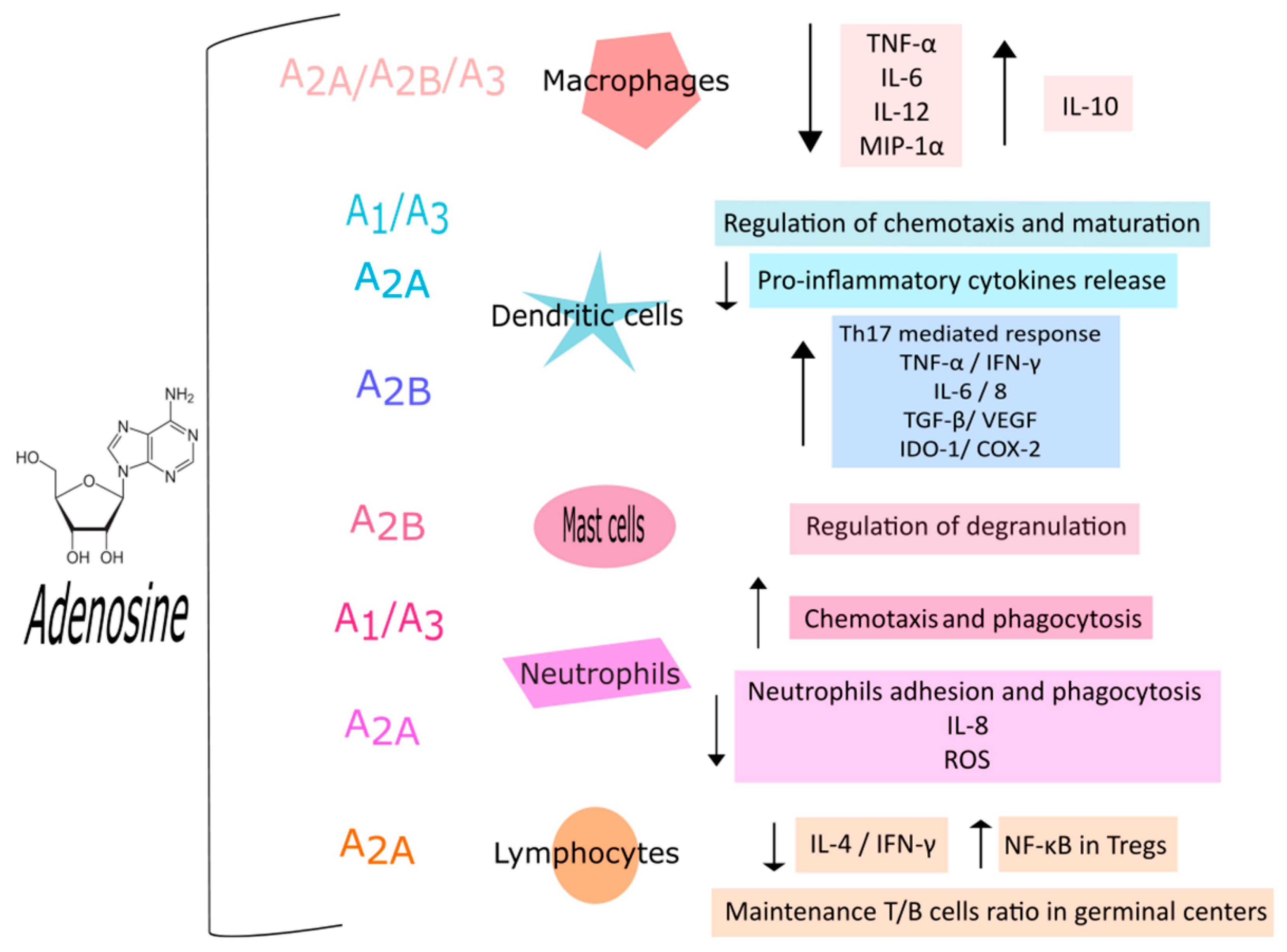{kind=link}
Table 1.
Current clinical trials of AR ligands (data from ClinicalTrials.gov, accessed 15 July 2021).
Table 1.
Current clinical trials of AR ligands (data from ClinicalTrials.gov, accessed 15 July 2021).
| Compound | Pharmacological Behavior | Condition | Phase | Identifier |
|---|---|---|---|---|
| Regadenoson | A2AAR agonist | Lung Transplantation | 1 | NCT03072589 |
| Regadenoson | A2AAR agonist | Lung Transplantation | 1 | NCT04521569 |
| Regadenoson | A2AAR agonist | Gliomas | 1 | NCT03971734 |
| Regadenoson | A2AAR agonist | COVID-19 | 1/2 | NCT04606069 |
| Regadenoson | A2AAR agonist | Heart Transplant | 4 | NCT03102125 |
| Ciforadenant | A2AAR antagonist | Incurable Cancers | 1 | NCT02655822 |
| Ciforadenant | A2AAR antagonist | Multiple Myeloma | 1 | NCT04280328 |
| Ciforadenant | A2AAR antagonist | Advanced Cancers | 1 | NCT03454451 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Metastatic Castrate Resistant Prostate Cancer | 1b/2 | NCT04381832 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Head and Neck Cancers | 1 | NCT04892875 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Non-Small Cell Lung Cancer | 2 | NCT04791839 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Non-Small Cell Lung Cancer | 2 | NCT04262856 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Advanced Cancers | 1 | NCT03629756 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Lung Cancer | 1 | NCT03846310 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Triple-Negative Breast Cancer or Gynecologic Malignancies | 1 | NCT03719326 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Colorectal Cancer | 1/2 | NCT04660812 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Prostate Cancer | 2 | NCT03821246 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Metastatic Pancreatic Ductal Adenocarcinoma | 1/2 | NCT03193190 |
| Etrumadenant (AB928) | A2A and A2B AR antagonist | Metastatic Colorectal Cancer | 1/2 | NCT03555149 |
| PBF-1129 | A2BAR antagonist | Advanced Non-Small Cell Lung Cancer | 1 | NCT03274479 |
| Namodenoson (CF102) | A3AR agonist | Non-Alcoholic Steatohepatitis | 2 | NCT04697810 |
| Piclidenoson (IB-MECA) | A3AR agonist | COVID-19 | 2 | NCT04333472 |
| CF101 (IB-MECA) | A3AR agonist | Plaque Psoriasis | 3 | NCT03168256 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pasquini, S.; Contri, C.; Borea, P.A.; Vincenzi, F.; Varani, K. Adenosine and Inflammation: Here, There and Everywhere. Int. J. Mol. Sci. 2021, 22, 7685. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147685
AMA Style
Pasquini S, Contri C, Borea PA, Vincenzi F, Varani K. Adenosine and Inflammation: Here, There and Everywhere. International Journal of Molecular Sciences. 2021; 22(14):7685. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147685
Chicago/Turabian StylePasquini, Silvia, Chiara Contri, Pier Andrea Borea, Fabrizio Vincenzi, and Katia Varani. 2021. "Adenosine and Inflammation: Here, There and Everywhere" International Journal of Molecular Sciences 22, no. 14: 7685. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147685
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.