
STAT1 Is Required for Decreasing Accumulation of Granulocytic Cells via IL-17 during Initial Steps of Colitis-Associated Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
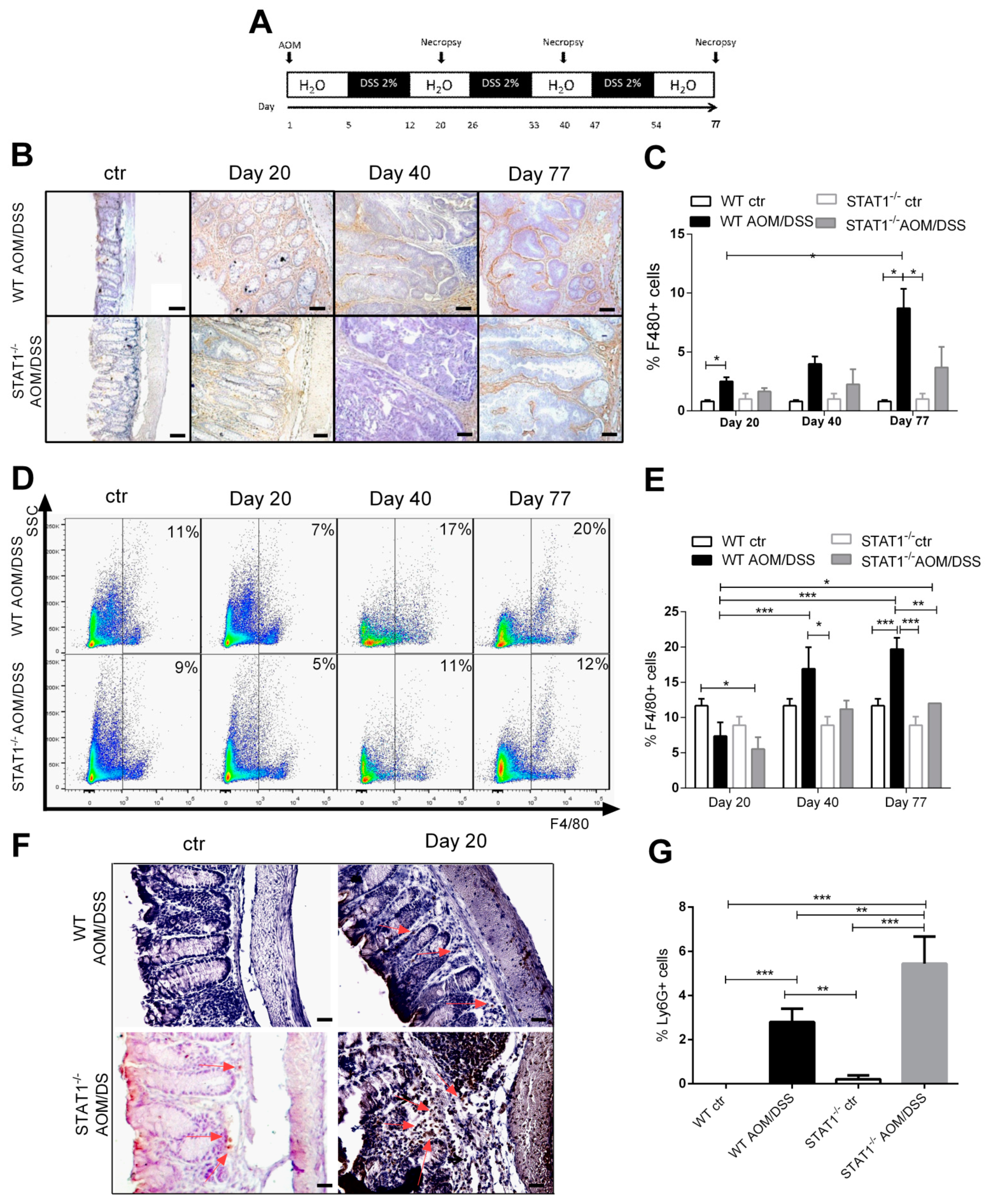
2.1. Deregulated Myeloid Cell Recruitment during Early Stages of Tumor Development in STAT1 Deficiency
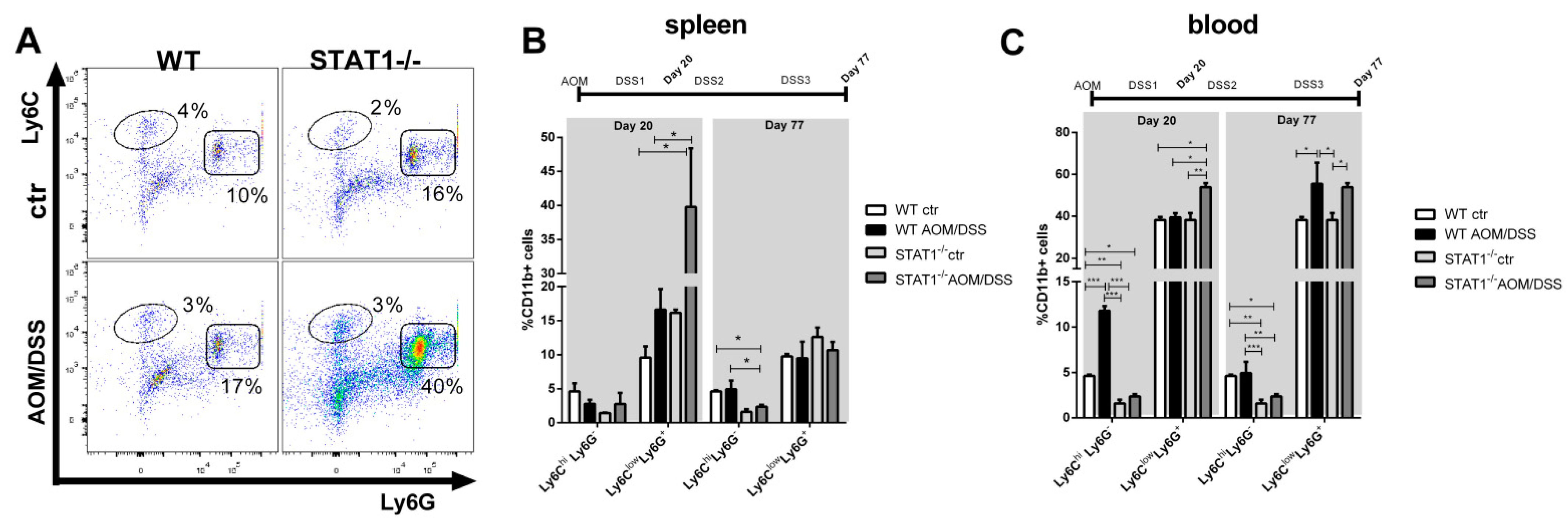
2.2. STAT1 Deficiency Increases the Accumulation of Myeloid-Derived Suppressor Cells (MDSCs) during Chronic Inflammation
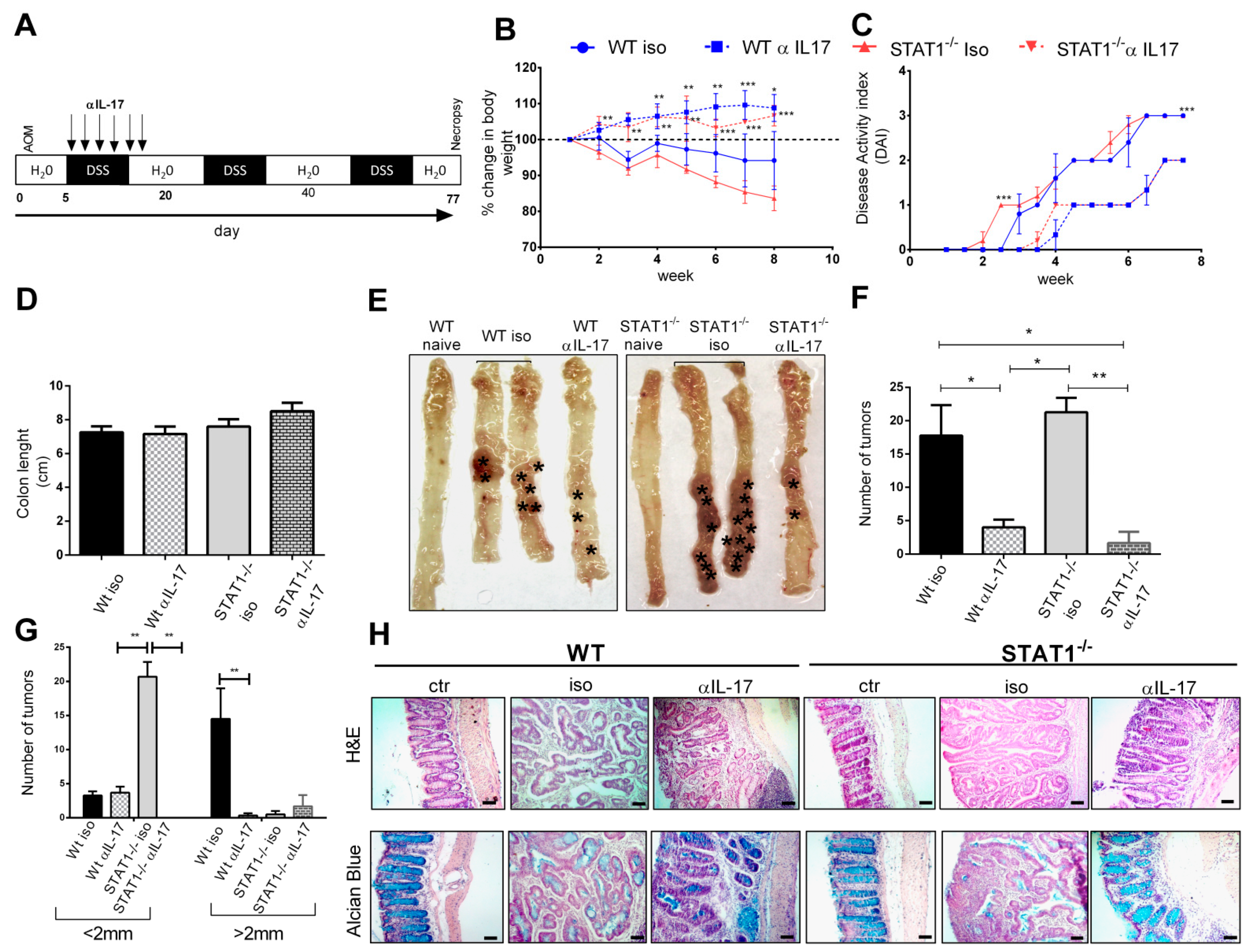
2.3. Early IL-17A Neutralization Inhibits Tumor Development in STAT1-/- Mice
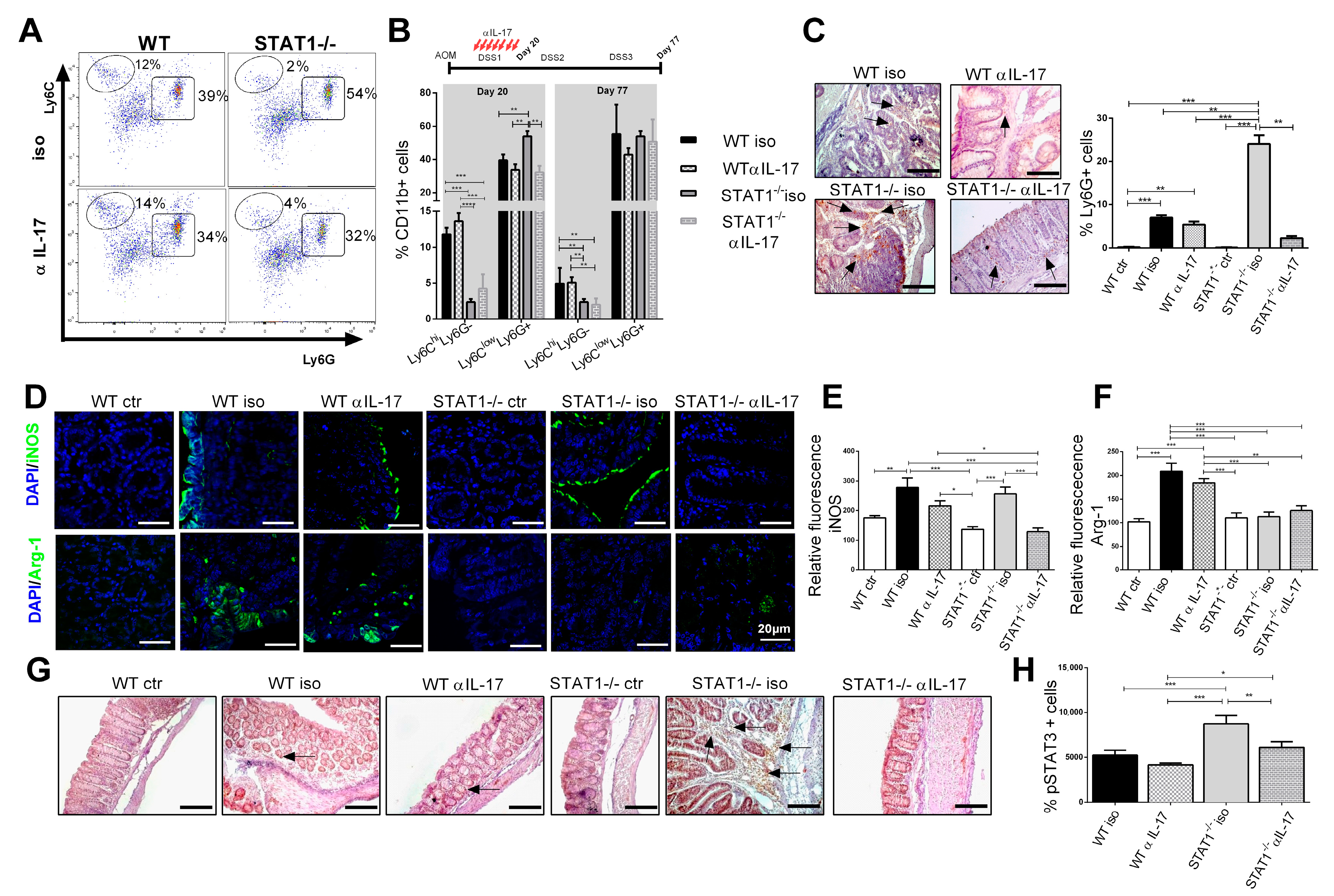
2.4. IL-17 Neutralization Decreases the Recruitment of Neutrophils and CD11b+Ly6ClowLy6G+ Cell Populations
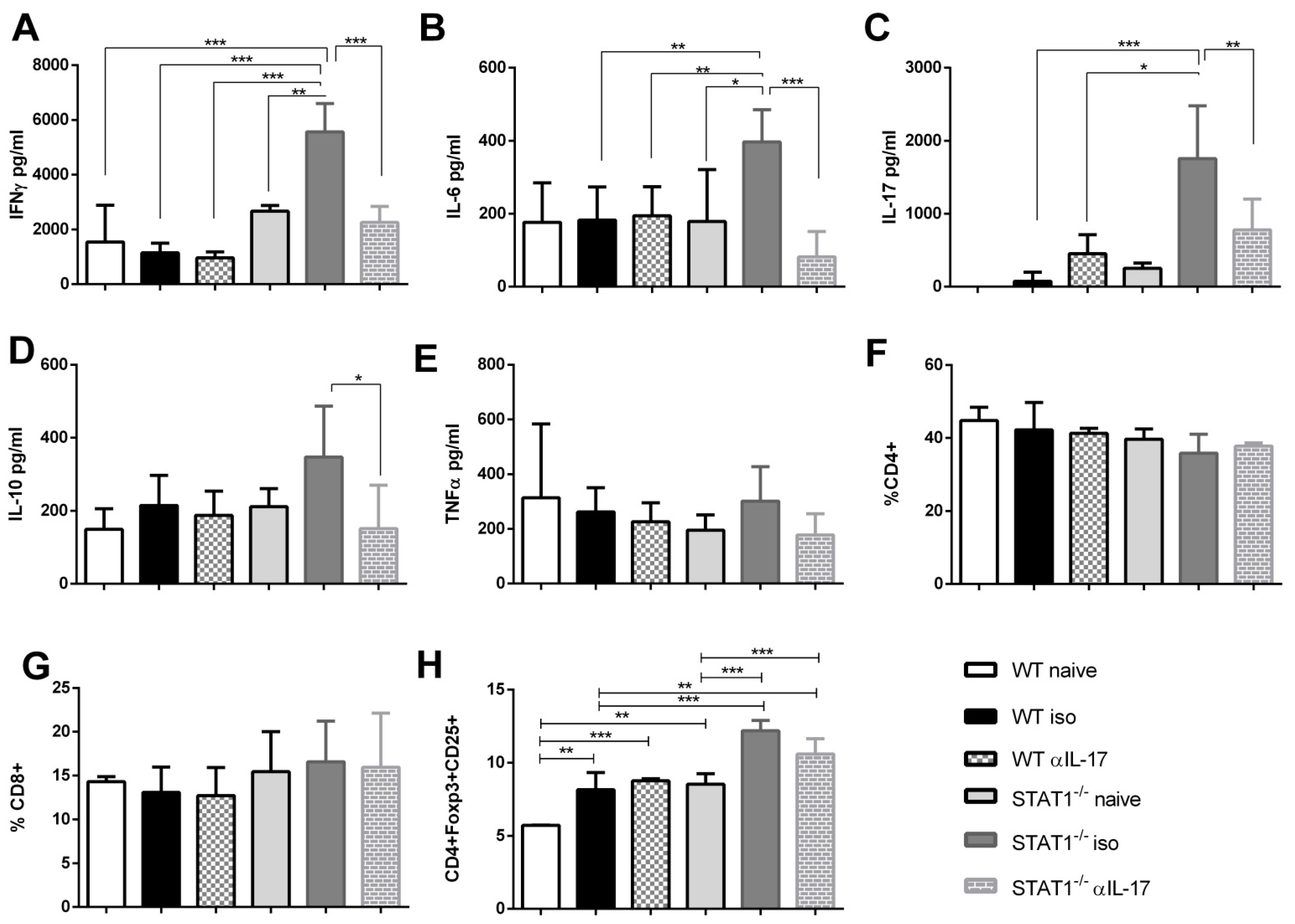
2.5. Enhanced Inflammatory Microenvironment in STAT1-/- Mice Is Controlled by IL-17 Blockade
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. CAC Induction
4.3. IL-17 Neutralization In Vivo
4.4. Histological Analysis
4.5. Flow Cytometry
4.6. Quantification of Cytokines
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Liu, Z. STAT1 in cancer: Friend or foe? Discov. Med. 2017, 24, 19–29. [Google Scholar] [PubMed]
- Meissl, K.; Macho-Maschler, S.; Muller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jess, T.; Winther, K.V.; Munkholm, P.; Langholz, E.; Binder, V. Mortality and causes of death in Crohn’s disease: Follow-up of a population-based cohort in Copenhagen County, Denmark. Gastroenterology 2002, 122, 1808–1814. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.A.D.; Al-Attar, A.; Watson, N.F.S.; Scholefield, J.H.; Ilyas, M.; Durrant, L.G. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 2010, 59, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Gordziel, C.; Bratsch, J.; Moriggl, R.; Knösel, T.; Friedrich, K. Both STAT1 and STAT3 are favourable prognostic determinants in colorectal carcinoma. Br. J. Cancer 2013, 109, 138–146. [Google Scholar] [CrossRef]
- Slattery, M.L.; Lundgreen, A.; Kadlubar, S.A.; Bondurant, K.L.; Wolff, R.K. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol. Carcinog. 2013, 52, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Nivarthi, H.; Gordziel, C.; Themanns, M.; Kramer, N.; Eberl, M.; Rabe, B.; Schlederer, M.; Rose-John, S.; Knösel, T.; Kenner, L.; et al. The ratio of STAT1 to STAT3 expression is a determinant of colorectal cancer growth. Oncotarget 2016, 7, 51096–51106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Cabrera, S.; Vázquez-Sandoval, A.; Molina-Guzman, E.; Delgado-Ramirez, Y.; Delgado-Buenrostro, N.L.; Callejas, B.E.; Chirino, Y.I.; Pérez-Plasencia, C.; Rodríguez-Sosa, M.; Olguín, J.E.; et al. Deficiency in STAT1 Signaling Predisposes Gut Inflammation and Prompts Colorectal Cancer Development. Cancers 2018, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Fu, X.Y.; Plate, J.; Chong, A.S. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosis: Requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998, 58, 2832–2837. [Google Scholar]
- Kim, H.S.; Lee, M.-S. STAT1 as a key modulator of cell death. Cell. Signal. 2007, 19, 454–465. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Yun, H.; Lai, R.; Su, M. Correlation of STAT1 with Apoptosis and Cell-Cycle Markers in Esophageal Squamous Cell Carcinoma. PLoS ONE 2014, 9, e113928. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.; Qiu, Y.; Yu, H.; Kong, Q.; Zhen, B. Interleukin-17 promotes the development of cisplatin resistance in colorectal cancer. Oncol. Lett. 2018, 17, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Razi, S.; Noveiry, B.B.; Keshavarz-Fathi, M.; Rezaei, N. IL-17 and colorectal cancer: From carcinogenesis to treatment. Cytokine 2019, 116, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Duan, Y.; Cheng, X.; Chen, X.; Xie, W.; Long, H.; Lin, Z.; Zhu, B. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem. Biophys. Res. Commun. 2011, 407, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Cheng, Q.; Cai, Y.; Gong, H.; Wu, Y.; Yu, X.; Shi, L.; Wu, D.; Dong, C.; Liu, H. IL-17A produced by gammadelta T cells promotes tumor growth in hepatocellular carcinoma. Cancer Res. 2014, 74, 1969–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, J.; Wang, W.; Tian, J.; Yin, K.; Tang, X.; Ma, J.; Xu, H.; Wang, S. IL-17A produced by peritoneal macrophages promote the accumulation and function of granulocytic myeloid-derived suppressor cells in the development of colitis-associated cancer. Tumor Biol. 2016, 37, 15883–15891. [Google Scholar] [CrossRef]
- Karin, N. The Development and Homing of Myeloid-Derived Suppressor Cells: From a Two-Stage Model to a Multistep Narrative. Front. Immunol. 2020, 11, 557586. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer. Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, M.L.; Kumar, V.; Martner, A.; Mony, S.; Donthireddy, L.; Condamine, T.; Seykora, J.; Knight, S.C.; Malietzis, G.; Lee, G.H.; et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17–producing CD4+ T cells. J. Exp. Med. 2015, 212, 351–367. [Google Scholar] [CrossRef]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6–Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufert, C.; Becker, C.; Wirtz, S.; Fantini, M.C.; Weigmann, B.; Galle, P.R.; Neurath, M.F. IL-27 controls the development of inducible regulatory T cells and Th17 cells via differential effects on STAT1. Eur. J. Immunol. 2007, 37, 1809–1816. [Google Scholar] [CrossRef]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 Promotes Tumor Development through the Induction of Tumor Promoting Microenvironments at Tumor Sites and Myeloid-Derived Suppressor Cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.; Yang, H.; Zhao, J.; Yuan, A.; Florholmen, J. Elevated Proinflammatory Cytokine IL-17A in the Adjacent Tissues Along the Adenoma-Carcinoma Sequence. Pathol. Oncol. Res. 2014, 21, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Qu, Y.; Leng, Y.; Sun, W.; Ma, S.; Wei, J.; Hu, J.; Zhang, X. Human colon carcinogenesis is associated with increased interleukin-17-driven inflammatory responses. Drug Des. Dev. Ther. 2015, 9, 1679–1689. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. Gammadeltat17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufman, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 Activation in Colon Cancer Is Associated with Enhanced Cell Proliferation and Tumor Growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, N.; Anderson, K.; Volpedo, G.; Hamza, O.; Varikuti, S.; Satoskar, A.R.; Oghumu, S. STAT1 inhibits T-cell exhaustion and myeloid derived suppressor cell accumulation to promote antitumor immune responses in head and neck squamous cell car-cinoma. Int. J. Cancer 2020, 146, 1717–1729. [Google Scholar] [CrossRef]
- Friedrich, J.; Heim, L.; Trufa, D.I.; Sirbu, H.; Rieker, R.J.; Chiriac, M.T.; Finotto, S. STAT1 deficiency supports PD-1/PD-L1 signaling resulting in dysfunctional TNFalpha mediated immune responses in a model of NSCLC. Oncotarget 2018, 9, 37157–37172. [Google Scholar] [CrossRef] [Green Version]
- Flood, B.; Manils, J.; Nulty, C.; Flis, E.; Kenealy, S.; Barber, G.; Fay, J.; Mills, K.; Kay, E.W.; Creagh, E.M. Caspase-11 regulates the tumour suppressor function of STAT1 in a murine model of colitis-associated carcinogenesis. Oncogene 2019, 38, 2658–2674. [Google Scholar] [CrossRef] [Green Version]
- A Choi, E.; Lei, H.; Maron, D.J.; Wilson, J.M.; Barsoum, J.; Fraker, D.L.; El-Deiry, W.; Spitz, F.R. Stat1-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand and the cell-surface death signaling pathway by interferon beta in human cancer cells. Cancer Res. 2003, 63, 5299–5307. [Google Scholar]
- Dimco, G.; Knight, R.A.; Latchman, D.S.; Stephanou, A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle 2010, 9, 4638–4649. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Darini, C.; Désaubry, L.; Koromilas, A.E. STAT1 Promotes KRAS Colon Tumor Growth and Susceptibility to Pharmacological Inhibition of Translation Initiation Factor eIF4A. Mol. Cancer Ther. 2016, 15, 3055–3063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, U.; Longley, D.B.; Galligan, L.; Allen, W.; Wilson, T.; Johnston, P.G. Effect of p53 status and STAT1 on chemo-therapy-induced, Fas-mediated apoptosis in colorectal cancer. Cancer Res. 2005, 65, 8951–8960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Zhou, Y.; Ogawa, M.; Shia, J.; Klimstra, D.S.; Wang, J.Y.; Roehrl, M.H. STAT1 as a potential prognosis marker for poor outcomes of early stage colorectal cancer with microsatellite instability. PLoS ONE 2020, 15, e0229252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallina, G.; Dolcetti, L.; Serafini, P.; De Santo, C.; Marigo, I.; Colombo, M.P.; Basso, G.; Brombacher, F.; Borrello, I.; Zanovello, P.; et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Investig. 2006, 116, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. STAT1 Signaling Regulates Tumor-Associated Macrophage-Mediated T Cell Deletion. J. Immunol. 2005, 174, 4880–4891. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.-Q.; Hu, X.-W.; Liu, Y.-L.; Ye, Z.-J.; Gui, Y.-H.; Zhou, D.-L.; Qi, C.-L.; He, X.-D.; Wang, H.; Wang, L.-J. CD11b deficiency suppresses intestinal tumor growth by reducing myeloid cell recruitment. Sci. Rep. 2015, 5, 15948. [Google Scholar] [CrossRef]
- Varikuti, S.; Oghumu, S.; Elbaz, M.; Volpedo, G.; Ahirwar, D.K.; Alarcon, P.C.; Sperling, R.H.; Moretti, E.; Pioso, M.S.; Kimble, J.; et al. STAT1 gene deficient mice develop accelerated breast cancer growth and metastasis which is reduced by IL-17 blockade. Oncoimmunology 2017, 6, e1361088. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, J.; Wang, J.; Vangundy, Z.; You, J.; Yildiz, V.; Yu, L.B.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4(+) T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7, 15424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Echeverz, J.; Haile, L.A.; Zhao, F.; Gamrekelashvili, J.; Ma, C.; Metais, J.Y.; Dunbar, C.E.; Kapoor, V.; Manns, M.P.; Korangy, F.; et al. IFN-gamma regulates survival and function of tumor-induced CD11b(+)Gr-1(high) myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur. J. Immunol. 2014, 44, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Hyun, Y.S.; Han, D.S.; Lee, A.; Eun, C.S.; Youn, J.; Kim, H.-Y. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012, 33, 931–936. [Google Scholar] [CrossRef]
- Wu, D.; Wu, P.; Huang, Q.; Liu, Y.; Ye, J.; Huang, J. Interleukin-17: A Promoter in Colorectal Cancer Progression. Clin. Dev. Immunol. 2013, 2013, 436307. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Kim, M.K.; Di Caro, G.; Wong, J.; Shalapour, S.; Wan, J.; Zhang, W.; Zhong, Z.; Sanchez-Lopez, E.; Wu, L.-W.; et al. Interleukin-17 Receptor A Signaling in Transformed Enterocytes Promotes Early Colorectal Tumorigenesis. Immunity 2014, 41, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consonni, F.M.; Porta, C.; Marino, A.; Pandolfo, C.; Mola, S.; Bleve, A.; Sica, A. Myeloid-Derived Suppressor Cells: Ductile Targets in Disease. Front. Immunol. 2019, 10, 949. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, A.M.; Bian, Z.; Culpepper, C.; Liu, Y. Induction of arginase-1 in MDSC requires exposure to CD3/CD28 activated T cells. J. Immunol. 2017, 198, 154. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Ramirez, Y.; Baltazar-Perez, I.; Martinez, Y.; Callejas, B.E.; Medina-Andrade, I.; Olguín, J.E.; Delgado-Buenrostro, N.L.; Chirino, Y.I.; Terrazas, L.I.; Leon-Cabrera, S. STAT1 Is Required for Decreasing Accumulation of Granulocytic Cells via IL-17 during Initial Steps of Colitis-Associated Cancer. Int. J. Mol. Sci. 2021, 22, 7695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147695
Delgado-Ramirez Y, Baltazar-Perez I, Martinez Y, Callejas BE, Medina-Andrade I, Olguín JE, Delgado-Buenrostro NL, Chirino YI, Terrazas LI, Leon-Cabrera S. STAT1 Is Required for Decreasing Accumulation of Granulocytic Cells via IL-17 during Initial Steps of Colitis-Associated Cancer. International Journal of Molecular Sciences. 2021; 22(14):7695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147695
Chicago/Turabian StyleDelgado-Ramirez, Yael, Itzel Baltazar-Perez, Yamileth Martinez, Blanca E. Callejas, Itzel Medina-Andrade, Jonadab E. Olguín, Norma L. Delgado-Buenrostro, Yolanda I. Chirino, Luis I. Terrazas, and Sonia Leon-Cabrera. 2021. "STAT1 Is Required for Decreasing Accumulation of Granulocytic Cells via IL-17 during Initial Steps of Colitis-Associated Cancer" International Journal of Molecular Sciences 22, no. 14: 7695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147695