
Investigating LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
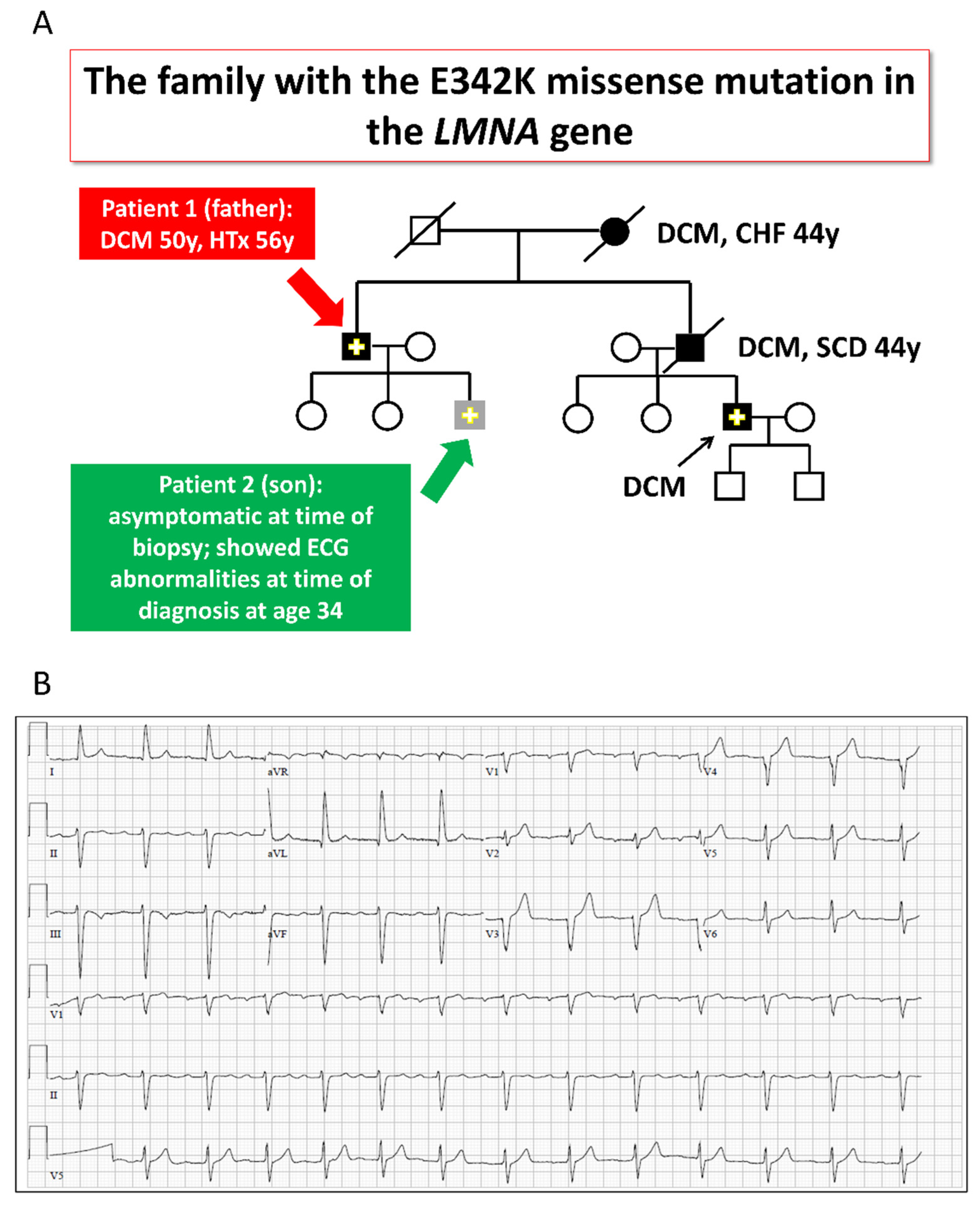
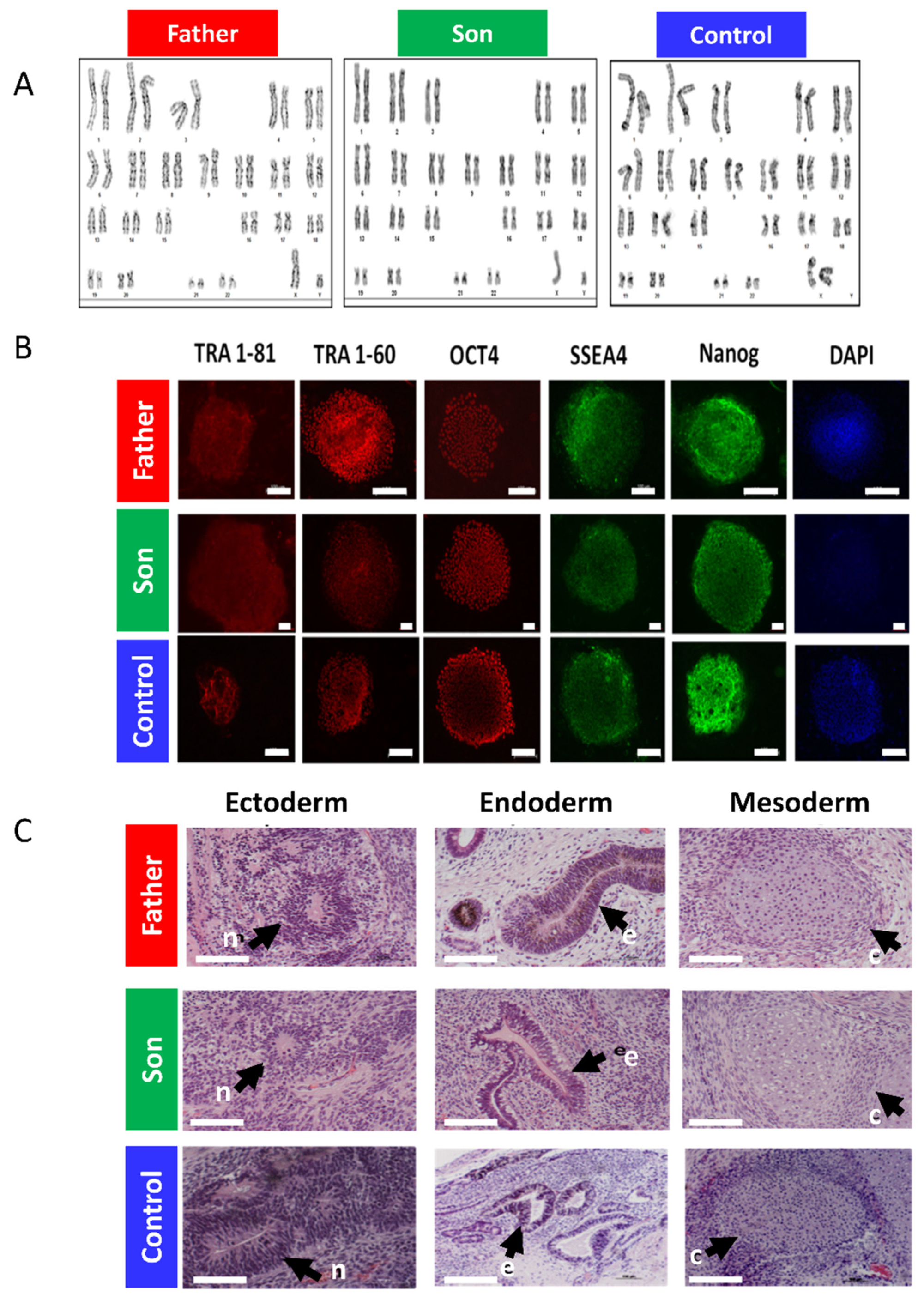
2.1. The LMNA-Mutated Patients and iPSCs Characteristics
2.2. Electrophysiological Abnormalities and Arrhythmias in LMNA-Mutated iPSC-CMs
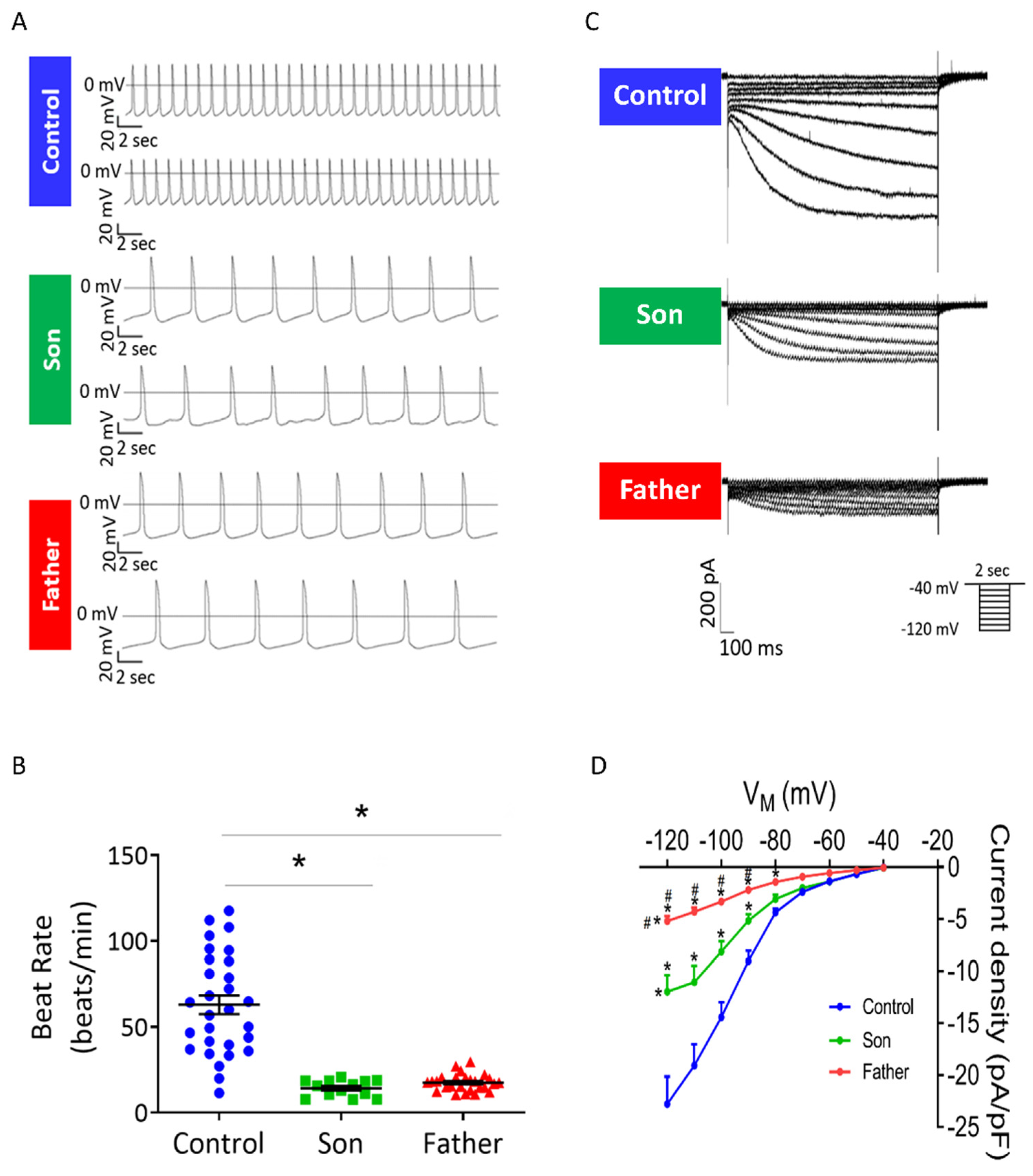
2.2.1. Decreased Spontaneous Beat Rate and If in LMNA-Mutated Cardiomyocytes
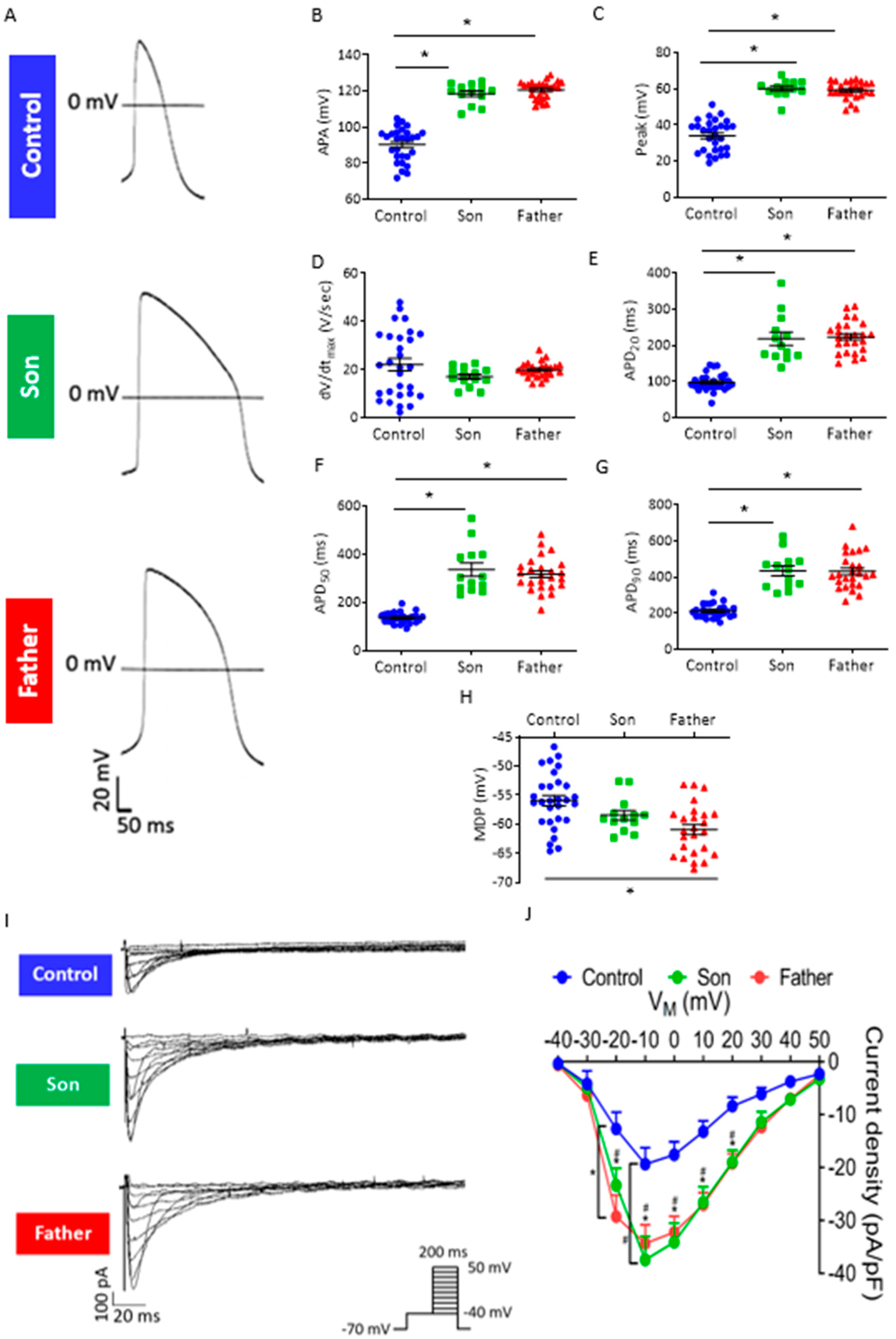
2.2.2. Prolonged Action Potential Duration and Increased ICa,L in LMNA-Mutated Cardiomyocytes
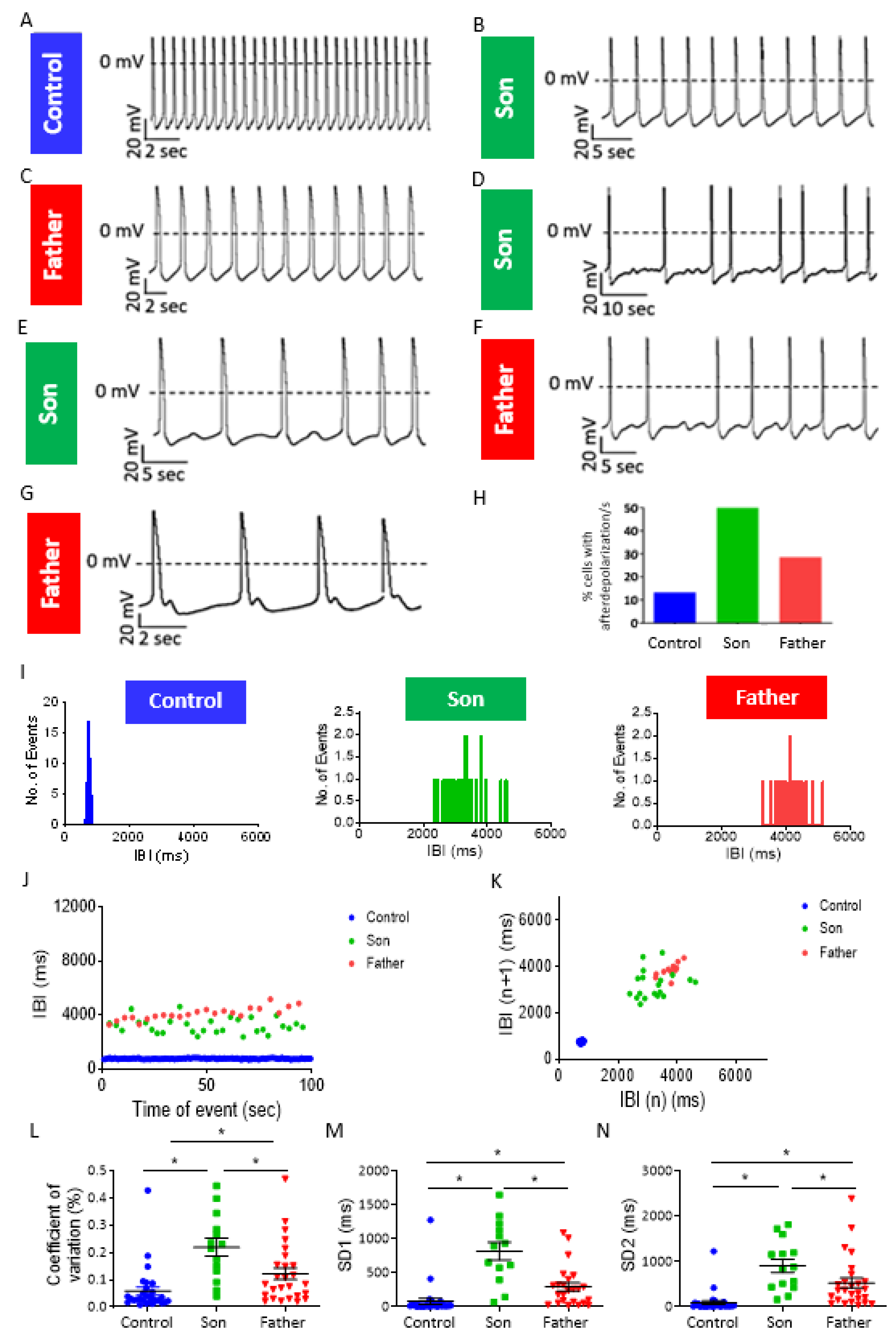
2.3. LMNA-Mutated Cardiomyocytes Are Arrhythmogenic
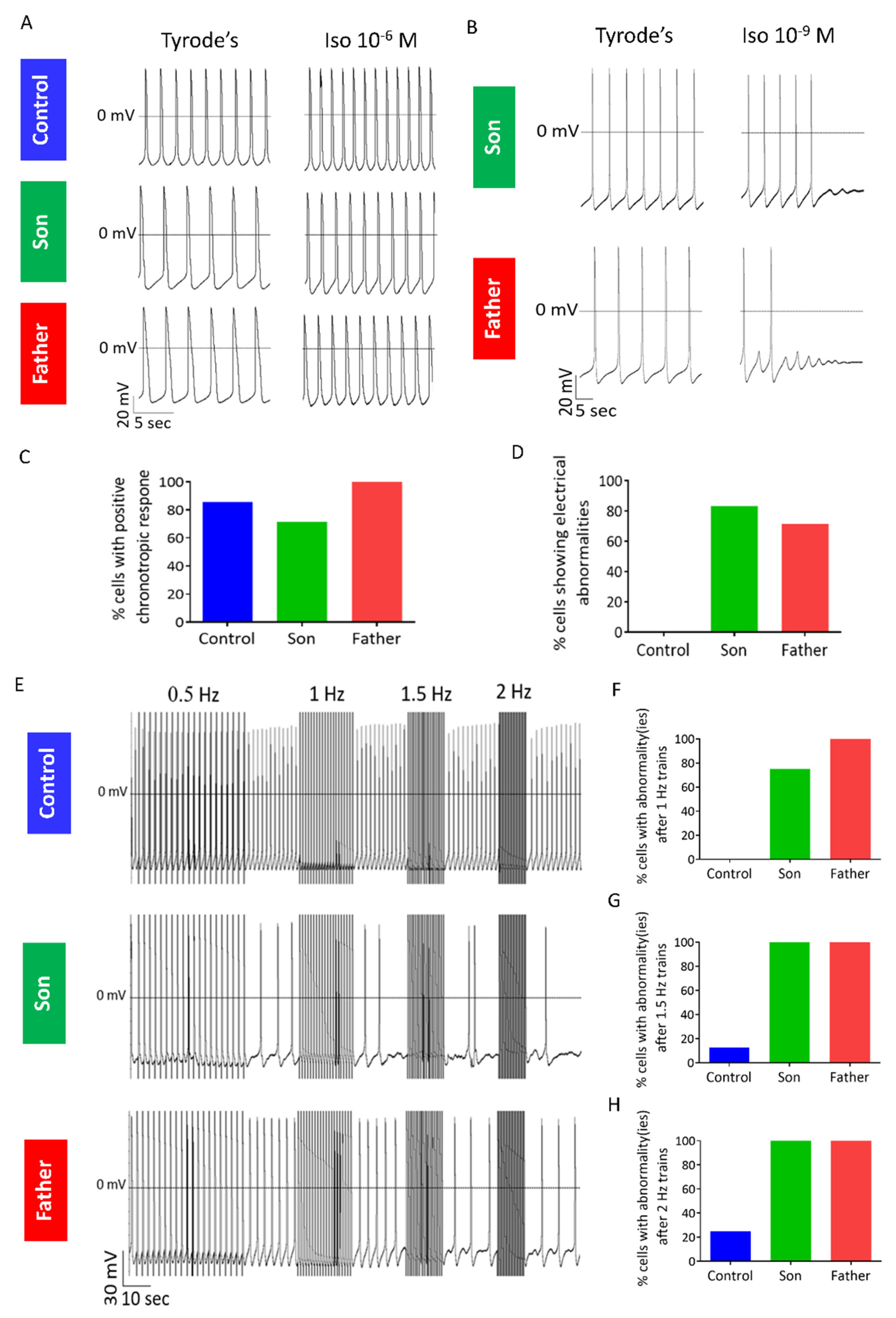
2.3.1. Abnormal Response to β-Adrenergic Stimulation in LMNA-Mutated iPSC-CMs
2.3.2. Abnormal Response to Rapid Pacing in LMNA-Mutated iPSC-CMs
2.3.3. Abnormal Caffeine-Induced Ca2+ Release in LMNA-Mutated Cardiomyocytes
2.3.4. KB-R7943 Eliminates DADs in LMNA-Mutated Cardiomyocytes
2.3.5. Investigating the Anti-Arrhythmic Efficacy of Ranolazine
2.4. Structural and Gene Expression Models in LMNA-Mutated iPSC-CMs
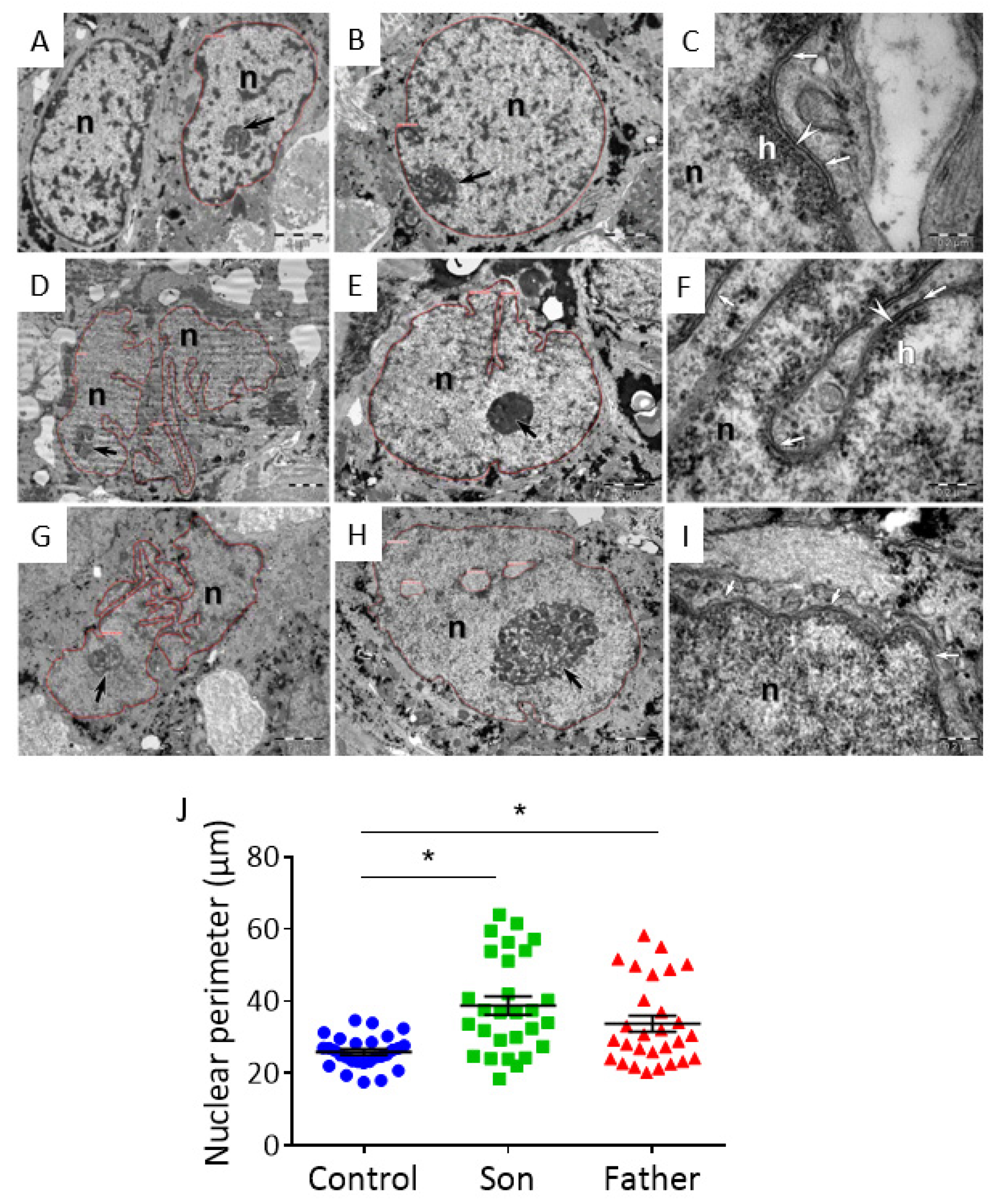
2.4.1. Ultrastructural Changes in LMNA-Mutated iPSC-CMs
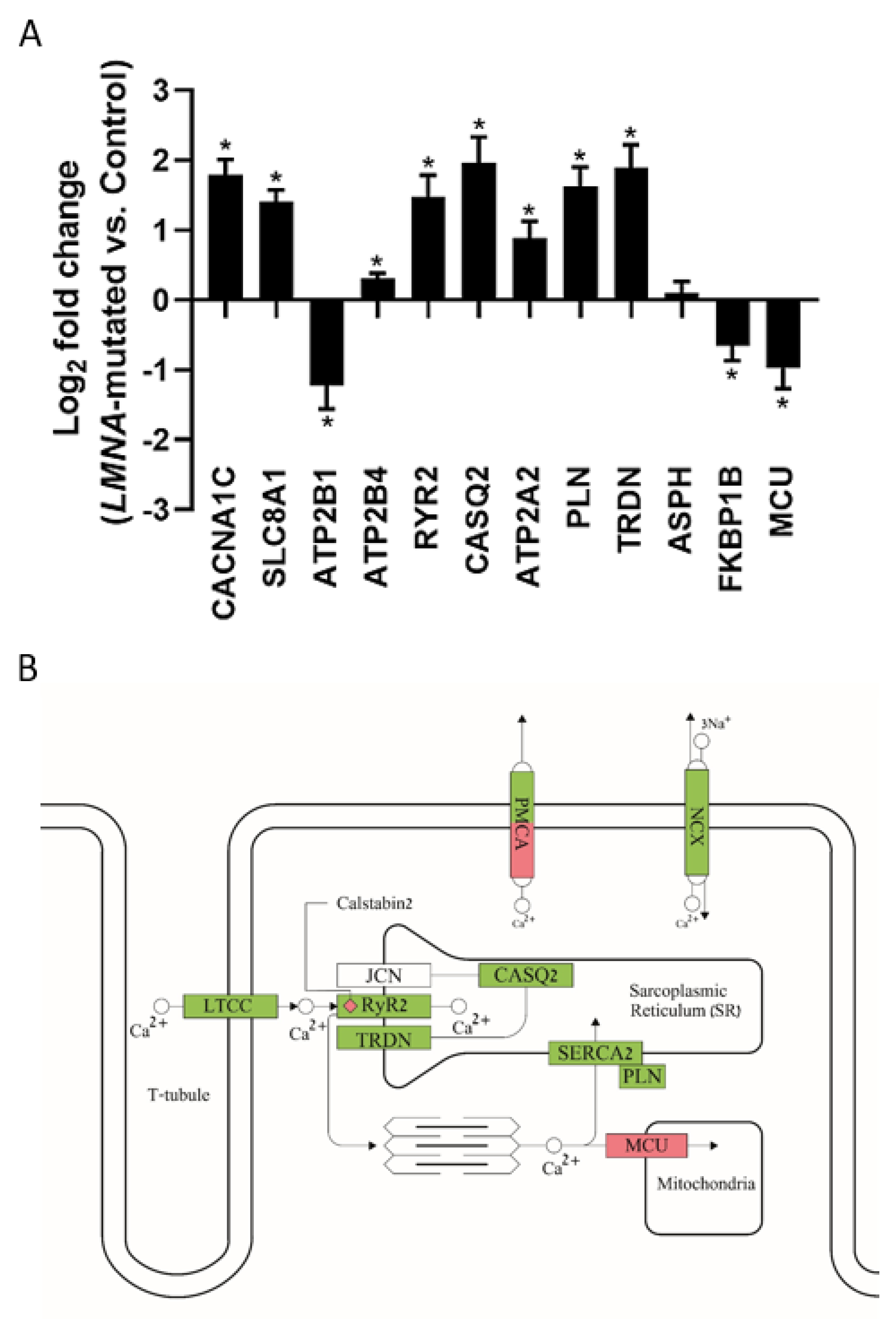
2.4.2. Gene Expression Alterations in LMNA-Mutated iPSC-CMs
3. Discussion
3.1. Electrophysiological Abnormalities in LMNA-Mutated Cardiomyocytes
3.1.1. Decreased Spontaneous Beat Rate and If Density
3.1.2. Altered Action Potential Parameters and Increased ICa,L Density
3.1.3. Arrhythmias and DADs
3.1.4. Altered Response to β-Adrenergic Stimulation, Rapid Pacing and Caffeine Application in LMNA-Mutated Cardiomyocytes
3.1.5. KB-R7943 Eliminates DADs in LMNA-Mutated Cardiomyocytes
3.1.6. Ranolazine Does Not Eliminate DADs and Arrhythmias in LMNA-Mutated Cardiomyocytes
3.2. The Probable Association between LMNA Mutations and Electrophysiological Abnormalities
3.2.1. Ultrastructural Abnormalities in LMNA-Mutated iPSC-CMs
3.2.2. Gene Expression Alterations in LMNA-Mutated iPSC-CMs
3.3. Proposed Mechanism of Arrhythmias in LMNA-Mutated iPSC-CMs
4. Materials and Methods
4.1. LMNA-Mutated Dermal Fibroblasts
4.2. Generation of LMNA-Mutated iPSCs
4.3. Karyotype Analysis
4.4. Genotyping
4.5. Immunofluorescence Staining
4.6. Teratomas Formation
4.7. Exome Sequencing
4.8. Culturing and Differentiation of iPSCs into Cardiomyocytes
4.9. Action Potentials, If and ICa,L Recording and Analysis
4.10. Micro-Electrode-Array (MEA) Recordings
4.11. Measurements of Intracellular Ca2+ Transients
4.12. Transmission Electron Microscopy (TEM)
4.13. RNA Sequencing
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamat, A.K.; Rocchi, M.; Smith, D.I.; Miller, O.J. Lamin A/C gene and a related sequence map to human chromosomes 1q12.1-q23 and 10. Somat. Cell Mol. Genet. 1993, 19, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Hutchison, C.J.; Worman, H.J. A-type lamins: Guardians of the soma? Nat. Cell. Biol. 2004, 6, 1062–1067. [Google Scholar] [CrossRef]
- Davidson, P.M.; Lammerding, J. Broken nuclei—Lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puckelwartz, M.J.; Depreux, F.F.S.; McNally, E.M. Gene expression, chromosome position and lamin A/C mutations. Nucleus 2011, 2, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.; Antoku, S.; Östlund, C.; Worman, H.J.; Gundersen, G.G. Linker of nucleoskeleton and cytoskeleton (LINC) complex-mediated actin-dependent nuclear positioning orients centrosomes in migrating myoblasts. Nucleus 2015, 6, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur. Heart J. 2012, 33, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Morales, A. LMNA-Related Dilated Cardiomyopathy. Available online: http://www.genetests.org (accessed on 1 July 2021).
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Cronk, L.B.; Ye, B.; Kaku, T.; Tester, D.J.; Vatta, M.; Makielski, J.C.; Ackerman, M.J. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007, 4, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghouse, J.; Have, C.T.; Weeke, P.; Bille Nielsen, J.; Ahlberg, G.; Balslev-Harder, M.; Appel, E.V.; Skaaby, T.; Olesen, S.P.; Grarup, N.; et al. Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. Eur. Heart J. 2015, 36, 2523–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedley, P.L.; Kanters, J.K.; Dembic, M.; Jespersen, T.; Skibsbye, L.; Aidt, F.H.; Eschen, O.; Graff, C.; Behr, E.R.; Schlamowitz, S.; et al. The role of CAV3 in long-QT syndrome: Clinical and functional assessment of a caveolin-3/Kv11.1 double heterozygote versus caveolin-3 single heterozygote. Circ. Cardiovasc. Genet. 2013, 6, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadafora, P.; Liguori, M.; Andreoli, V.; Quattrone, A.; Gambardella, A. CAV3 T78M mutation as polymorphic variant in South Italy. Neuromuscul. Disord. 2012, 22, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.; Dickenson, D.R.; Beatch, G.N. Kinetics of rate-dependent shortening of action potential duration in guinea-pig ventricle; effects of IK1 and IKr blockade. Br. J. Pharmacol. 1999, 126, 1426–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jost, N.; Acsai, K.; Horváth, B.; Bányász, T.; Baczkó, I.; Bitay, M.; Bogáts, G.; Nánási, P.P. Contribution of IKr and IK1 to ventricular repolarization in canine and human myocytes: Is there any influence of action potential duration? Basic Res. Cardiol. 2009, 104, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Michler, K.; Johnston, P.; MacFarlane, P.W. A comparison of commonly used QT correction formulae: The effect of heart rate on the QTc of normal ECGs. J. Electrocardiol. 2004, 37, 81–90. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of β-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Naga Prasad, S.V.; Nienaber, J.; Rockman, H.A. β-Adrenergic axis and heart disease. Trends Genet. 2001, 17, S44–S49. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Matsa, E.; Rajamohan, D.; Dick, E.; Young, L.; Mellor, I.; Staniforth, A.; Denning, C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur. Heart J. 2011, 32, 952–962. [Google Scholar] [CrossRef] [Green Version]
- Novak, A.; Barad, L.; Zeevi-Levin, N.; Shick, R.; Shtrichman, R.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to $β$-adrenergic stimulation. J. Cell. Mol. Med. 2012, 16, 468–482. [Google Scholar] [CrossRef]
- Johnson, D.M.; Antoons, G. Arrhythmogenic mechanisms in heart failure: Linking β-adrenergic stimulation, stretch, and calcium. Front. Physiol. 2018, 9, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Trafford, A.W.; Hutchings, D.C. The control of diastolic calcium in the heart: Basic mechanisms and functional implications. Circ. Res. 2020, 126, 395–412. [Google Scholar] [CrossRef]
- Ottolia, M.; Torres, N.; Bridge, J.H.B.; Philipson, K.D.; Goldhaber, J.I. Na/Ca exchange and contraction of the heart. J Mol Cell Cardiol 2013, 61, 28–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pott, C.; Eckardt, L.; Goldhaber, J.I. Triple threat: The Na+/Ca2+ exchanger in the pathophysiology of cardiac arrhythmia, ischemia and heart failure. Curr. Drug Targets 2011, 12, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markandeya, Y.S.; Tsubouchi, T.; Hacker, T.A.; Wolff, M.R.; Belardinelli, L.; Balijepalli, R.C. Inhibition of late sodium current attenuates ionic arrhythmia mechanism in ventricular myocytes expressing LaminA-N195K mutation. Heart Rhythm 2016, 13, 2228–2236. [Google Scholar] [CrossRef] [Green Version]
- Worman, H.J.; Courvalin, J.-C. How do mutations in lamins A and C cause disease? J. Clin. Invest. 2004, 113, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Lammerding, J. Lamins at a glance. J. Cell Sci. 2012, 125, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Baruscotti, M.; Barbuti, A.; Bucchi, A. The cardiac pacemaker current. J. Mol. Cell Cardiol. 2010, 48, 55–64. [Google Scholar] [CrossRef]
- Milanesi, R.; Baruscotti, M.; Gnecchi-Ruscone, T.; DiFrancesco, D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 2006, 354, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nof, E.; Luria, D.; Brass, D.; Marek, D.; Lahat, H.; Reznik-Wolf, H.; Pras, E.; Dascal, N.; Eldar, M.; Glikson, M. Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation 2007, 116, 463–470. [Google Scholar] [CrossRef] [Green Version]
- DiFrancesco, D. Funny channel-based pacemaking. Heart Rhythm 2010, 7, 276–279. [Google Scholar] [CrossRef]
- Baruscotti, M.; Bucchi, A.; Viscomi, C.; Mandelli, G.; Consalez, G.; Gnecchi-Rusconi, T.; Montano, N.; Casali, K.R.; Micheloni, S.; Barbuti, A.; et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc. Natl. Acad. Sci. USA 2011, 108, 1705–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.M.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson–Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, A.; Fields, P.A.; Smith, A.S.T.; Leonard, A.; Beussman, K.; Sniadecki, N.J.; Kim, D.H.; Tse, H.F.; Pabon, L.; Shendure, J.; et al. Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J. Cell Biol. 2019, 218, 2919–2944. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Termglinchan, V.; Diecke, S.; Itzhaki, I.; Lam, C.K.; Garg, P.; Lau, E.; Greenhaw, M.; Seeger, T.; Wu, H.; et al. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 2019, 572, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Schillinger, W.; Fiolet, J.W.; Schlotthauer, K.; Hasenfuss, G. Relevance of Na+–Ca2+ exchange in heart failure. Cardiovasc. Res. 2003, 57, 921–933. [Google Scholar] [CrossRef]
- Shryock, J.C.; Song, Y.; Rajamani, S.; Antzelevitch, C.; Belardinelli, L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc. Res 2013, 99, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Grewal, S.I.S.; Moazed, D. Heterochromatin and epigenetic control of gene expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Yehezkel, S.; Rebibo-Sabbah, A.; Segev, Y.; Tzukerman, M.; Shaked, R.; Huber, I.; Gepstein, L.; Skorecki, K.; Selig, S. Reprogramming of telomeric regions during the generation of human induced pluripotent stem cells and subsequent differentiation into fibroblast-like derivatives. Epigenetics 2011, 6, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, A.; Barad, L.; Lorber, A.; Gherghiceanu, M.; Reiter, I.; Eisen, B.; Eldor, L.; Itskovitz-Eldor, J.; Eldar, M.; Arad, M.; et al. Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J. Cell. Mol. Med. 2015, 19, 2006–2018. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujala, K.; Paavola, J.; Lahti, A.; Larsson, K.; Pekkanen-Mattila, M.; Viitasalo, M.; Lahtinen, A.M.; Toivonen, L.; Kontula, K.; Swan, H.; et al. Cell model of catecholaminergic polymorphic ventricular tachycardia reveals early and delayed afterdepolarizations. PLoS ONE 2012, 7, e44660. [Google Scholar] [CrossRef]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Shemer, Y.; Baskin, P.; Reiter, I.; Willi, L.; Freimark, D.; Gherghiceanu, M.; et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. J. Cell. Mol. Med. 2019, 23, 2125–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallas, T.; Eisen, B.; Shemer, Y.; Ben Jehuda, R.; Mekies, L.N.; Naor, S.; Schick, R.; Eliyahu, S.; Reiter, I.; Vlodavsky, E.; et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell–derived cardiomyocytes. J. Cell. Mol. Med. 2018, 22, 913–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm 2018, 15, 267–276. [Google Scholar] [CrossRef]
- Schick, R.; Mekies, L.N.; Shemer, Y.; Eisen, B.; Hallas, T.; Ben Jehuda, R.; Ben-Ari, M.; Szantai, A.; Willi, L.; Shulman, R.; et al. Functional abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS ONE 2018, 13, e0205719. [Google Scholar] [CrossRef] [PubMed]
- Mekies, L.N.; Regev, D.; Eisen, B.; Fernandez-Gracia, J.; Baskin, P.; Ben Jehuda, R.; Shulman, R.; Reiter, I.; Palty, R.; Arad, M.; et al. Depressed β-adrenergic inotropic responsiveness and intracellular calcium handling abnormalities in Duchenne Muscular Dystrophy patients’ induced pluripotent stem cell–derived cardiomyocytes. J. Cell. Mol. Med. 2021, 25, 3922–3934. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shemer, Y.; Mekies, L.N.; Ben Jehuda, R.; Baskin, P.; Shulman, R.; Eisen, B.; Regev, D.; Arbustini, E.; Gerull, B.; Gherghiceanu, M.; et al. Investigating LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 7874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157874
Shemer Y, Mekies LN, Ben Jehuda R, Baskin P, Shulman R, Eisen B, Regev D, Arbustini E, Gerull B, Gherghiceanu M, et al. Investigating LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. International Journal of Molecular Sciences. 2021; 22(15):7874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157874
Chicago/Turabian StyleShemer, Yuval, Lucy N. Mekies, Ronen Ben Jehuda, Polina Baskin, Rita Shulman, Binyamin Eisen, Danielle Regev, Eloisa Arbustini, Brenda Gerull, Mihaela Gherghiceanu, and et al. 2021. "Investigating LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes" International Journal of Molecular Sciences 22, no. 15: 7874. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22157874