
Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia


Abstract
:
1. Introduction
2. Results
2.1. Susceptibility of AML Cell Lines Grown in the Absence or Presence of HS-5 Stroma Cells to AC-4-130 and Venetoclax
2.2. AC-4-130 Combination Treatment in AML Cell Lines
2.3. Changed Susceptibility of AML Cell Lines to Combination Treatment with AC-4-130 in the Presence of Bone Marrow Stroma
2.4. Induction of Apoptosis and Cell Death in AML Cell Lines
2.5. AC-4-140 Combination Treatments in Leukemic Cells In Vitro
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Cell Lines and Cell Culture
4.3. Cytotoxicity Assays
4.4. Measurement of mRNA Expression by qPCR
4.5. Antibodies and Cytometry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal Transduction Mediated by the Ras/Raf/MEK/ERK Pathway from Cytokine Receptors to Transcription Factors: Potential Targeting for Therapeutic Intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Takahashi, S. Downstream Molecular Pathways of FLT3 in the Pathogenesis of Acute Myeloid Leukemia: Biology and Therapeutic Implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Spiekermann, K.; Bagrintseva, K.; Schwab, R.; Schmieja, K.; Hiddemann, W. Overexpression and Constitutive Activation of FLT3 Induces STAT5 Activation in Primary Acute Myeloid Leukemia Blast Cells. Clin. Cancer Res. 2003, 9, 2140–2150. [Google Scholar]
- Lam, S.S.Y.; Leung, A.Y.H. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. [Google Scholar] [CrossRef] [Green Version]
- Orlova, A.; Neubauer, H.A.; Moriggl, R. The Stromal Microenvironment Provides an Escape Route from FLT3 Inhibitors through the GAS6-AXL-STAT5 Axis. Haematologica 2019, 104, 1907–1909. [Google Scholar] [CrossRef]
- Dumas, P.-Y.; Naudin, C.; Martin-Lannerée, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossée, M.; Giese, A.; et al. Hematopoietic Niche Drives FLT3-ITD Acute Myeloid Leukemia Resistance to Quizartinib via STAT5-and Hypoxia-Dependent Upregulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. JAK/STAT Pathways in Cytokine Signaling and Myeloproliferative Disorders. Genes Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 Signaling on Gene Regulation and Chromatin Remodeling in Hematopoietic Cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, H.; Wierenga, A.T.J.; Vellenga, E.; Schuringa, J.J. STAT5-Mediated Self-Renewal of Normal Hematopoietic and Leukemic Stem Cells. JAK-STAT 2012, 1, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Wierenga, A.T.J.; Rozenveld-Geugien, M.; van de Lande, K.; Vellenga, E.; Schuringa, J.J. Single-Cell STAT5 Signal Transduction Profiling in Normal and Leukemic Stem and Progenitor Cell Populations Reveals Highly Distinct Cytokine Responses. PLoS ONE 2009, 4, e7989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Jin, Q.; Fu, Q.; You, P.; Jiang, X.; Yuan, Q.; Huang, H. Induction of Multidrug Resistance of Acute Myeloid Leukemia Cells by Cocultured Stromal Cells via Upregulation of the PI3K/Akt Signaling Pathway. Oncol. Res. 2016, 24, 215–223. [Google Scholar] [CrossRef]
- Hou, D.; Wang, B.; You, R.; Wang, X.; Liu, J.; Zhan, W.; Chen, P.; Qin, T.; Zhang, X.; Huang, H. Stromal Cells Promote Chemoresistance of Acute Myeloid Leukemia Cells via Activation of the IL-6/STAT3/OXPHOS Axis. Ann. Transl. Med. 2020, 8, 1346. [Google Scholar] [CrossRef] [PubMed]
- Brachet-Botineau, M.; Polomski, M.; Neubauer, H.A.; Juen, L.; Hédou, D.; Viaud-Massuard, M.-C.; Prié, G.; Gouilleux, F. Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers. Cancers 2020, 12, 240. [Google Scholar] [CrossRef] [Green Version]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic Inhibition of STAT5 in Acute Myeloid Leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debierre-Grockiego, F. Anti-Apoptotic Role of STAT5 in Haematopoietic Cells and in the Pathogenesis of Malignancies. Apoptosis 2004, 9, 717–728. [Google Scholar] [CrossRef]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-Regulates MCL-1 to Promote Survival of Stem Cells in Acute Myeloid Leukemia via FLT3-ITD-Specific STAT5 Activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipel, K.; Schmitter, K.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy Consisting of MCL1- and MEK-Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipel, K.; Kopp, B.; Bacher, U.; Pabst, T. BMI1-Inhibitor PTC596 in Combination with MCL1 Inhibitor S63845 or MEK Inhibitor Trametinib in the Treatment of Acute Leukemia. Cancers 2021, 13, 581. [Google Scholar] [CrossRef]
- Seipel, K.; Marques, M.A.T.; Sidler, C.; Mueller, B.U.; Pabst, T. MDM2- and FLT3-Inhibitors in the Treatment of FLT3-ITD Acute Myeloid Leukemia, Specificity and Efficacy of NVP-HDM201 and Midostaurin. Haematologica 2018, 103, 1862–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipel, K.; Marques, M.A.T.; Sidler, C.; Mueller, B.U.; Pabst, T. The Cellular P53 Inhibitor MDM2 and the Growth Factor Receptor FLT3 as Biomarkers for Treatment Responses to the MDM2-Inhibitor Idasanutlin and the MEK1 Inhibitor Cobimetinib in Acute Myeloid Leukemia. Cancers 2018, 10, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasca, S.; Jurj, A.; Zdrenghea, M.; Tomuleasa, C. The Potential Equivalents of TET2 Mutations. Cancers 2021, 13, 1499. [Google Scholar] [CrossRef]
- Basu, S.; Hubbard, B.; Shevach, E.M. Foxp3-Mediated Inhibition of Akt Inhibits Glut1 (Glucose Transporter 1) Expression in Human T Regulatory Cells. J. Leukoc. Biol. 2015, 97, 279–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-J.; Huang, C.-H.; Shi, Y.-J.; Lee, Y.-C.; Wang, L.-J.; Chang, L.-S. The Suppressive Effect of Arsenic Trioxide on TET2-FOXP3-Lyn-Akt Axis-Modulated MCL1 Expression Induces Apoptosis in Human Leukemia Cells. Toxicol. Appl. Pharmacol. 2018, 358, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, D.; Qing, W.; Ma, C.; Zhang, Z.; Wei, H.; Wu, G. Bone Marrow Stromal-Cell Line HS-5 Affects Apoptosis of Acute Myeloid Leukemia Cells HL-60 through GLI1 Activation. Biomed. Res. 2018, 29, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Gordon, P.M.; Dias, S.; Williams, D.A. Cytokines Secreted by Bone Marrow Stromal Cells Protect C-KIT Mutant AML Cells from c-KIT Inhibitor-Induced Apoptosis. Leukemia 2014, 28, 2257–2260. [Google Scholar] [CrossRef] [Green Version]
- Podszywalow-Bartnicka, P.; Kominek, A.; Wolczyk, M.; Kolba, M.D.; Swatler, J.; Piwocka, K. Characteristics of Live Parameters of the HS-5 Human Bone Marrow Stromal Cell Line Cocultured with the Leukemia Cells in Hypoxia, for the Studies of Leukemia-Stroma Cross-Talk. Cytom. A 2018, 93, 929–940. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Gao, W.; Sun, X.; Wang, W. STAT5 and TET2 Cooperate to Regulate FOXP3-TSDR Demethylation in CD4+ T Cells of Patients with Colorectal Cancer. J. Immunol. Res. 2018, 2018, 6985031. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Qu, C.; Zhou, Y.; Konkel, J.E.; Shi, S.; Liu, Y.; Chen, C.; Liu, S.; Liu, D.; Chen, Y.; et al. Hydrogen Sulfide Promotes Tet1- and Tet2-Mediated Foxp3 Demethylation to Drive Regulatory T Cell Differentiation and Maintain Immune Homeostasis. Immunity 2015, 43, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) Gene in Hematopoiesis and Hematopoietic Diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, M.; Chen, X.; Chen, L.; Xu, Y.; Lv, L.; Wang, P.; Yang, H.; Ma, S.; Lin, H.; et al. WT1 Recruits TET2 to Regulate Its Target Gene Expression and Suppress Leukemia Cell Proliferation. Mol. Cell 2015, 57, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Belickova, M.; Vesela, J.; Jonasova, A.; Pejsova, B.; Votavova, H.; Merkerova, M.D.; Zemanova, Z.; Brezinova, J.; Mikulenkova, D.; Lauermannova, M.; et al. TP53 Mutation Variant Allele Frequency Is a Potential Predictor for Clinical Outcome of Patients with Lower-Risk Myelodysplastic Syndromes. Oncotarget 2016, 7, 36266–36279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallman, D.A.; Komrokji, R.; Vaupel, C.; Cluzeau, T.; Geyer, S.M.; McGraw, K.L.; Al Ali, N.H.; Lancet, J.; McGinniss, M.J.; Nahas, S.; et al. Impact of TP53 Mutation Variant Allele Frequency on Phenotype and Outcomes in Myelodysplastic Syndromes. Leukemia 2016, 30, 666–673. [Google Scholar] [CrossRef]
- Shumilov, E.; Flach, J.; Kohlmann, A.; Banz, Y.; Bonadies, N.; Fiedler, M.; Pabst, T.; Bacher, U. Current Status and Trends in the Diagnostics of AML and MDS. Blood Rev. 2018, 32, 508–519. [Google Scholar] [CrossRef] [PubMed]






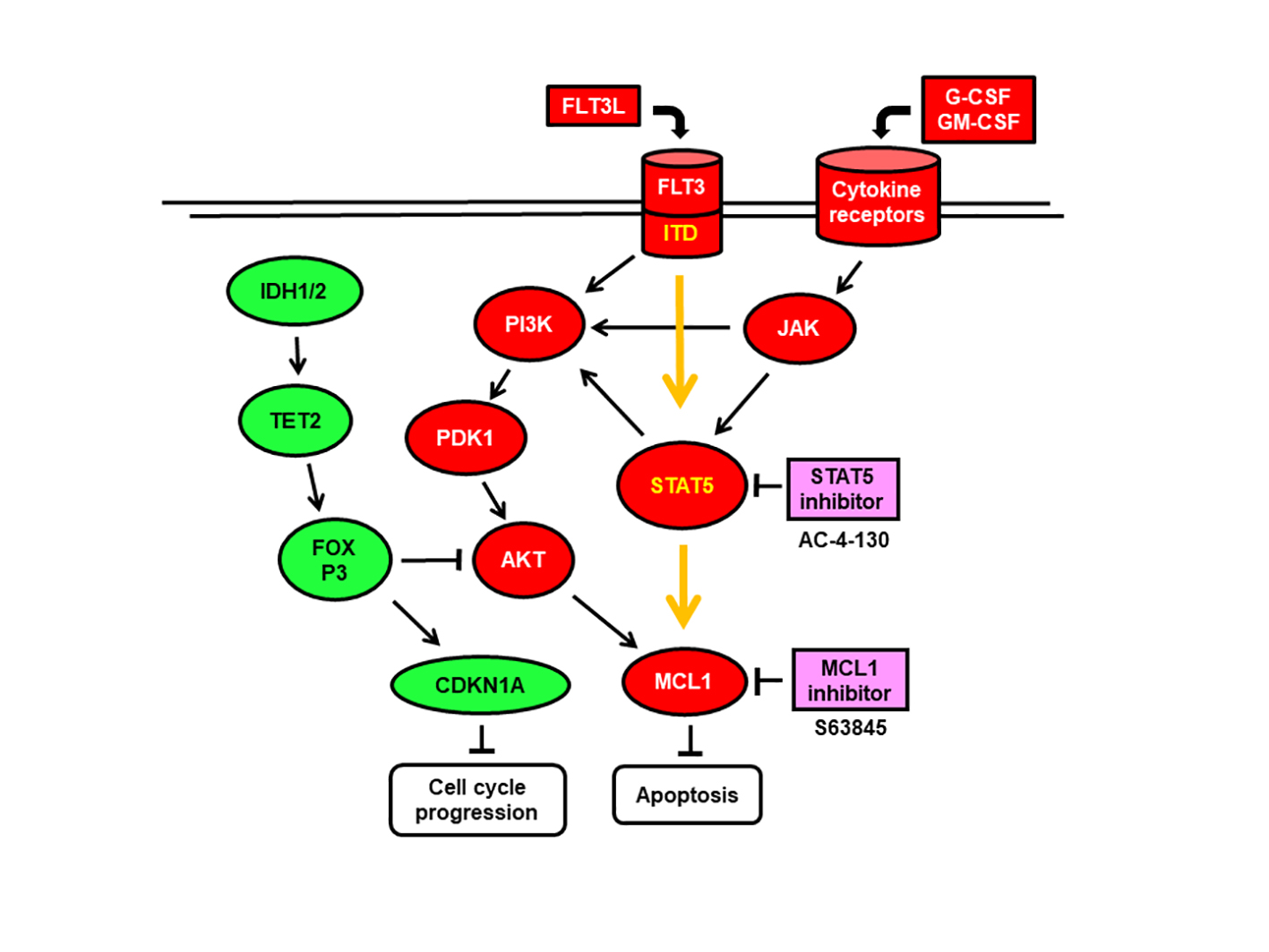{kind=link}
{kind=link}
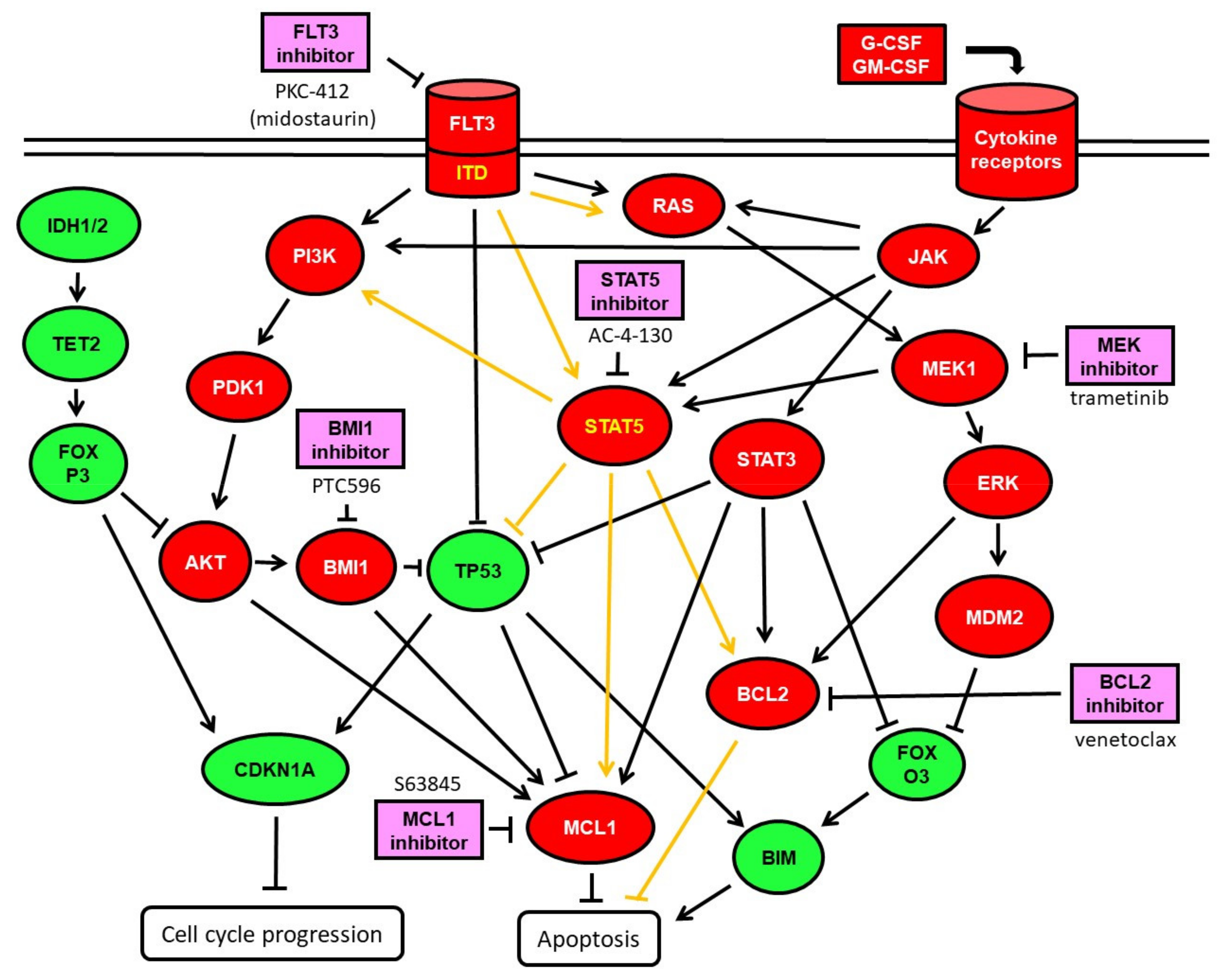{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | FAB | Origin | FLT3 | TP53 | Gene Variants | Karyotype |
|---|---|---|---|---|---|---|
| HL-60 | M2 | de novo | wt | null | CDKN2A R80X NRAS Q61L | hypotetraploid |
| ML-2 | M4 | de novo | wt | wt | KMT2A-AFDN KRAS A146T | t(6;11) |
| MOLM-13 | M5a, relapse | MDS | ITD | wt | KMT2A-MLLT3 | t(9;11) |
| MOLM-16 | M0, relapse | de novo | wt | V173M C238S | MLL V1368L MTOR T571K | hypotetraploid |
| OCI-AML3 | M4 | de novo | wt | wt | NRAS Q61L NPM1 L287fs DNMT3A R882C | +1, +5, +8 |
| SKM-1 | M5, refractory | MDS | wt | R248Q R248Q | ASXL1 Y591X KRAS K117N TET2 C1419fs | del9q12 |
| AML Cell Line | HS-5 Stroma | AC-4-130 +PTC596 | AC-4-130 +Trametinib | AC-4-130 +S63845 | AC-4-130 +PKC412 | AC-4-130 +Venetoclax |
|---|---|---|---|---|---|---|
| SKM-1 | absent | 0.6–0.8 | 0.6–0.8 | 0.7–0.9 | nd | 0.4–0.6 |
| present | 1.0–1.2 | 0.8–1.0 | 1.1–1.3 | nd | 0.5–0.7 | |
| MOLM-13 | absent | 0.9–1.1 | 1.2–1.7 | 0.8–1.0 | 0.4–0.6 | 0.6–0.8 |
| present | 0.6–0.8 | nc | 0.8–1.0 | 0.4–0.6 | 0.8–0.9 |
| ID | Disease | FAB | Antecedent | Gene Variants | Karyotype | AC+S * |
|---|---|---|---|---|---|---|
| AML1 | sAML | M0 | MDS | EVI1 (overexpressed) | −7 | NR |
| AML2 | AML | M0 | TP53 (VAF 92%) | complex | NR | |
| AML3 | sAML | MDS | CEPBA, ASXL1, EZH2, RUNX1 | normal | NR | |
| AML4 | sAML | ET | ASXL1, CALR, KMT2A (amp), TP53 (VAF 50%), | 49, +der(11) | NR | |
| AML5 | AML | M5 | FLT3-TKD (0.63), NPM1, DNMT3A | normal | SR | |
| AML6 | sAML | M4 | bicytopenia | NPM1,FLT3-TKD (0.57), TET2, TP53 (VAF 5%), SRSF2 | +8 | SR |
| AML7 | AML | M5 | ASXL1, TET2, KRAS, SH2B3, U2AF1 | −7 | SR | |
| AML8 | AML | M5 | FLT3-ITD (0.58), NPM1, DNMT3A | normal | SR | |
| AML9 | sAML | M1 | MDS | NPM1, TET2, DNMT3A | normal | SR |
| AML10 | AML | M1 | FLT3-ITD (0.833), NPM1 | normal | SR | |
| AML11 | AML | M2 | normal | complex, −7,−9 | NR | |
| AML12 | sAML | MPN | JAK2 | normal | NR | |
| AML13 | sAML | M4 | breast cancer | NPM1, TET2 | normal | SR |
| AML14 | AML | M1 | FLT3-ITD (>1.0), NPM1 | normal | SR | |
| AML15 | AML | M4 | FLT3-ITD (0.504), NPM1 | normal | SR | |
| AML16 | AML | M2 | NPM1, IDH2, DNMT3A | normal | SR | |
| AML17 | AML | M1 | FLT-3-ITD (0.783), BCOR, TET2, U2AF1 | del20q11, +8 | SR | |
| AML18 | AML | M4 | FLT3-TKD (0.487), NRAS, KRAS, KMT2A-MLLT10 | t(10;11), +8 | SR | |
| MDS1 | MDS | TET2, ETV6, KRAS, SRSF2, CBL | del7q | SR | ||
| BA1 | B-ALL | IGH (rearranged) | normal | NR | ||
| BA2 | B-ALL | normal | normal | SR | ||
| CML1 | CML | BCR-ABL1 | t(9;22) | NR | ||
| MM1 | MM | normal | NR | |||
| MM2 | MM | t(4;14) | NR | |||
| HD | n.a. | normal | normal | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seipel, K.; Graber, C.; Flückiger, L.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 8092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158092
Seipel K, Graber C, Flückiger L, Bacher U, Pabst T. Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2021; 22(15):8092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158092
Chicago/Turabian StyleSeipel, Katja, Carolyn Graber, Laura Flückiger, Ulrike Bacher, and Thomas Pabst. 2021. "Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia" International Journal of Molecular Sciences 22, no. 15: 8092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158092