
Aloysia Citrodora Essential Oil Inhibits Melanoma Cell Growth and Migration by Targeting HB-EGF-EGFR Signaling



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
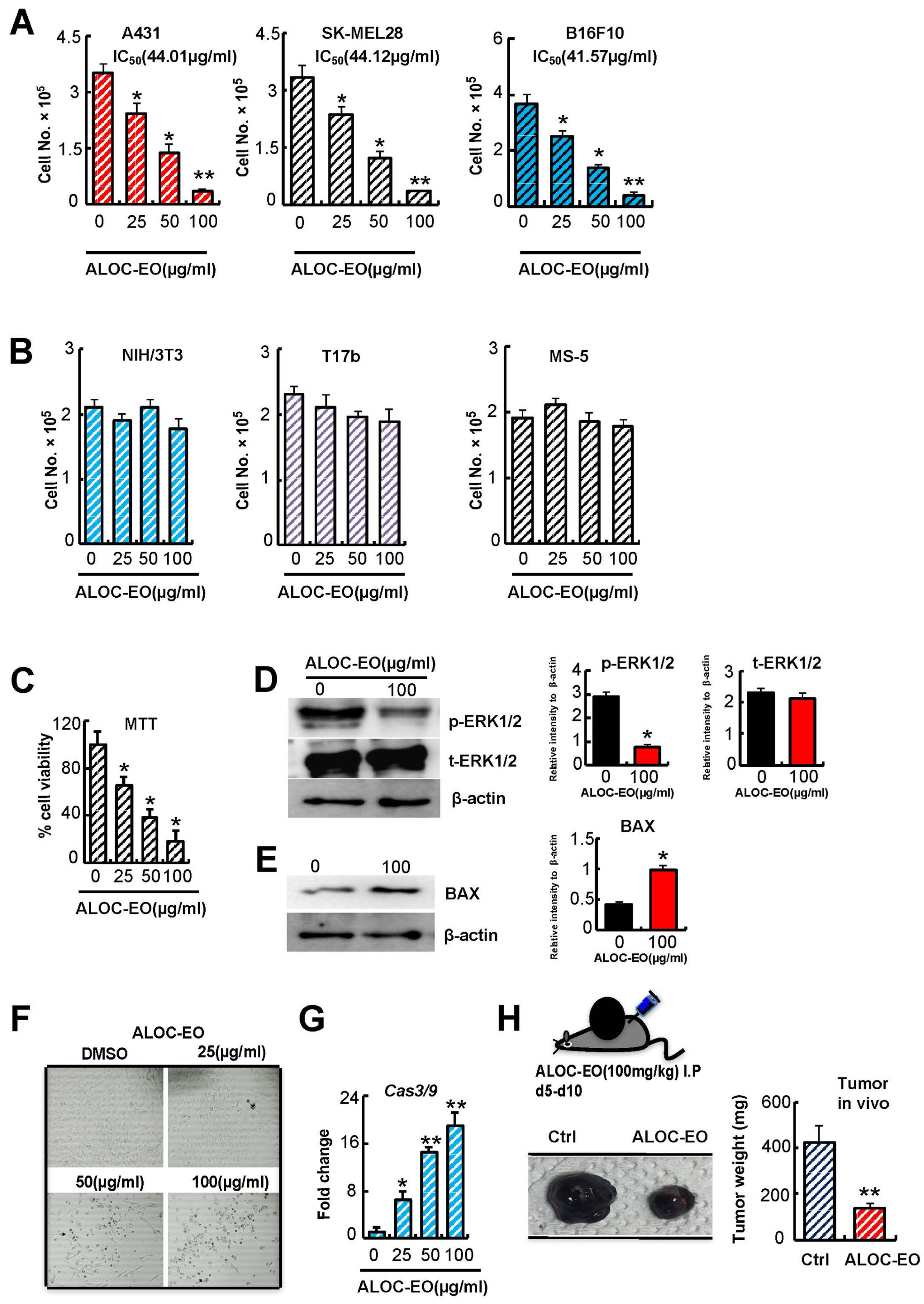
2.1. Growth Inhibition and Induction of Apoptosis by ALOC-EO in Melanoma Cells
2.2. ALOC_EO Blocks Melanoma Cell Proliferation and ERK1/2 Phosphorylation
2.3. ALOC-EO Inhibits Tumor Cell Migration and MMP Expression
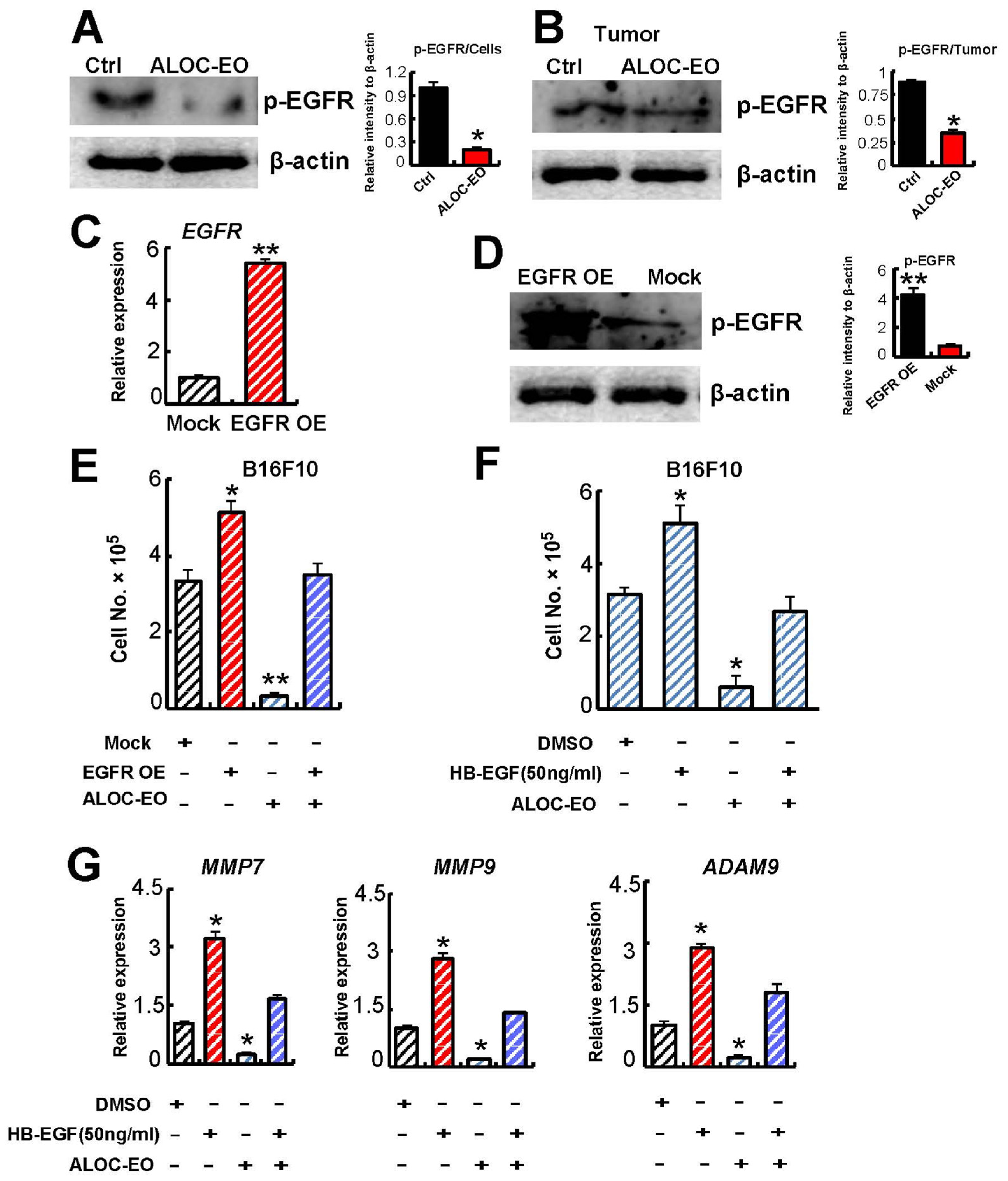
2.4. ALOC-EO Blocks EGFR-Mediated Melanoma Cell Growth and MMP Production
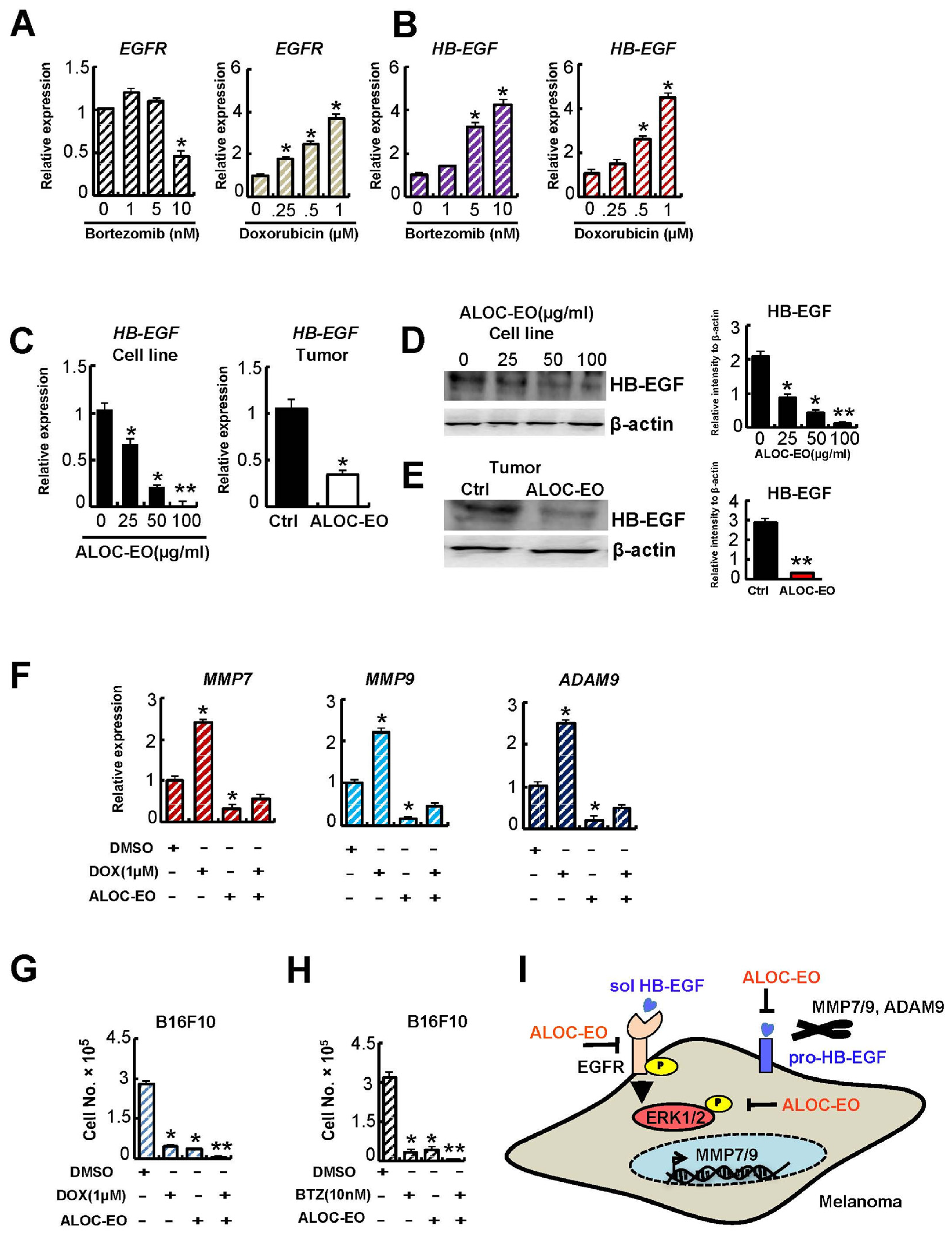
2.5. ALOC-EO Improves Chemosensitivity by Targeting EGFR Signaling
3. Discussion
4. Material and Methods
4.1. Chemicals and Cytokines
4.2. Generation of ALOC-EO
4.3. Cell Line and Cell Culture
4.4. Cell Proliferation Assay
4.5. Wound Healing (Scratch) Assay
4.6. Cell Culture
4.7. Cloning of Mouse EGF Receptor
4.8. EGF Receptor Lentivirus Generation for Overexpression
4.9. siRNA-Based Gene Knockdown
4.10. In Vivo Melanoma Model
4.11. Immunoassay
4.12. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qPCR)
- qPCR Forward; Reverse
- mMMP7 5′-TAATTGGCTTCGCAAGGAGA-′3; 5′-AAGGCATGACCTAGAGTGTTCC-′3
- mMMP9 5′-AGACGACATAGACGGCATCC-′3; 5′-TCGGCTGTGGTTCAGTTGT-′3
- mβ-actin 5′-CTAAGGCCAACCGTGAAAAG-′3; 5′-ACCAGAGGCATACAGGGACA-′3
- mHB-EGF 5′-TCTTCTTGTCATCGTGGGACT-′3; 5′-CACGCCCAACTTCACTTTCT-′3
- mEGFR 5′-ACGCATTCCTCCCTGTACCT-′3; 5′-GCAGGGGCTGATTGTGATAG-′3
4.13. Western Blotting Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Dong, J.; Xie, J.; Xing, M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle 2009, 8, 2122–2124. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2011, 44, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Heissig, B.; Salama, Y.; Osada, T.; Okumura, K.; Hattori, K. The Multifaceted Role of Plasminogen in Cancer. Int. J. Mol. Sci. 2021, 22, 2304. [Google Scholar] [CrossRef]
- Gautam, N.; Mantha, A.K.; Mittal, S. Essential oils and their constituents as anticancer agents: A mechanistic view. Biomed Res. Int. 2014, 2014, 154106. [Google Scholar] [CrossRef] [Green Version]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of plant defense against insect herbivores. Plant Signal. Behav. 2012, 7, 1306–1320. [Google Scholar] [CrossRef] [Green Version]
- Dhifi, W.; Bellili, S.; Jazi, S.; Bahloul, N.; Mnif, W. Essential Oils’ Chemical Characterization and Investigation of Some Biological Activities: A Critical Review. Medicines 2016, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Bahramsoltani, R.; Rostamiasrabadi, P.; Shahpiri, Z.; Marques, A.M.; Rahimi, R.; Farzaei, M.H. Aloysia citrodora Paláu (Lemon verbena): A review of phytochemistry and pharmacology. J. Ethnopharmacol. 2018, 222, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Najar, B.; Shortrede, J.E.; Pistelli, L.; Buhagiar, J. Chemical Composition and In Vitro Cytotoxic Screening of Sixteen Commercial Essential Oils on Five Cancer Cell Lines. Chem. Biodivers. 2020, 17, e1900478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, S.; Kapur, A.; Patankar, M.S.; Xiong, M.P. Formulation, Characterization, and Antitumor Properties of Trans- and Cis-Citral in the 4T1 Breast Cancer Xenograft Mouse Model. Pharm. Res. 2015, 32, 2548–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivisaari, A.K.; Kallajoki, M.; Ala-aho, R.; McGrath, J.A.; Bauer, J.W.; Königová, R.; Medvecz, M.; Beckert, W.; Grénman, R.; Kähäri, V.M. Matrix metalloproteinase-7 activates heparin-binding epidermal growth factor-like growth factor in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2010, 163, 726–735. [Google Scholar] [CrossRef]
- Felli, N.; Felicetti, F.; Lustri, A.M.; Errico, M.C.; Bottero, L.; Cannistraci, A.; De Feo, A.; Petrini, M.; Pedini, F.; Biffoni, M.; et al. miR-126&126* restored expressions play a tumor suppressor role by directly regulating ADAM9 and MMP7 in melanoma. PLoS ONE 2013, 8, e56824. [Google Scholar]
- Pines, G.; Köstler, W.J.; Yarden, Y. Oncogenic mutant forms of EGFR: Lessons in signal transduction and targets for cancer therapy. FEBS Lett. 2010, 584, 2699–2706. [Google Scholar] [CrossRef] [Green Version]
- Meierjohann, S.; Wende, E.; Kraiss, A.; Wellbrock, C.; Schartl, M. The oncogenic epidermal growth factor receptor variant Xiphophorus melanoma receptor kinase induces motility in melanocytes by modulation of focal adhesions. Cancer Res. 2006, 66, 3145–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tworkoski, K.; Singhal, G.; Szpakowski, S.; Zito, C.I.; Bacchiocchi, A.; Muthusamy, V.; Bosenberg, M.; Krauthammer, M.; Halaban, R.; Stern, D.F. Phosphoproteomic Screen Identifies Potential Therapeutic Targets in Melanoma. Mol. Cancer Res. 2011, 9, 801. [Google Scholar] [CrossRef] [Green Version]
- Kreß, J.K.; Jessen, C.; Marquardt, A.; Hufnagel, A.; Meierjohann, S. NRF2 Enables EGFR Signaling in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 3803. [Google Scholar] [CrossRef]
- Reschke, M.; Mihic-Probst, D.; van der Horst, E.H.; Knyazev, P.; Wild, P.J.; Hutterer, M.; Meyer, S.; Dummer, R.; Moch, H.; Ullrich, A. HER3 is a determinant for poor prognosis in melanoma. Clin. Cancer Res. 2008, 14, 5188–5197. [Google Scholar] [CrossRef] [Green Version]
- Prickett, T.D.; Agrawal, N.S.; Wei, X.; Yates, K.E.; Lin, J.C.; Wunderlich, J.R.; Cronin, J.C.; Cruz, P.; Rosenberg, S.A.; Samuels, Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 2009, 41, 1127–1132. [Google Scholar] [CrossRef]
- Bardeesy, N.; Kim, M.; Xu, J.; Kim, R.S.; Shen, Q.; Bosenberg, M.W.; Wong, W.H.; Chin, L. Role of epidermal growth factor receptor signaling in RAS-driven melanoma. Mol. Cell Biol. 2005, 25, 4176–4188. [Google Scholar] [CrossRef] [Green Version]
- Rákosy, Z.; Vízkeleti, L.; Ecsedi, S.; Vokó, Z.; Bégány, Á.; Barok, M.; Krekk, Z.; Gallai, M.; Szentirmay, Z.; Ádány, R.; et al. EGFR gene copy number alterations in primary cutaneous malignant melanomas are associated with poor prognosis. Int. J. Cancer 2007, 121, 1729–1737. [Google Scholar] [CrossRef]
- Udart, M.; Utikal, J.; Krähn, G.M.; Peter, R.U. Chromosome 7 aneusomy. A marker for metastatic melanoma? Expression of the epidermal growth factor receptor gene and chromosome 7 aneusomy in nevi, primary malignant melanomas and metastases. Neoplasia 2001, 3, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, K.; Kawakami, T.; Watabe, H.; Itoh, F.; Mizoguchi, M.; Soma, Y. Expression of matrilysin (matrix metalloproteinase-7) in primary cutaneous and metastatic melanoma. Br. J. Dermatol. 2007, 156, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Salama, Y.; Lin, S.Y.; Dhahri, D.; Hattori, K.; Heissig, B. The fibrinolytic factor tPA drives LRP1-mediated melanoma growth and metastasis. FASEB J. 2019, 33, 3465–3480. [Google Scholar] [CrossRef] [PubMed]
- Zigrino, P.; Nischt, R.; Mauch, C. The disintegrin-like and cysteine-rich domains of ADAM-9 mediate interactions between melanoma cells and fibroblasts. J. Biol. Chem. 2011, 286, 6801–6807. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.; So, I.; Chun, J.N.; Jeon, J.-H. The antitumor effects of geraniol: Modulation of cancer hallmark pathways (Review). Int. J. Oncol. 2016, 48, 1772–1782. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, S.C.; Siddiqui, M.S.; Athar, M.; Alam, M.S. Geraniol inhibits murine skin tumorigenesis by modulating COX-2 expression, Ras-ERK1/2 signaling pathway and apoptosis. J. Appl. Toxicol. 2013, 33, 828–837. [Google Scholar] [CrossRef]
- Connor, M.J. Modulation of tumor promotion in mouse skin by the food additive citral (3,7-dimethyl-2,6-octadienal). Cancer Lett. 1991, 56, 25–28. [Google Scholar] [CrossRef]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [Green Version]
- Yotsumoto, F.; Yagi, H.; Suzuki, S.O.; Oki, E.; Tsujioka, H.; Hachisuga, T.; Sonoda, K.; Kawarabayashi, T.; Mekada, E.; Miyamoto, S. Validation of HB-EGF and amphiregulin as targets for human cancer therapy. Biochem. Biophys. Res. Commun. 2008, 365, 555–561. [Google Scholar] [CrossRef]
- Hudson, L.G.; Moss, N.M.; Stack, M.S. EGF-receptor regulation of matrix metalloproteinases in epithelial ovarian carcinoma. Future Oncol. 2009, 5, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Liu, R.; Lee, S.W.; Sloss, C.M.; Couget, J.; Cusack, J.C. Heparin-binding EGF-like growth factor is an early response gene to chemotherapy and contributes to chemotherapy resistance. Oncogene 2007, 26, 2006–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.; Niemetz-Rahn, A.; Nonnenmacher, A.; Tucholski, J.; Keilholz, U.; Fusi, A. Expression and activity of EGFR in human cutaneous melanoma cell lines and influence of vemurafenib on the EGFR pathway. Target. Oncol. 2015, 10, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Simiczyjew, A.; Pietraszek-Gremplewicz, K.; Dratkiewicz, E.; Podgórska, M.; Matkowski, R.; Ziętek, M.; Nowak, D. Combination of Selected MET and EGFR Inhibitors Decreases Melanoma Cells’ Invasive Abilities. Front. Pharmacol. 2019, 10, 1116. [Google Scholar] [CrossRef]
- Takács, A.; Lajkó, E.; Láng, O.; Istenes, I.; Kőhidai, L. Alpha-lipoic acid alters the antitumor effect of bortezomib in melanoma cells in vitro. Sci. Rep. 2020, 10, 14287. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [Green Version]
- Heissig, B.; Hattori, K.; Friedrich, M.; Rafii, S.; Werb, Z. Angiogenesis: Vascular remodeling of the extracellular matrix involves metalloproteinases. Curr. Opin. Hematol. 2003, 10, 136–141. [Google Scholar] [CrossRef]
- Villanueva, J.; Herlyn, M. Melanoma and the tumor microenvironment. Curr. Oncol. Rep. 2008, 10, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Dhahri, D.; Eiamboonsert, S.; Salama, Y.; Shimazu, H.; Munakata, S.; Hattori, K. Role of mesenchymal stem cell-derived fibrinolytic factor in tissue regeneration and cancer progression. Cell. Mol. Life Sci. 2015, 72, 4759–4770. [Google Scholar] [CrossRef] [PubMed]
- Meierjohann, S.; Hufnagel, A.; Wende, E.; Kleinschmidt, M.A.; Wolf, K.; Friedl, P.; Gaubatz, S.; Schartl, M. MMP13 mediates cell cycle progression in melanocytes and melanoma cells: In vitro studies of migration and proliferation. Mol. Cancer 2010, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Mader, C.C.; Oser, M.; Magalhaes, M.A.; Bravo-Cordero, J.J.; Condeelis, J.; Koleske, A.J.; Gil-Henn, H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011, 71, 1730–1741. [Google Scholar] [CrossRef] [Green Version]
- Khokha, R. Suppression of the tumorigenic and metastatic abilities of murine B16-F10 melanoma cells in vivo by the overexpression of the tissue inhibitor of the metalloproteinases-1. J. Natl Cancer Inst. 1994, 86, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Hicks, M.J.; Nemechek, A.J.; Mehta, R.; O’Malley, B.W., Jr.; Goepfert, H.; Flaitz, C.M.; Boyd, D. PD 098059, an inhibitor of ERK1 activation, attenuates the in vivo invasiveness of head and neck squamous cell carcinoma. Br. J. Cancer 1999, 80, 1412–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honjo, K.; Munakata, S.; Tashiro, Y.; Salama, Y.; Shimazu, H.; Eiamboonsert, S.; Dhahri, D.; Ichimura, A.; Dan, T.; Miyata, T.; et al. Plasminogen activator inhibitor-1 regulates macrophage-dependent postoperative adhesion by enhancing EGF-HER1 signaling in mice. FASEB J. 2017, 31, 2625–2637. [Google Scholar] [CrossRef] [Green Version]
- Rocha-Lima, C.M.; Soares, H.P.; Raez, L.E.; Singal, R. EGFR targeting of solid tumors. Cancer Control. 2007, 14, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Cohen, M.; Sridhara, R.; Chen, Y.-F.; Williams, G.M.; Duan, J.; Gobburu, J.; Booth, B.; Benson, K.; Leighton, J.; et al. Approval Summary for Erlotinib for Treatment of Patients with Locally Advanced or Metastatic Non–Small Cell Lung Cancer after Failure of at Least One Prior Chemotherapy Regimen. Clin. Cancer Res. 2005, 11, 6414. [Google Scholar] [CrossRef] [Green Version]
- Klein, B.; Gottfried, M. Targeted agents to improve treatment results in colon cancer: Bevacizumab and cetuximab. J. BU ON Off. J. Balk. Union Oncol. 2007, 12, S127–S136. [Google Scholar]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Chen, Y.E.; Kumar, R.; Taylor, M.; Njauw, C.-N.J.; Miao, B.; Frederick, D.T.; Wargo, J.A.; Flaherty, K.T.; Jönsson, G.; et al. MITF Modulates Therapeutic Resistance through EGFR Signaling. J. Investig. Dermatol. 2015, 135, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Jaradat, N.; Hawash, M.; Abualhasan, M.N.; Qadi, M.; Ghanim, M.; Massarwy, E.; Ammar, S.A.; Zmero, N.; Arar, M.; Hussein, F.; et al. Spectral characterization, antioxidant, antimicrobial, cytotoxic, and cyclooxygenase inhibitory activities of Aloysia citriodora essential oils collected from two Palestinian regions. BMC Complementary Med. Ther. 2021, 21, 143. [Google Scholar]
- Dhahri, D.; Sato-Kusubata, K.; Ohki-Koizumi, M.; Nishida, C.; Tashiro, Y.; Munakata, S.; Shimazu, H.; Salama, Y.; Eiamboonsert, S.; Nakauchi, H.; et al. Fibrinolytic factor-mediated crosstalk with endothelial cells expands murine bone marrow mesenchymal stromal cells. Blood 2016, 128, 1063–1075. [Google Scholar] [CrossRef]
- Kanegae, Y.; Makimura, M.; Saito, I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn. J. Med. Sci. Biol. 1994, 47, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salama, Y.; Jaradat, N.; Hattori, K.; Heissig, B. Aloysia Citrodora Essential Oil Inhibits Melanoma Cell Growth and Migration by Targeting HB-EGF-EGFR Signaling. Int. J. Mol. Sci. 2021, 22, 8151. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158151
Salama Y, Jaradat N, Hattori K, Heissig B. Aloysia Citrodora Essential Oil Inhibits Melanoma Cell Growth and Migration by Targeting HB-EGF-EGFR Signaling. International Journal of Molecular Sciences. 2021; 22(15):8151. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158151
Chicago/Turabian StyleSalama, Yousef, Nidal Jaradat, Koichi Hattori, and Beate Heissig. 2021. "Aloysia Citrodora Essential Oil Inhibits Melanoma Cell Growth and Migration by Targeting HB-EGF-EGFR Signaling" International Journal of Molecular Sciences 22, no. 15: 8151. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158151