
Phospholipids: Identification and Implication in Muscle Pathophysiology

 , , and
, , and
Abstract
:1. Introduction
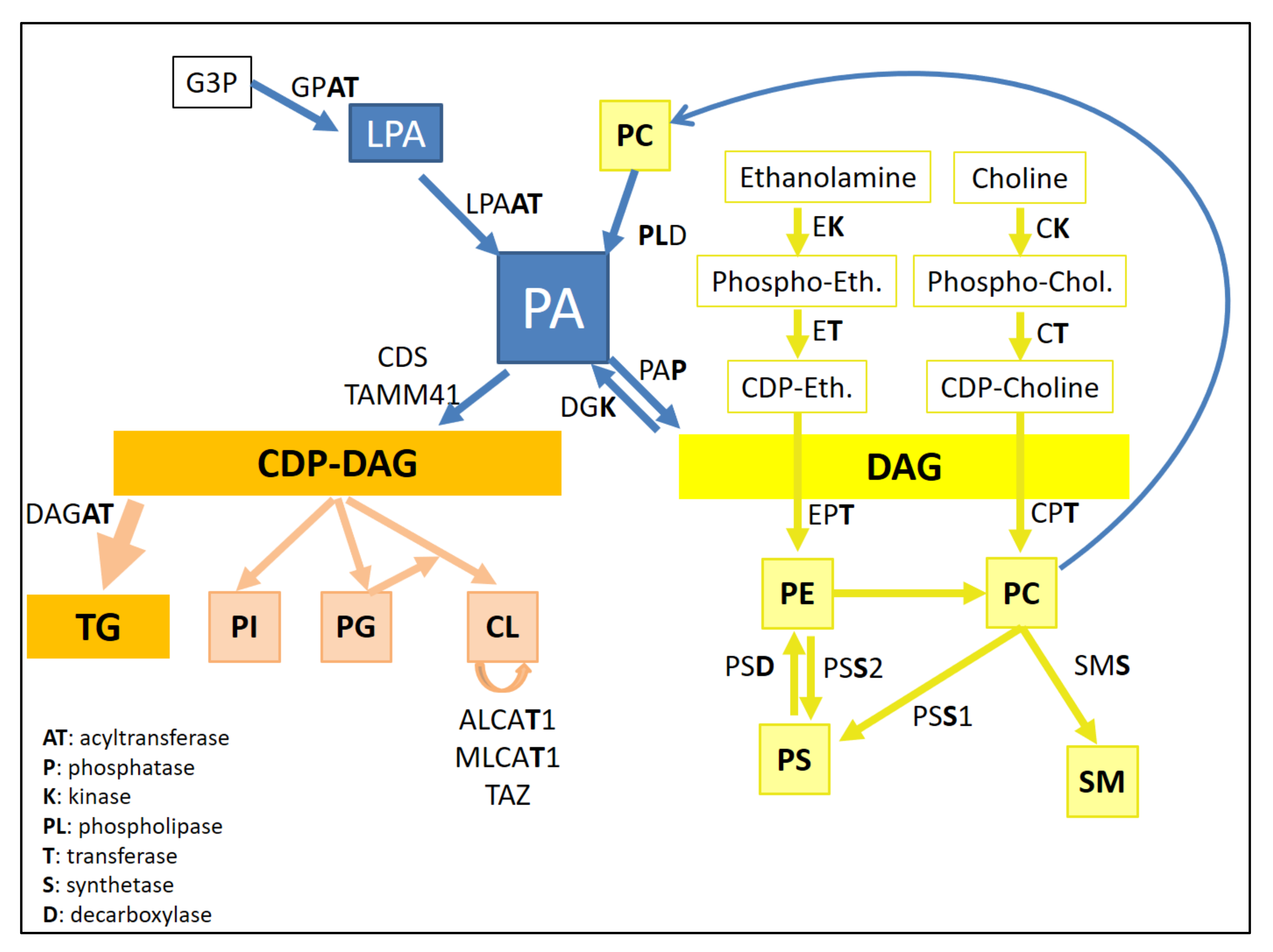
2. Basic Concepts on PLs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phospholipid Group | % of Total Lipids in Eukaryotic Cells [27] | Main Biosynthesis Site | Main Characteristics |
|---|---|---|---|
| Phosphatidic acid PA | 1–2 | ER | The simplest PL Main precursor for PL synthesis Inverse cone |
| Phosphatidylglycerol PG | <1 | Mitochondria | Precursor of cardiolipin |
| Cardiolipin CL | 2–5 | Mitochondria | Present only in mitochondrial membranes Contains four fatty acyl chains Inverse cone |
| Phosphatidylinositol PI | 10–15 | ER | Source of inositol 1,4,5 triphosphate (IP3) Cylinder shape |
| Phosphatidylethanolamine PE | 15–25 | ER and also mitochondria from PS | Second most abundant PL Inverse cone |
| Phosphatidylserine PS | 5–10 | ER | Mostly present in the inner leaflet of the cell membrane, when externalized they became a signal of recognition for cell phagocytosis by macrophages (characteristic of apoptosis) Cylinder shape |
| Lysophosphatidylethanolamine LPE | ND | Obtained from phospholipase action | Contains one fatty acyl chain Hydrolysis of one fatty acid of a PE by a phospholipase |
| Phosphatidylcholine PC | 45–55 | ER | The most abundant (outer leaflet of cell membrane) Constitutes a reserve of choline and methyl groups Formerly called lecithin Cylinder shape |
| Sphingomyelin SM | 5–10 | Golgi | Phosphocholine bound to ceramide Abundant on the outer leaflet of the membrane Cylinder shape |
| Lysophosphatidylcholine LPC | ND | Obtained from phospholipase action | Contains one fatty acyl chain Hydrolysis of one fatty acid of a PC by a phospholipase |
3. Main Methods for PL Analysis
4. Phospholipids in Biological Membranes
5. Importance of PLs in Striated Muscle Functions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bloor, W.R. Outline of a classification of the lipids. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1920, 17, 138–140. [Google Scholar] [CrossRef] [Green Version]
- Zumbuehl, A. Artificial Phospholipids and Their Vesicles. Langmuir 2019, 35, 10223–10232. [Google Scholar] [CrossRef]
- Robertson, J.L. The lipid bilayer membrane and its protein constituents. J. Gen. Physiol. 2018, 150, 1472–1483. [Google Scholar] [CrossRef] [Green Version]
- Christie, W.W.; Han, X. Lipid Analysis—Isolation, Separation, Identification and Lipidomic Analysis, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2010; p. 446. [Google Scholar]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [Green Version]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Kofeler, H.; et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jennings, W.; Epand, R.M. Roles of specific lipid species in the cell and their molecular mechanism. Prog. Lipid Res. 2016, 62, 75–92. [Google Scholar] [CrossRef]
- Wenk, M.R. Lipidomics: New tools and applications. Cell 2010, 143, 888–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.J. MolView: A program for analyzing and displaying atomic structures on the Macintosh personal computer. J. Mol. Graph. 1995, 13, 122–125, 115. [Google Scholar] [CrossRef]
- Moulin, M.; Solgadi, A.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Chaminade, P. Sex-specific cardiac cardiolipin remodelling after doxorubicin treatment. Biol. Sex Differ. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhukovsky, M.A.; Filograna, A.; Luini, A.; Corda, D.; Valente, C. Phosphatidic acid in membrane rearrangements. FEBS Lett. 2019, 593, 2428–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutkewitte, A.J.; Finck, B.N. Regulation of Signaling and Metabolism by Lipin-mediated Phosphatidic Acid Phosphohydrolase Activity. Biomolecules 2020, 10, 1386. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Zierath, J.R. Role of Diacylglycerol Kinases in Glucose and Energy Homeostasis. Trends Endocrinol. Metab. 2019, 30, 603–617. [Google Scholar] [CrossRef]
- Maguire, J.J.; Tyurina, Y.Y.; Mohammadyani, D.; Kapralov, A.A.; Anthonymuthu, T.S.; Qu, F.; Amoscato, A.A.; Sparvero, L.J.; Tyurin, V.A.; Planas-Iglesias, J.; et al. Known unknowns of cardiolipin signaling: The best is yet to come. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 8–24. [Google Scholar] [CrossRef] [Green Version]
- Furse, S. Is phosphatidylglycerol essential for terrestrial life? J. Chem. Biol. 2017, 10, 1–9. [Google Scholar] [CrossRef]
- Blunsom, N.J.; Cockcroft, S. Phosphatidylinositol synthesis at the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158471. [Google Scholar] [CrossRef]
- Blunsom, N.J.; Cockcroft, S. CDP-Diacylglycerol Synthases (CDS): Gateway to Phosphatidylinositol and Cardiolipin Synthesis. Front. Cell Dev. Biol. 2020, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Pennington, E.R.; Funai, K.; Brown, D.A.; Shaikh, S.R. The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1039–1052. [Google Scholar] [CrossRef]
- Pangborn, M.C. Isolation and purification of a serologically acitve phospholipid from beef heart. J. Biol. Chem. 1942, 143, 247–256. [Google Scholar] [CrossRef]
- Traynor-Kaplan, A.; Kruse, M.; Dickson, E.J.; Dai, G.; Vivas, O.; Yu, H.; Whittington, D.; Hille, B. Fatty-acyl chain profiles of cellular phosphoinositides. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 513–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859 Pt B, 1558–1572. [Google Scholar] [CrossRef]
- Kimura, A.K.; Kimura, T. Phosphatidylserine biosynthesis pathways in lipid homeostasis: Toward resolution of the pending central issue for decades. FASEB J. 2021, 35, e21177. [Google Scholar] [CrossRef] [PubMed]
- Futerman, A.H.; Riezman, H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Funai, K.; Summers, S.A.; Rutter, J. Reign in the membrane: How common lipids govern mitochondrial function. Curr. Opin. Cell Biol. 2020, 63, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Aloulou, A.; Rahier, R.; Arhab, Y.; Noiriel, A.; Abousalham, A. Phospholipases: An Overview. Methods Mol. Biol. 2018, 1835, 69–105. [Google Scholar] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Lofgren, L.; Forsberg, G.B.; Stahlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Sci. Rep. 2016, 6, 27688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaian, M.; Kramer, G.; Poorthuis, B.J. Quantification of sulfatides and lysosulfatides in tissues and body fluids by liquid chromatography-tandem mass spectrometry. J. Lipid Res. 2015, 56, 936–943. [Google Scholar] [CrossRef] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Chen, S.; Syed, I.; Stahlman, M.; Kolar, M.J.; Homan, E.A.; Chu, Q.; Smith, U.; Boren, J.; Kahn, B.B.; et al. A LC-MS-based workflow for measurement of branched fatty acid esters of hydroxy fatty acids. Nat. Protoc. 2016, 11, 747–763. [Google Scholar] [CrossRef] [Green Version]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Zhang, J.L. Mass-spectrometry-based lipidomics. J. Sep. Sci. 2018, 41, 351–372. [Google Scholar] [CrossRef]
- Essaid, D.; Rosilio, V.; Daghildjian, K.; Solgadi, A.; Vergnaud, J.; Kasselouri, A.; Chaminade, P. Artificial plasma membrane models based on lipidomic profiling. Biochim. Biophys. Acta 2016, 1858, 2725–2736. [Google Scholar] [CrossRef]
- Schlotterbeck, J.; Chatterjee, M.; Gawaz, M.; Lammerhofer, M. Comprehensive MS/MS profiling by UHPLC-ESI-QTOF-MS/MS using SWATH data-independent acquisition for the study of platelet lipidomes in coronary artery disease. Anal. Chim. Acta 2019, 1046, 1–15. [Google Scholar] [CrossRef]
- Holcapek, M.; Liebisch, G.; Ekroos, K. Lipidomic Analysis. Anal. Chem. 2018, 90, 4249–4257. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Yokomizo, T. Applications of mass spectrometry-based targeted and non-targeted lipidomics. Biochem. Biophys. Res. Commun. 2018, 504, 576–581. [Google Scholar] [CrossRef]
- Ekroos, K.; Ejsing, C.S.; Bahr, U.; Karas, M.; Simons, K.; Shevchenko, A. Charting molecular composition of phosphatidylcholines by fatty acid scanning and ion trap MS3 fragmentation. J. Lipid Res. 2003, 44, 2181–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Han, X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, S.; El Banna, N.; Tfaili, S.; Chaminade, P. A study of inter-species ion suppression in electrospray ionization-mass spectrometry of some phospholipid classes. Anal. Bioanal. Chem. 2016, 408, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Nicolson, G.L. The Fluid-Mosaic Model of Membrane Structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta 2014, 1838, 1451–1466. [Google Scholar] [CrossRef] [Green Version]
- Doktorova, M.; Symons, J.L.; Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 2020, 16, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Doktorova, M. On the Long and Winding Road to a Perfect Membrane Model. Biophys. J. 2020, 118, 273–275. [Google Scholar] [CrossRef]
- Steck, T.L.; Lange, Y. Transverse distribution of plasma membrane bilayer cholesterol: Picking sides. Traffic 2018, 19, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Vance, J.E. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 1991, 266, 89–97. [Google Scholar] [CrossRef]
- Boncompagni, S.; Pozzer, D.; Viscomi, C.; Ferreiro, A.; Zito, E. Physical and Functional Cross Talk Between Endo-Sarcoplasmic Reticulum and Mitochondria in Skeletal Muscle. Antioxid. Redox Signal. 2019, 32, 873–883. [Google Scholar] [CrossRef]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Heden, T.D.; Neufer, P.D.; Funai, K. Looking Beyond Structure: Membrane Phospholipids of Skeletal Muscle Mitochondria. Trends Endocrinol. Metab. 2016, 27, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd; Maack, C. SR and mitochondria: Calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 2013, 55, 42–49. [Google Scholar] [CrossRef]
- Piquereau, J.; Novotova, M.; Fortin, D.; Garnier, A.; Ventura-Clapier, R.; Veksler, V.; Joubert, F. Postnatal development of mouse heart: Formation of energetic microdomains. J. Physiol. 2010, 588 Pt 13, 2443–2454. [Google Scholar] [CrossRef]
- Yang, Y.; Lee, M.; Fairn, G.D. Phospholipid subcellular localization and dynamics. J. Biol. Chem. 2018, 293, 6230–6240. [Google Scholar] [CrossRef] [Green Version]
- Adhyapak, P.; Kapoor, S. Membrane Dynamics in Health and Disease: Impact on Cellular Signalling. J. Membr. Biol. 2019, 252, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, E.; Crea, E.; Torelli, L.; Bergamo, A.; Reggiani, C.; Sava, G.; Toniolo, L. Age Dependent Modification of the Metabolic Profile of the Tibialis Anterior Muscle Fibers in C57BL/6J Mice. Int. J. Mol. Sci. 2020, 21, 3923. [Google Scholar] [CrossRef] [PubMed]
- Nohara, K.; Kim, E.; Wirianto, M.; Mileykovskaya, E.; Dowhan, W.; Chen, Z.; Yoo, S.H. Cardiolipin Synthesis in Skeletal Muscle Is Rhythmic and Modifiable by Age and Diet. Oxidative Med. Cell. Longev. 2020, 2020, 5304768. [Google Scholar] [CrossRef]
- Janikiewicz, J.; Szymanski, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszynski, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Yaffe, M.P. Dynamic mitochondria. Nat. Cell Biol. 1999, 1, E149–E150. [Google Scholar] [CrossRef]
- Bozdaganyan, M.E.; Lokhmatikov, A.V.; Voskoboynikova, N.; Cherepanov, D.A.; Steinhoff, H.J.; Shaitan, K.V.; Mulkidjanian, A.Y. Proton leakage across lipid bilayers: Oxygen atoms of phospholipid ester linkers align water molecules into transmembrane water wires. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, D.; Valentine, W.J.; Iizuka-Hishikawa, Y.; Shindou, H.; Shimizu, T. Metabolism and functions of docosahexaenoic acid-containing membrane glycerophospholipids. FEBS Lett. 2017, 591, 2730–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124 Pt 1, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Tyler, A.I.I.; Greenfield, J.L.; Seddon, J.M.; Brooks, N.J.; Purushothaman, S. Coupling Phase Behavior of Fatty Acid Containing Membranes to Membrane Bio-Mechanics. Front. Cell Dev. Biol. 2019, 7, 187. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Steinhardt, R.A. Plasma membrane disruption: Repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Witt, S.N. Ethanolamine and Phosphatidylethanolamine: Partners in Health and Disease. Oxidative Med. Cell. Longev. 2017, 2017, 4829180. [Google Scholar] [CrossRef] [Green Version]
- Lamari, F.; Mochel, F.; Sedel, F.; Saudubray, J.M. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: Toward a new category of inherited metabolic diseases. J. Inherit. Metab. Dis. 2013, 36, 411–425. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Espeel, M.; Almeida, L.; Reimer, A.; Bosboom, D.; Roels, F.; de Brouwer, A.P.; Wevers, R.A. Inborn errors of metabolism in the biosynthesis and remodelling of phospholipids. J. Inherit. Metab. Dis. 2015, 38, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.G.; Valianpour, F.; Bowen, V.M.; Lam, J.; Duran, M.; Vaz, F.M.; Wanders, R.J. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update. Am. J. Med. Genet. A 2004, 126A, 349–354. [Google Scholar] [CrossRef]
- Damschroder, D.; Reynolds, C.; Wessells, R. Drosophila tafazzin mutants have impaired exercise capacity. Physiol. Rep. 2018, 6, e13604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegallai, H.M.; Hatch, G.M. Barth syndrome: Cardiolipin, cellular pathophysiology, management, and novel therapeutic targets. Mol. Cell. Biochem. 2021, 476, 1605–1629. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Sengers, R.C.; Trijbels, J.M.; Willems, J.L.; Daniels, O.; Stadhouders, A.M. Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise. J. Pediatrics 1975, 86, 873–880. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, Z.; Ding, N.; Yang, M.; Zhang, L.; Fan, X.; Zhou, Y.; Zou, Q.; Hou, J.; Zheng, J.; et al. The role of AGK in thrombocytopoiesis and possible therapeutic strategies. Blood 2020, 136, 119–129. [Google Scholar] [CrossRef]
- Allali, S.; Dorboz, I.; Samaan, S.; Slama, A.; Rambaud, C.; Boespflug-Tanguy, O.; Sarret, C. Mutation in the AGK gene in two siblings with unusual Sengers syndrome. Metab. Brain Dis. 2017, 32, 2149–2154. [Google Scholar] [CrossRef]
- Zeharia, A.; Shaag, A.; Houtkooper, R.H.; Hindi, T.; de Lonlay, P.; Erez, G.; Hubert, L.; Saada, A.; de Keyzer, Y.; Eshel, G.; et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am. J. Hum. Genet. 2008, 83, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Donkor, J.; Sariahmetoglu, M.; Dewald, J.; Brindley, D.N.; Reue, K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 2007, 282, 3450–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csaki, L.S.; Reue, K. Lipins: Multifunctional lipid metabolism proteins. Annu. Rev. Nutr. 2010, 30, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, T.; Nemazanyy, I.; Paolini, C.; Tatsuta, T.; Crespin, P.; de Villeneuve, D.; Brodesser, S.; Benit, P.; Rustin, P.; Baraibar, M.A.; et al. Lipin1 deficiency causes sarcoplasmic reticulum stress and chaperone-responsive myopathy. EMBO J. 2019, 38, e99576. [Google Scholar] [CrossRef]
- Reilich, P.; Horvath, R.; Krause, S.; Schramm, N.; Turnbull, D.M.; Trenell, M.; Hollingsworth, K.G.; Gorman, G.S.; Hans, V.H.; Reimann, J. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J. Neurol. 2011, 258, 1987–1997. [Google Scholar] [CrossRef]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Guo, G.; Li, M.; Chen, S.; Chen, K.; Chen, X.; Song, J.; Hu, S. The homozygous variant c.245G > A/p.G82D in PNPLA2 is associated with arrhythmogenic cardiomyopathy phenotypic manifestations. Clin. Genet. 2019, 96, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hirano, K.I.; Ikeda, Y.; Higashi, M.; Hashimoto, C.; Zhang, B.; Kozawa, J.; Sugimura, K.; Miyauchi, H.; Suzuki, A.; et al. Triglyceride deposit cardiomyovasculopathy: A rare cardiovascular disorder. Orphanet J Rare Dis 2019, 14, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sher, R.B.; Aoyama, C.; Huebsch, K.A.; Ji, S.; Kerner, J.; Yang, Y.; Frankel, W.N.; Hoppel, C.L.; Wood, P.A.; Vance, D.E.; et al. A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 2006, 281, 4938–4948. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, S.; Hatakeyama, H.; Karahashi, M.; Koumura, T.; Nonaka, I.; Hayashi, Y.K.; Noguchi, S.; Sher, R.B.; Nakagawa, Y.; Manfredi, G.; et al. Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy. Hum. Mol. Genet. 2011, 20, 3841–3851. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Ohkuma, A.; Talim, B.; Karahashi, M.; Koumura, T.; Aoyama, C.; Kurihara, M.; Quinlivan, R.; Sewry, C.; Mitsuhashi, H.; et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am. J. Hum. Genet. 2011, 88, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Sayed-Zahid, A.A.; Sher, R.B.; Sukoff Rizzo, S.J.; Anderson, L.C.; Patenaude, K.E.; Cox, G.A. Functional rescue in a mouse model of congenital muscular dystrophy with megaconial myopathy. Hum. Mol. Genet. 2019, 28, 2635–2647. [Google Scholar] [CrossRef]
- Blondelle, J.; Ohno, Y.; Gache, V.; Guyot, S.; Storck, S.; Blanchard-Gutton, N.; Barthelemy, I.; Walmsley, G.; Rahier, A.; Gadin, S.; et al. HACD1, a regulator of membrane composition and fluidity, promotes myoblast fusion and skeletal muscle growth. J. Mol. Cell Biol. 2015, 7, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Prola, A.; Blondelle, J.; Vandestienne, A.; Piquereau, J.; Denis, R.G.P.; Guyot, S.; Chauvin, H.; Mourier, A.; Maurer, M.; Henry, C.; et al. Cardiolipin content controls mitochondrial coupling and energetic efficiency in muscle. Sci. Adv. 2021, 7, eabd6322. [Google Scholar] [CrossRef] [PubMed]
- Abbasi-Moheb, L.; Westenberger, A.; Alotaibi, M.; Alghamdi, M.A.; Hertecant, J.L.; Ariamand, A.; Beetz, C.; Rolfs, A.; Bertoli-Avella, A.M.; Bauer, P. Biallelic loss-of-function HACD1 variants are a bona fide cause of congenital myopathy. Clin. Genet. 2021, 99, 513–518. [Google Scholar] [CrossRef]
- Gregersen, N. The acyl-CoA dehydrogenation deficiencies. Recent advances in the enzymic characterization and understanding of the metabolic and pathophysiological disturbances in patients with acyl-CoA dehydrogenation deficiencies. Scand. J. Clin. Lab. Investig. Suppl. 1985, 174, 1–60. [Google Scholar]
- Strauss, A.W.; Powell, C.K.; Hale, D.E.; Anderson, M.M.; Ahuja, A.; Brackett, J.C.; Sims, H.F. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proc. Natl. Acad. Sci. USA 1995, 92, 10496–10500. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Flogel, U.; Spiekerkoetter, U. Sexual dimorphism of lipid metabolism in very long-chain acyl-CoA dehydrogenase deficient (VLCAD-/-) mice in response to medium-chain triglycerides (MCT). Biochim. Biophys. Acta 2015, 1852, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Alatibi, K.I.; Wehbe, Z. Altered Metabolic Flexibility in Inherited Metabolic Diseases of Mitochondrial Fatty Acid Metabolism. Int. J. Mol. Sci. 2021, 22, 3799. [Google Scholar] [CrossRef]
- Vengalil, S.; Preethish-Kumar, V.; Polavarapu, K.; Christopher, R.; Gayathri, N.; Natarajan, A.; Manjunath, M.; Nashi, S.; Prasad, C.; Nalini, A. Fatty acid oxidation defects presenting as primary myopathy and prominent dropped head syndrome. Neuromuscul. Disord. 2017, 27, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.A. New genetic defects in mitochondrial fatty acid oxidation and carnitine deficiency. Adv. Pediatrics 1987, 34, 59–88. [Google Scholar]
- Tolwani, R.J.; Hamm, D.A.; Tian, L.; Sharer, J.D.; Vockley, J.; Rinaldo, P.; Matern, D.; Schoeb, T.R.; Wood, P.A. Medium-chain acyl-CoA dehydrogenase deficiency in gene-targeted mice. PLoS Genet. 2005, 1, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.C.; Tajika, M.; Shimura, M.; Carey, K.T.; Stroud, D.A.; Murayama, K.; Ohtake, A.; McKenzie, M. Loss of the Mitochondrial Fatty Acid beta-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function. Sci. Rep. 2018, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Scheuerman, O.; Wanders, R.J.; Waterham, H.R.; Dubnov-Raz, G.; Garty, B.Z. Mitochondrial trifunctional protein deficiency with recurrent rhabdomyolysis. Pediatric Neurol. 2009, 40, 465–467. [Google Scholar] [CrossRef]
- Ibdah, J.A.; Paul, H.; Zhao, Y.; Binford, S.; Salleng, K.; Cline, M.; Matern, D.; Bennett, M.J.; Rinaldo, P.; Strauss, A.W. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Investig. 2001, 107, 1403–1409. [Google Scholar] [CrossRef] [Green Version]
- Fraser, H.; Geppert, J.; Johnson, R.; Johnson, S.; Connock, M.; Clarke, A.; Taylor-Phillips, S.; Stinton, C. Evaluation of earlier versus later dietary management in long-chain 3-hydroxyacyl-CoA dehydrogenase or mitochondrial trifunctional protein deficiency: A systematic review. Orphanet J. Rare Dis. 2019, 14, 258. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, M.L.; Hershko, C.; Eisenberg, S.; Sagher, F. Ichthyosiform dermatosis with systemic lipidosis. Arch. Dermatol. 1974, 110, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, C.; Jobard, F.; Caux, F.; Bouadjar, B.; Karaduman, A.; Heilig, R.; Lakhdar, H.; Wollenberg, A.; Verret, J.L.; Weissenbach, J.; et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 2001, 69, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberer, M.; Boeszoermenyi, A.; Nagy, H.M.; Zechner, R. Recent insights into the structure and function of comparative gene identification-58. Curr. Opin. Lipidol. 2011, 22, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jebessa, Z.H.; Shanmukha, K.D.; Dewenter, M.; Lehmann, L.H.; Xu, C.; Schreiter, F.; Siede, D.; Gong, X.M.; Worst, B.C.; Federico, G.; et al. The lipid droplet-associated protein ABHD5 protects the heart through proteolysis of HDAC4. Nat. Metab. 2019, 1, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Radner, F.P.; Streith, I.E.; Schoiswohl, G.; Schweiger, M.; Kumari, M.; Eichmann, T.O.; Rechberger, G.; Koefeler, H.C.; Eder, S.; Schauer, S.; et al. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58). J. Biol. Chem. 2010, 285, 7300–7311. [Google Scholar] [CrossRef] [Green Version]
- North, K.N.; Hoppel, C.L.; De Girolami, U.; Kozakewich, H.P.; Korson, M.S. Lethal neonatal deficiency of carnitine palmitoyltransferase II associated with dysgenesis of the brain and kidneys. J. Pediatrics 1995, 127, 414–420. [Google Scholar] [CrossRef]
- Corti, S.; Bordoni, A.; Ronchi, D.; Musumeci, O.; Aguennouz, M.; Toscano, A.; Lamperti, C.; Bresolin, N.; Comi, G.P. Clinical features and new molecular findings in Carnitine Palmitoyltransferase II (CPT II) deficiency. J. Neurol. Sci. 2008, 266, 97–103. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Hasek, L.Y.; Harris, K.L.; Berman, A.G.; Damen, F.W.; Goergen, C.J.; Ellis, J.M. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J. Biol. Chem. 2017, 292, 18443–18456. [Google Scholar] [CrossRef] [Green Version]
- Land, J.M.; Mistry, S.; Squier, M.; Hope, P.; Ghadiminejad, I.; Orford, M.; Saggerson, D. Neonatal carnitine palmitoyltransferase-2 deficiency: A case presenting with myopathy. Neuromuscul. Disord. 1995, 5, 129–137. [Google Scholar] [CrossRef]
- Begam, M.; Roche, R.; Hass, J.J.; Basel, C.A.; Blackmer, J.M.; Konja, J.T.; Samojedny, A.L.; Collier, A.F.; Galen, S.S.; Roche, J.A. The effects of concentric and eccentric training in murine models of dysferlin-associated muscular dystrophy. Muscle Nerve 2020, 62, 393–403. [Google Scholar] [CrossRef]
- Kobayashi, K.; Izawa, T.; Kuwamura, M.; Yamate, J. Dysferlin and animal models for dysferlinopathy. J. Toxicol. Pathol. 2012, 25, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Roche, J.A.; Ru, L.W.; Bloch, R.J. Distinct effects of contraction-induced injury in vivo on four different murine models of dysferlinopathy. J. Biomed. Biotechnol. 2012, 2012, 134031. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.M.L.; Ong, C.C.; Tan, K.B.; Chin, H.L.; Paliwal, P.R.; Ng, K.W.P.; Lin, W. Subclinical Cardiomyopathy in Miyoshi Myopathy Detected by Late Gadolinium Enhancement Cardiac Magnetic Resonance Imaging. Int. Heart J. 2021, 62, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Benabdellah, F.; Yu, H.; Brunelle, A.; Laprevote, O.; De La Porte, S. MALDI reveals membrane lipid profile reversion in MDX mice. Neurobiol. Dis. 2009, 36, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabaj, I.; Ferey, J.; Marguet, F.; Gilard, V.; Basset, C.; Bahri, Y.; Brehin, A.C.; Vanhulle, C.; Leturcq, F.; Marret, S.; et al. Muscle metabolic remodelling patterns in Duchenne muscular dystrophy revealed by ultra-high-resolution mass spectrometry imaging. Sci. Rep. 2021, 11, 1906. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Models Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [Green Version]
- Senoo, N.; Miyoshi, N.; Kobayashi, E.; Morita, A.; Tanihata, J.; Takeda, S.; Miura, S. Glycerophospholipid profile alterations are associated with murine muscle-wasting phenotype. Muscle Nerve 2020, 62, 413–418. [Google Scholar] [CrossRef]
- Touboul, D.; Piednoel, H.; Voisin, V.; De La Porte, S.; Brunelle, A.; Halgand, F.; Laprevote, O. Changes of phospholipid composition within the dystrophic muscle by matrix-assisted laser desorption/ionization mass spectrometry and mass spectrometry imaging. Eur. J. Mass Spectrom. 2004, 10, 657–664. [Google Scholar] [CrossRef]
- Tuazon, M.A.; Henderson, G.C. Fatty acid profile of skeletal muscle phospholipid is altered in mdx mice and is predictive of disease markers. Metabolism 2012, 61, 801–811. [Google Scholar] [CrossRef]
- Woodman, S.E.; Park, D.S.; Cohen, A.W.; Cheung, M.W.; Chandra, M.; Shirani, J.; Tang, B.; Jelicks, L.A.; Kitsis, R.N.; Christ, G.J. Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J. Biol. Chem. 2002, 277, 38988–38997. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.S.; Nisr, R.B.; Stretton, C.; Krasteva-Christ, G.; Hundal, H.S. Caveolin-3 deficiency associated with the dystrophy P104L mutation impairs skeletal muscle mitochondrial form and function. J. Cachexia Sarcopenia Muscle 2020, 11, 838–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, W.; Han, C.; Wu, J.; Zhang, H.; Tong, X. Mutation in the caveolin-3 gene causes asymmetrical distal myopathy. Neuropathology 2016, 36, 485–489. [Google Scholar] [CrossRef]
- Pradhan, B.S.; Proszynski, T.J. A Role for Caveolin-3 in the Pathogenesis of Muscular Dystrophies. Int. J. Mol. Sci. 2020, 21, 8736. [Google Scholar] [CrossRef] [PubMed]
- Garlid, A.O.; Schaffer, C.T.; Kim, J.; Bhatt, H.; Guevara-Gonzalez, V.; Ping, P. TAZ encodes tafazzin, a transacylase essential for cardiolipin formation and central to the etiology of Barth syndrome. Gene 2020, 726, 144148. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Kelley, R.I.; Feigenbaum, A.; Towbin, J.A.; Heerdt, P.M.; Schieble, T.; Wanders, R.J.; DiMauro, S.; Blanck, T.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003, 42, 1994–1999. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, V.A.; Mikhaeil, J.S.; Leveille, C.F.; Saint, C.; LeBlanc, P.J. Cardiolipin content, linoleic acid composition, and tafazzin expression in response to skeletal muscle overload and unload stimuli. Sci. Rep. 2017, 7, 2060. [Google Scholar] [CrossRef] [Green Version]
- Ikon, N.; Ryan, R.O. Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy. Lipids 2017, 52, 99–108. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J. Mol. Biol. 2006, 361, 462–469. [Google Scholar] [CrossRef]
- Wasmus, C.; Dudek, J. Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies. Life 2020, 10, 277. [Google Scholar] [CrossRef]
- Bektas, M.; Payne, S.G.; Liu, H.; Goparaju, S.; Milstien, S.; Spiegel, S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J. Cell Biol. 2005, 169, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Vukotic, M.; Nolte, H.; Konig, T.; Saita, S.; Ananjew, M.; Kruger, M.; Tatsuta, T.; Langer, T. Acylglycerol Kinase Mutated in Sengers Syndrome Is a Subunit of the TIM22 Protein Translocase in Mitochondria. Mol. Cell 2017, 67, 471–483.e7. [Google Scholar] [CrossRef]
- Kang, Y.; Stroud, D.A.; Baker, M.J.; De Souza, D.P.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.R. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470.e5. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Nishino, I. Phospholipid synthetic defect and mitophagy in muscle disease. Autophagy 2011, 7, 1559–1561. [Google Scholar] [CrossRef]
- Marchet, S.; Invernizzi, F.; Blasevich, F.; Bruno, V.; Dusi, S.; Venco, P.; Fiorillo, C.; Baranello, G.; Pallotti, F.; Lamantea, E.; et al. Alteration of mitochondrial membrane inner potential in three Italian patients with megaconial congenital muscular dystrophy carrying new mutations in CHKB gene. Mitochondrion 2019, 47, 24–29. [Google Scholar] [CrossRef]
- Muhammad, E.; Reish, O.; Ohno, Y.; Scheetz, T.; Deluca, A.; Searby, C.; Regev, M.; Benyamini, L.; Fellig, Y.; Kihara, A.; et al. Congenital myopathy is caused by mutation of HACD1. Hum. Mol. Genet. 2013, 22, 5229–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paran, C.W.; Zou, K.; Ferrara, P.J.; Song, H.; Turk, J.; Funai, K. Lipogenesis mitigates dysregulated sarcoplasmic reticulum calcium uptake in muscular dystrophy. Biochim. Et Biophys. Acta 2015, 1851, 1530–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grounds, M.D.; Terrill, J.R.; Radley-Crabb, H.G.; Robertson, T.; Papadimitriou, J.; Spuler, S.; Shavlakadze, T. Lipid accumulation in dysferlin-deficient muscles. Am. J. Pathol. 2014, 184, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Haynes, V.R.; Keenan, S.N.; Bayliss, J.; Lloyd, E.M.; Meikle, P.J.; Grounds, M.D.; Watt, M.J. Dysferlin deficiency alters lipid metabolism and remodels the skeletal muscle lipidome in mice. J. Lipid Res. 2019, 60, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, K.R.; McMillan, J.D.; Kumar, A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J. Cell. Physiol. 2018, 233, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signaling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef] [PubMed]



| Fatty Acid | 18:1 | 18:2 | 18:3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mass and Formula | 282.26 and C18H34O2 | 280.24 and C18H32O2 | 278.22 and C18H30O2 | ||||||
| Systemic Name | Octadecenoic acid | Octadecadienoic acid | Octadecatrienoic acid | ||||||
| Number of Lipid Records | 43 | 101 | 63 | ||||||
| Common Name | Double Bond Position | Number of PubMed References | Common Name | Double Bond Position | Number of PubMed References | Common Name | Double Bond Position | Number of PubMed References | |
| Example | Oleic acid | 9Z | 23,694 | Linoleic acid | 9Z, 12Z | 24,310 | α-Linolenic acid | 9Z, 12Z, 15Z | 7036 |
| Trans- vaccenic acid | 11E | 325 | Rumenic acid | 9Z, 11E | 316 | γ-Linolenic acid | 6Z, 9Z, 12Z | 3145 | |
| Cis-vaccenic acid | 11Z | 202 | Linoelaidic acid | 9E, 12E | 25 | Punicic acid | 9Z, 11E, 13Z | 111 | |
| Petroselinic acid | 6Z | 67 | Sebaleic acid | 5Z, 8Z | 9 | Pinolenic acid | 5Z, 9Z, 12Z | 56 | |
| Petroselaidic acid | 6E | 4 | Vaccelenic acid | 11E, 15Z | 2 | Jacaric acid | 8Z, 10E, 12Z | 13 | |
| Gene Mutated | Protein and Function | Disease | Muscular Symptoms | Lipid Disorders | Animal Experimental Models | Ref. | |
|---|---|---|---|---|---|---|---|
| ORPHA 352312: group of disorders of PL, sphingolipids and FA biosynthesis with skeletal muscle characteristics | TAZ chromosome X | Tafazzin Phospholipid-lysophospholipid transacylase Catalyzes remodeling of immature CL to its mature composition | Barth syndrome | Dilated cardiomyopathy Skeletal muscle weakness | Reduced level of CL Increased level of mono-lysocardiolipin |
| [71,72,73] |
| AGK chromosome 7 | AGK Acylglycerol kinase Phosphorylates MAG and DAG to form LPA and PA | Sengers syndrome | Hypertrophic cardiomyopathy Skeletal myopathy Congenital cataract | Defects in mitochondria and storage of lipids in muscle (skeletal and heart) |
| [74,75,76,77] | |
| LPIN 1 chromosome 2 | Lipin-1 Phosphatidate phosphatase Catalyzes the conversion of PA to DAG | Myoglobinuria | Rhabdomyolysis Muscle fiber loss | Hyper-triglyceridemia Accumulation of PL intermediates |
| [78,79,80,81] | |
| PNPLA2 chromosome 11 | ATGL Adipose triglyceride lipase Catalyses the hydrolysis of TAG into DAG | Slowly progressive myopathy: neutral lipid storage disease with myopathy (NLSDM) Arrhythmogenic cardiomyopathy | Distal muscle weakness Cardiomyopathy | Accumulation of lipid droplets(TAG) in multiple tissues Altered energy metabolism Lipidosis of internal organs |
| [82,83,84,85] | |
| CHKB chromosome 22 | CHKB Choline kinase β Catalyzes the first step in PE biosynthesis | Megaconial congenital muscular dystrophy | Mitochondrial structural (enlargement) and functional abnormalities | Decreased level of PC |
| [86,87,88,89] | |
| Disease not listed in ORPHA 352312 despite gene mutation in gene involved in PL biosynthesis, sphingolipids, and FA synthesis | HACD1 chromosome 10 | HACD1 3-Hydroxyacyl-CoA dehydratase 1 Catalyzes the third of the four reactions of the long-chain FA elongation cycle | Congenital fiber-type disproportion, rare internalized nuclei | Congenital myopathy with proximal ± distal muscle weakness, progressively improving over time | Increased concentrations of ≥ C18 and mono-unsaturated FA |
| [90,91,92] |
| ACADVL chromosome 17 | VLCAD Very long chain Acyl-coA dehydrogenase Involved in the β-oxidation pathway (FA longer than C16) | VLCADD (VLCAD deficiency) | Cardiomyopathy Myopathy with intermittent myalgia and rhabdomyolysis, progressive muscle weakness, often dropped head | Defective long-chain FA oxidation |
| [93,94,95,96,97] | |
| ACADM chromosome 1 | MCAD Medium-chain acyl-coenzyme A dehydrogenase Involved in the β-oxidation pathway (C4 to C12 acyl chain) | Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) | Myopathy exertion induced myalgia, progressive proximal limb weakness Secondary carnitine deficiency | Accumulation of FA intermediates |
| [98,99,100] | |
| HADHA chromosome 2 | MTP Mitochondrial trifunctional protein Catalyzes three out of the four steps in beta oxidation | Mitochondrial trifunctional protein deficiency (MTP deficiency or MTPD) | Skeletal myopathy, episodic rhabdomyolysis, peripheral neuropathy, cardiomyopathy in severe cases | Decreased long-chain FA oxidation |
| [101,102,103] | |
| CGI-58 chromosome 3 | ABHD5 Alpha/beta hydrolase domain containing 5 Lipid droplet-associated protein that activates ATGL | Chanarin–Dorfman syndrome (neutral lipid storage disease) | Moderate myopathy Possible cardiomyopathy | Accumulation of TAG droplets |
| [104,105,106,107,108] | |
| CPT2 chromosome 1 | CPT2 Carnitine palmitoyltransferase 2 Catalyzes the transfer of FA from cytoplasm to mitochondria | Carnitine palmitoyl-transferase II deficiency | Episodic myalgia, muscle weakness, and rhabdomyolysis with myoglobinuria | Reduced the rates of long-chain FA oxidation into carbon dioxide |
| [109,110,111,112] | |
| Alteration of PL in inherited form of myopathies due to mutation in genes without a direct link to PL biosynthesis | DYSF chromosome 2 | Dysferlin Involved in the sarcolemma repair mechanism, interacts with caveolin 3 | Dysferlinopathy, including: limb girdle muscular dystrophy type 2B (LGMD2B) Miyoshi myopathy | Proximal, distal, or proximo-distal myopathy, predominantly involving lower limbs Rhabdomyolysis | Early intra-myocellular lipid accumulation (sphingolipids, PLs, cholesterol) |
| [113,114,115,116] |
| DMD chromosome X | Dystrophin Involved in the connection of the cytoskeleton of a muscle fiber to the extracellular matrix through the cell membrane | Duchenne muscular dystrophy Becker muscular dystrophy | Cardiomyopathy with myocardial fibrosis, muscle weakness and muscle fiber necrosis, respiratory failure | PL dyshomeostasis Increased PC and SM |
| [117,118,119,120,121,122,123] | |
| CAV3 chromosome 3 | Caveolin3 Component of caveolae plasma membrane (possible scaffolding role for caveolin-interacting molecules) | Rippling muscle disease LGMD Isolated familial hypertrophic cardiomyopathy HyperCKemia and myalgia | Skeletal muscle weakness Muscle hyperirritability triggered by stretch | Decreased CL |
| [124,125,126,127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bargui, R.; Solgadi, A.; Prost, B.; Chester, M.; Ferreiro, A.; Piquereau, J.; Moulin, M. Phospholipids: Identification and Implication in Muscle Pathophysiology. Int. J. Mol. Sci. 2021, 22, 8176. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158176
Bargui R, Solgadi A, Prost B, Chester M, Ferreiro A, Piquereau J, Moulin M. Phospholipids: Identification and Implication in Muscle Pathophysiology. International Journal of Molecular Sciences. 2021; 22(15):8176. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158176
Chicago/Turabian StyleBargui, Rezlène, Audrey Solgadi, Bastien Prost, Mélanie Chester, Ana Ferreiro, Jérôme Piquereau, and Maryline Moulin. 2021. "Phospholipids: Identification and Implication in Muscle Pathophysiology" International Journal of Molecular Sciences 22, no. 15: 8176. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158176