
Features of Lipid Metabolism in Humanized ApoE Knockin Rat Models

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Expression of Rat ApoE and Human ApoE in WT, hApoE KI, and ApoE KO Rats
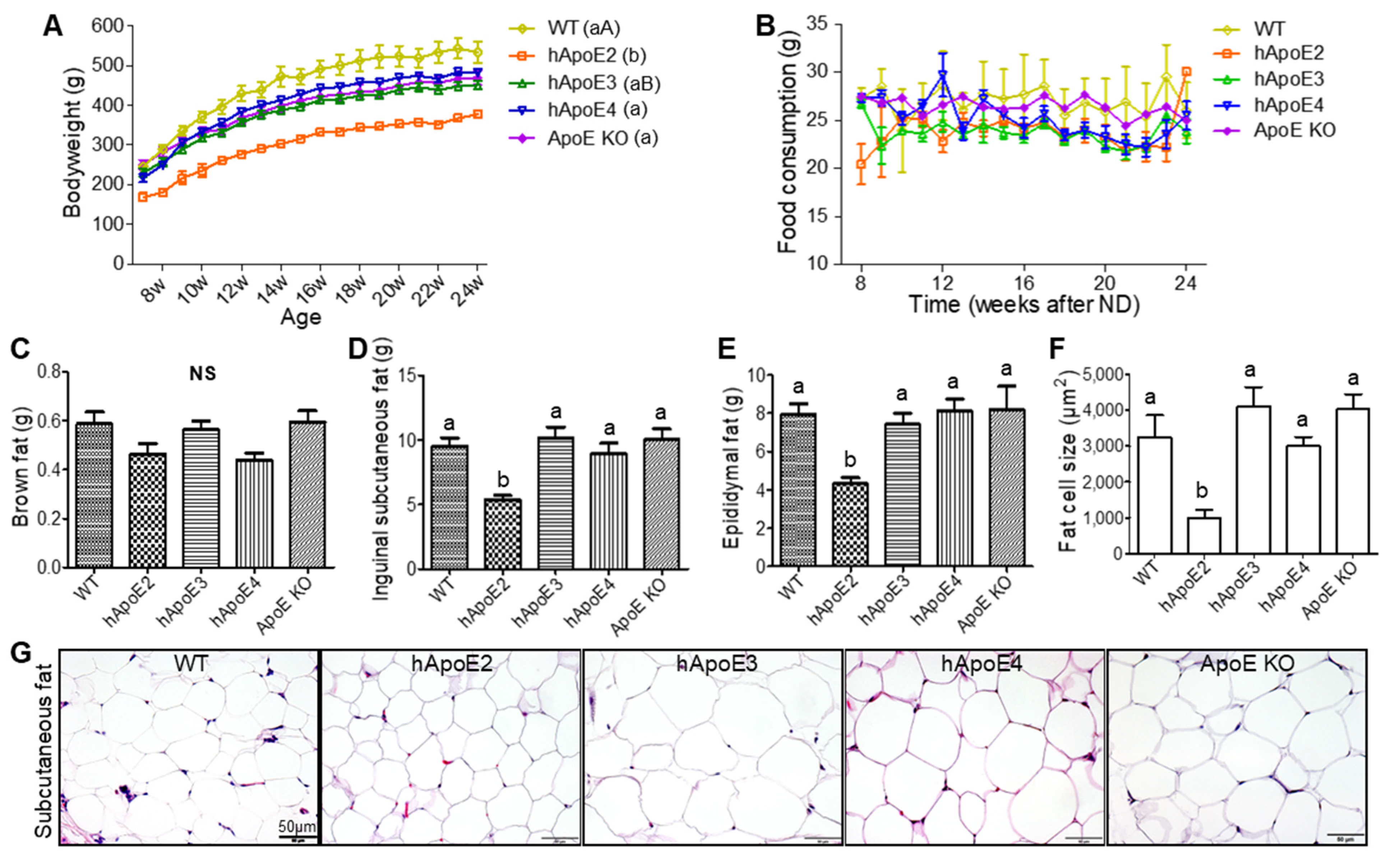
2.2. Bodyweight, Food Consumption, and Fat Mass
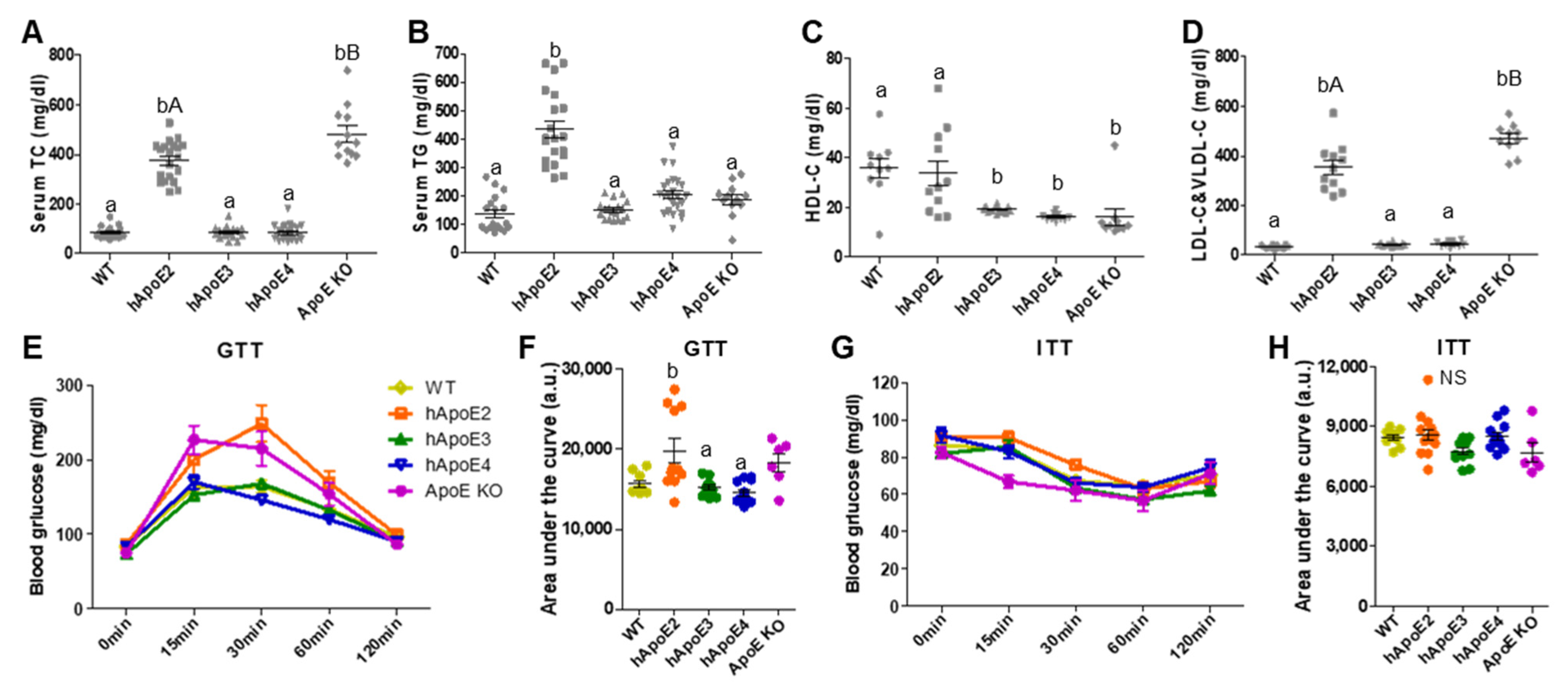
2.3. Serum Lipid and Glucose Metabolism Profiles
2.4. Ectopic Lipid Deposition In Vital Organs and Hepatic Nodular Cirrhosis in hApoE2 Rats
2.5. Mild Atherosclerotic Lesion in hApoE2 Rats
2.6. Premature Death of hApoE2 and ApoE KO Rats after a Short Period of PD
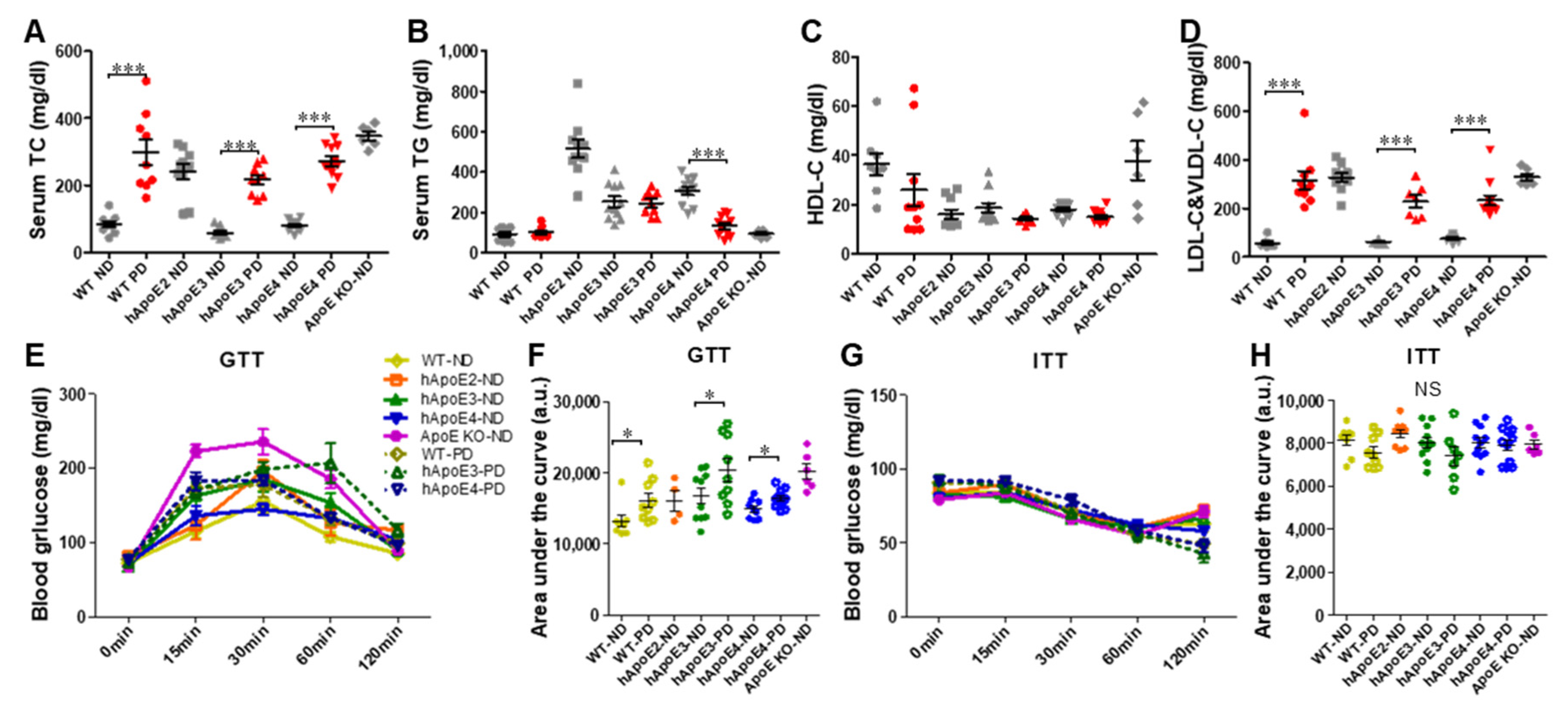
2.7. Metabolic Profiles of WT, hApoE3, and E4 Rats after 4-Month PD
2.8. Thickening of Small Arteries in hApoE4 Rats Fed with 4-Month PD
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Genotyping and Western Blotting
4.3. Diet, Bodyweight, and Food Consumption
4.4. Lifespan
4.5. Lipid Measurement
4.6. Blood Glucose Tests
4.7. Histology
4.8. Randomization/Double Blind
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CVDs | cardiovascular diseases |
| CHD | coronary heart disease |
| AD | Alzheimer’s disease |
| WT | wild-type |
| ApoE | apolipoprotein E |
| ApoE KO | ApoE knockout |
| hApoE KI | humanized ApoE knockin |
| PD | Paigen diet |
| ND | normal diet |
| TC | total cholesterol |
| TG | total triglyceride |
| GTT | glucose tolerance test |
| ITT | insulin tolerance test |
References
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E—A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Bock, H.H. Lipoprotein receptors in the nervous system. Annu. Rev. Biochem. 2002, 71, 405–434. [Google Scholar] [CrossRef]
- Herz, J.; Chen, Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 2006, 7, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.M.; Perez-Tilve, D.; Greer, T.M.; Coburn, B.A.; Grant, E.; Basford, J.E.; Tschop, M.H.; Hui, D.Y. Defective lipid delivery modulates glucose tolerance and metabolic response to diet in apolipoprotein E-deficient mice. Diabetes 2008, 57, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid. Res. 2009, 50 (Suppl. 1), 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.T.; Kuzawa, C.W.; Hayes, M.G. Worldwide allele frequencies of the human apolipoprotein E gene: Climate, local adaptations, and evolutionary history. Am. J. Phys. Anthropol. 2010, 143, 100–111. [Google Scholar] [CrossRef]
- Phillips, M.C. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeren, J.; Beisiegel, U.; Grewal, T. Apolipoprotein E recycling: Implications for dyslipidemia and atherosclerosis. Arterioscler. Thromb Vasc. Biol. 2006, 26, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Corbo, R.M.; Scacchi, R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele? Ann. Hum. Genet. 1999, 63 Pt 4, 301–310. [Google Scholar] [CrossRef]
- Wilson, P.W.; Myers, R.H.; Larson, M.G.; Ordovas, J.M.; Wolf, P.A.; Schaefer, E.J. Apolipoprotein E alleles, dyslipidemia, and coronary heart disease. The Framingham Offspring Study. JAMA 1994, 272, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.; Mevissen, V.; Krantz, C.; Kimmel, M.; Erdmann, J.; Hoffmann, R.; Zerres, K.; Ortlepp, J.R. Robust association of the APOE epsilon4 allele with premature myocardial infarction especially in patients without hypercholesterolaemia: The Aachen study. Eur. J. Clin. Investig. 2007, 37, 106–108. [Google Scholar] [CrossRef]
- Piedrahita, J.A.; Zhang, S.H.; Hagaman, J.R.; Oliver, P.M.; Maeda, N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. USA 1992, 89, 4471–4475. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.M.; Mezdour, H.; Aratani, Y.; Knouff, C.; Najib, J.; Reddick, R.L.; Quarfordt, S.H.; Maeda, N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 1997, 272, 17972–17980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.M.; Mezdour, H.; Quarfordt, S.H.; Maeda, N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J. Clin. Investig. 1998, 102, 130–135. [Google Scholar] [CrossRef]
- Knouff, C.; Hinsdale, M.E.; Mezdour, H.; Altenburg, M.K.; Watanabe, M.; Quarfordt, S.H.; Sullivan, P.M.; Maeda, N. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J. Clin. Investig. 1999, 103, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Marino, A.; Zhang, Y.; Rubinelli, L.; Riemma, M.A.; Ip, J.E.; Di Lorenzo, A. Pressure overload leads to coronary plaque formation, progression, and myocardial events in ApoE-/- mice. JCI Insight 2019, 4, e128220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellenbroek, B.; Youn, J. Rodent models in neuroscience research: Is it a rat race? Dis. Model. Mech. 2016, 9, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Iannaccone, P.M.; Jacob, H.J. Rats! Dis. Model. Mech. 2009, 2, 206–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rune, I.; Rolin, B.; Lykkesfeldt, J.; Nielsen, D.S.; Krych, L.; Kanter, J.E.; Bornfeldt, K.E.; Kihl, P.; Buschard, K.; Josefsen, K.; et al. Long-term Western diet fed apolipoprotein E-deficient rats exhibit only modest early atherosclerotic characteristics. Sci. Rep. 2018, 8, 5416. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, A.; Simsekyilmaz, S.; Liehn, E.; Rusu, M.; Schaaps, N.; Afify, M.; Florescu, R.; Almalla, M.; Borinski, M.; Vogt, F. Apolipoprotein E deficient rats generated via zinc-finger nucleases exhibit pronounced in-stent restenosis. Sci. Rep. 2019, 9, 18153. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.S.; Verghese, P.B.; Chakraborty, C.; Joung, J.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekuni, D.; Endo, Y.; Tomofuji, T.; Azuma, T.; Irie, K.; Kasuyama, K.; Morita, M. Effects of apoE deficiency and occlusal disharmony on amyloid-beta production and spatial memory in rats. PLoS ONE 2013, 8, e74966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, P.; Grant, S.; Morrison, T.R.; Cai, X.; Iriah, S.; Kristal, B.S.; Honeycutt, J.; Brenhouse, H.; Hartner, J.C.; Madularu, D.; et al. Characterizing the human APOE epsilon 4 knock-in transgene in female and male rats with multimodal magnetic resonance imaging. Brain Res. 2020, 1747, 147030. [Google Scholar] [CrossRef]
- Sharabi, Y.; Eldad, A. Nonalcoholic fatty liver disease is associated with hyperlipidemia and obesity. Am. J. Med. 2000, 109, 171. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef]
- Zaragoza, C.; Gomez-Guerrero, C.; Martin-Ventura, J.L.; Blanco-Colio, L.; Lavin, B.; Mallavia, B.; Tarin, C.; Mas, S.; Ortiz, A.; Egido, J. Animal models of cardiovascular diseases. J. Biomed. Biotechnol. 2011, 2011, 497841. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Zadelaar, S.; Kleemann, R.; Verschuren, L.; de Vries-Van der Weij, J.; van der Hoorn, J.; Princen, H.M.; Kooistra, T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1706–1721. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Carballo-Jane, E.; McLaren, D.G.; Mendoza, V.H.; Gagen, K.; Geoghagen, N.S.; McNamara, L.A.; Gorski, J.N.; Eiermann, G.J.; Petrov, A.; et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J. Lipid. Res. 2012, 53, 51–65. [Google Scholar]
- Paigen, B.; Morrow, A.; Brandon, C.; Mitchell, D.; Holmes, P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis 1985, 57, 65–73. [Google Scholar] [CrossRef]
- Afroze, D.; Yousuf, A.; Tramboo, N.A.; Shah, Z.A.; Ahmad, A. ApoE gene polymorphism and its relationship with coronary artery disease in ethnic Kashmiri population. Clin. Exp. Med. 2016, 16, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Likidlilid, A.; Peerapatdit, T.; Tresukosol, D.; Srisuma, S.; Ratanamaneechat, S.; Sriratanasathavorn, C. Apolipoprotein E gene polymorphism: Effects on plasma lipids and risk of type 2 diabetes and coronary artery disease. Cardiovasc. Diabetol. 2012, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.H.; Maeda, N.; Mazzone, T. Expression of the human apoE2 isoform in adipocytes: Altered cellular processing and impaired adipocyte lipogenesis. J. Lipid. Res. 2011, 52, 1733–1741. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, J.V.; Kwok, M.K.; Schooling, C.M. Age and sex specific effects of APOE genotypes on ischemic heart disease and its risk factors in the UK Biobank. Sci. Rep. 2021, 11, 9229. [Google Scholar] [CrossRef]
- Lumsden, A.L.; Mulugeta, A.; Zhou, A.; Hypponen, E. Apolipoprotein E (APOE) genotype-associated disease risks: A phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine 2020, 59, 102954. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Kuchel, G.A.; Melzer, D. ApoE e2 and aging-related outcomes in 379,000 UK Biobank participants. Aging 2020, 12, 12222–12233. [Google Scholar] [CrossRef]
- Cash, J.G.; Kuhel, D.G.; Basford, J.E.; Jaeschke, A.; Chatterjee, T.K.; Weintraub, N.L.; Hui, D.Y. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 27876–27884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef]
- Martinez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.P.; Nielsen, H.M. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643. [Google Scholar] [CrossRef]
- Wildsmith, K.R.; Basak, J.M.; Patterson, B.W.; Pyatkivskyy, Y.; Kim, J.; Yarasheski, K.E.; Wang, J.X.; Mawuenyega, K.G.; Jiang, H.; Parsadanian, M.; et al. In vivo human apolipoprotein E isoform fractional turnover rates in the CNS. PLoS ONE 2012, 7, e38013. [Google Scholar]
- Rasmussen, K.L. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: A review. Atherosclerosis 2016, 255, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Kuhel, D.G.; Konaniah, E.S.; Basford, J.E.; McVey, C.; Goodin, C.T.; Chatterjee, T.K.; Weintraub, N.L.; Hui, D.Y. Apolipoprotein E2 accentuates postprandial inflammation and diet-induced obesity to promote hyperinsulinemia in mice. Diabetes 2013, 62, 382–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.S.; Watson, K.Q.; Rebeck, G.W. Metabolic Disturbances of a High-Fat Diet Are Dependent on APOE Genotype and Sex. eNeuro 2019, 6, ENEURO-0267. [Google Scholar] [CrossRef]
- Gao, J.; Katagiri, H.; Ishigaki, Y.; Yamada, T.; Ogihara, T.; Imai, J.; Uno, K.; Hasegawa, Y.; Kanzaki, M.; Yamamoto, T.T.; et al. Involvement of apolipoprotein E in excess fat accumulation and insulin resistance. Diabetes 2007, 56, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Xin, G.; Qiu, X.; Wang, Y.; Liu, G. Establishment of a rat model with diet-induced coronary atherosclerosis. J. Biomed. Res. 2016, 31, 47–55. [Google Scholar]
- Lepisto, P.V. Post-traumatic blood lipid changes and fat embolism. Relation of post-traumatic blood lipid changes and fat embolism syndrome. J. Trauma 1976, 16, 52–57. [Google Scholar] [CrossRef]
- Luo, J.Q.; Ren, H.; Banh, H.L.; Liu, M.Z.; Xu, P.; Fang, P.F.; Xiang, D.X. The Associations between Apolipoprotein E Gene Epsilon2/Epsilon3/Epsilon4 Polymorphisms and the Risk of Coronary Artery Disease in Patients with Type 2 Diabetes Mellitus. Front. Physiol. 2017, 8, 1031. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, P.N.; Wu, L.L.; Hunt, S.C.; Brinton, E.A. Plasma triglycerides and type III hyperlipidemia are independently associated with premature familial coronary artery disease. J. Am. Coll. Cardiol. 2005, 45, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- McBride, P. Triglycerides and risk for coronary artery disease. Curr. Atheroscler. Rep. 2008, 10, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, N.; Danesh, J.; Eiriksdottir, G.; Sigurdsson, G.; Wareham, N.; Bingham, S.; Boekholdt, S.M.; Khaw, K.T.; Gudnason, V. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation 2007, 115, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.L.; Evans, G.W.; Riley, W.A.; Sharrett, A.R.; Howard, G.; Barnes, R.W.; Rosamond, W.; Crow, R.S.; Rautaharju, P.M.; Heiss, G. Arterial wall thickness is associated with prevalent cardiovascular disease in middle-aged adults. The Atherosclerosis Risk in Communities (ARIC) Study. Stroke 1995, 26, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guallar, E.; Malhotra, S.; Astor, B.C.; Polak, J.F.; Qiao, Y.; Gomes, A.S.; Herrington, D.M.; Sharrett, A.R.; Bluemke, D.A.; et al. Carotid Artery Wall Thickness and Incident Cardiovascular Events: A Comparison between US and MRI in the Multi-Ethnic Study of Atherosclerosis (MESA). Radiology 2018, 289, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Gnasso, A.; Pujia, A.; Irace, C.; Mattioli, P.L. Increased carotid arterial wall thickness in common hyperlipidemia. Coron. Artery Dis. 1995, 6, 57–63. [Google Scholar] [CrossRef]
- Paigen, B.; Morrow, A.; Holmes, P.A.; Mitchell, D.; Williams, R.A. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 1987, 68, 231–240. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brown Fat (g) | Inguinal Subcutaneous Fat (g) | Epididymal Fat (g) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Average | n | SEM | Average | n | SEM | Average | n | SEM | |
| WT-ND | 0.59 | 9 | 0.05 | 9.53 | 9 | 0.62 | 7.96 | 9 | 0.53 |
| WT-PD | 0.77 * | 9 | 0.05 | 9.86 | 9 | 0.6 | 10.51 | 9 | 1.07 |
| hApoE2-ND | 0.46 | 9 | 0.04 | 5.4 | 9 | 0.3 | 4.33 | 9 | 0.32 |
| hApoE3-ND | 0.56 | 11 | 0.04 | 10.17 | 11 | 0.83 | 7.43 | 11 | 0.56 |
| hApoE3-PD | 0.66 | 9 | 0.03 | 11.72 | 9 | 0.86 | 9.05 | 9 | 0.76 |
| hApoE4-ND | 0.44 | 10 | 0.03 | 8.93 | 10 | 0.81 | 8.13 | 10 | 0.61 |
| hApoE4-PD | 0.51 | 11 | 0.03 | 11.17 * | 11 | 0.71 | 12.93 *** | 11 | 0.82 |
| ApoE KO-ND | 0.59 | 6 | 0.05 | 10.02 | 6 | 0.84 | 8.19 | 6 | 1.22 |
| Primers | Sequence (5′-3′) | WT Band | E2/3/4 Band | KO Band |
|---|---|---|---|---|
| Rat ApoE F | CGAGGGAGAGCTGGAGGT | 150 bp | 134 bp | |
| Rat ApoE R | TGTGTGACTTGGGAGCTCTG | |||
| Human ApoE F | CTGGAGGAACAACTGACCCC | 437 bp | ||
| Human ApoE R | CTGCCCATCTCCTCCATC | |||
| 5’ Cel-1 F | CACCCCGGGGTGCTGAGATAGAGAT | 455 bp | 455 bp | |
| 5’ Cel-1 R | TTCCACCATGTTGGGCTCCG | |||
| 3’ Cel-1 F | GTGCGCTCCAAGATGGAGGA | 388 bp | 388 bp | |
| 3’ Cel-1 R | GGCCATGGAATGTGTGCTATGTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Johnson, G.; Zhao, F.; Wu, Y.; Zhao, G.; Brown, A.; You, S.; Zou, M.-H.; Song, P. Features of Lipid Metabolism in Humanized ApoE Knockin Rat Models. Int. J. Mol. Sci. 2021, 22, 8262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158262
Wu Y, Johnson G, Zhao F, Wu Y, Zhao G, Brown A, You S, Zou M-H, Song P. Features of Lipid Metabolism in Humanized ApoE Knockin Rat Models. International Journal of Molecular Sciences. 2021; 22(15):8262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158262
Chicago/Turabian StyleWu, Yang, Gem Johnson, Fujie Zhao, Yin Wu, Guojun Zhao, Andrew Brown, Shaojin You, Ming-Hui Zou, and Ping Song. 2021. "Features of Lipid Metabolism in Humanized ApoE Knockin Rat Models" International Journal of Molecular Sciences 22, no. 15: 8262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158262