
Adjusting the Molecular Clock: The Importance of Circadian Rhythms in the Development of Glioblastomas and Its Intervention as a Therapeutic Strategy


Abstract
:1. Introduction
2. GBM General Hallmarks
3. GBM Classification
4. GBM Treatment
5. Current Standard of Care Treatment
6. Novel Therapeutic Strategies for GBM
7. Circadian Rhythms
8. The Molecular Clock
9. Circadian Disruption and Its Implication in Cancer Biology
10. Epidemiological Studies
11. Laboratory Evidence
12. Clock Genes and Their Incidence in GBM Development, Progression and Prognosis
13. The Positive Arm of the Molecular Clock
13.1. Bmal1 Gene
13.2. Clock Gene
14. The Negative Arm of the Molecular Clock
14.1. Period 1 Gene
14.2. Period 2 Gene
14.3. Period 3 Gene
14.4. Cryptochrome 1 Gene
14.5. Cryptochrome 2 Gene
14.6. Rev-Erb Genes
14.7. Other Clock Pathways Related Genes
15. Chronotherapy as a Promising Strategy for GBM Treatment
16. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S.; Meyer, M.A. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcella, A.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Oliva, M.A.; Storto, M.; Fanelli, M.; Gambardella, S.; Fornai, F. Dissecting molecular features of gliomas: Genetic loci and validated biomarkers. Int. J. Mol. Sci. 2020, 21, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesseling, P.; Capper, D. WHO 2016 Classification of Gliomas; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2018; Volume 44, pp. 139–150. [Google Scholar]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro. Oncol. 2012, 14 (Suppl. S5), v1–v49. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- De Vleeschouwer, S. (Ed.) Glioblastoma; Codon Publications: Brisbane, Australia, 2017; ISBN 9780994438126. [Google Scholar]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, F.R.S.; Kahn, S.A.; Soletti, R.C.; Biasoli, D.; Alves, T.; da Fonseca, A.C.C.; Garcia, C.; Romão, L.; Brito, J.; Holanda-Afonso, R.; et al. Glioblastoma: Therapeutic challenges, what lies ahead. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 338–349. [Google Scholar] [CrossRef]
- Jackson, R.J.; Fuller, G.N.; Abi-Said, D.; Lang, F.F.; Gokaslan, Z.L.; Shi, W.M.; Wildrick, D.M.; Sawaya, R. Limitations of stereotactic biopsy in the initial management of gliomas. Neuro. Oncol. 2001, 3, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Marko, N.F.; Weil, R.J.; Schroeder, J.L.; Lang, F.F.; Suki, D.; Sawaya, R.E. Extent of resection of glioblastoma revisited: Personalized survival modeling facilitates more accurate survival prediction and supports a maximum-safe-resection approach to surgery. J. Clin. Oncol. 2014, 32, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieber, D.; Golebiewska, A.; Evers, L.; Lenkiewicz, E.; Brons, N.H.C.; Nicot, N.; Oudin, A.; Bougnaud, S.; Hertel, F.; Bjerkvig, R.; et al. Glioblastomas are composed of genetically divergent clones with distinct tumourigenic potential and variable stem cell-associated phenotypes. Acta Neuropathol. 2014, 127, 203–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesche-Starnecker, F.; Mayer, K.; Kofler, F.; Baur, S.; Schmidt-Graf, F.; Kempter, J.; Prokop, G.; Pfarr, N.; Wei, W.; Gempt, J.; et al. Immunohistochemically characterized intratumoral heterogeneity is a prognostic marker in human glioblastoma. Cancers 2020, 12, 2964. [Google Scholar] [CrossRef]
- Aubry, M.; de Tayrac, M.; Etcheverry, A.; Clavreul, A.; Saikali, S.; Menei, P.; Mosser, J. From the core to beyond the margin: A genomic picture of glioblastoma intratumor heterogeneity. Oncotarget 2015, 6, 12094–12109. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Dakup, P.P.; Porter, K.I.; Little, A.A.; Gajula, R.P.; Zhang, H.; Skornyakov, E.; Kemp, M.G.; Van Dongen, H.P.A.; Gaddameedhi, S. The circadian clock regulates cisplatin-induced toxicity and tumor regression in melanoma mouse and human models. Oncotarget 2018, 9, 14524–14538. [Google Scholar] [CrossRef] [Green Version]
- Iurisci, I.; Filipski, E.; Reinhardt, J.; Bach, S.; Gianella-Borradori, A.; Iacobelli, S.; Meijer, L.; Lévi, F. Improved tumor control through circadian clock induction by seliciclib, a cyclin-dependent kinase inhibitor. Cancer Res. 2006, 66, 10720–10728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlap, J.C.; Loros, J.J.; DeCoursey, P.J. (Eds.) Chronobiology: Biological Timekeeping; Sinauer Associates: Sunderland, MA, USA, 2004; ISBN 087893149X. [Google Scholar]
- Wagner, P.M.; Prucca, C.G.; Velazquez, F.N.; Sosa Alderete, L.G.; Caputto, B.L.; Guido, M.E. Temporal regulation of tumor growth in nocturnal mammals: In vivo studies and chemotherapeutical potential. FASEB J. 2021, 35, e21231. [Google Scholar] [CrossRef] [PubMed]
- Slat, E.A.; Sponagel, J.; Marpegan, L.; Simon, T.; Kfoury, N.; Kim, A.; Binz, A.; Herzog, E.D.; Rubin, J.B. Cell-intrinsic, Bmal1-dependent Circadian Regulation of Temozolomide Sensitivity in Glioblastoma. J. Biol. Rhythm. 2017, 32, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasmita, A.O.; Wong, Y.P.; Ling, A.P.K. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia. Pac. J. Clin. Oncol. 2018, 14, 40–51. [Google Scholar] [CrossRef] [Green Version]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laezza, C.; D’Alessandro, A.; Di Croce, L.; Picardi, P.; Ciaglia, E.; Pisanti, S.; Malfitano, A.M.; Comegna, M.; Faraonio, R.; Gazzerro, P.; et al. P53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis. 2015, 6, e1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussière, M.; Marie, Y.; et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokoe, D. PTEN. Curr. Biol. 2001, 11, R502. [Google Scholar] [CrossRef] [Green Version]
- El-Khayat, S.M.; Arafat, W.O. Therapeutic strategies of recurrent glioblastoma and its molecular pathways “Lock up the beast”. Ecancermedicalscience 2021, 15, 1176. [Google Scholar] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Picca, A.; Berzero, G.; Di Stefano, A.L.; Sanson, M. The clinical use of IDH1 and IDH2 mutations in gliomas. Expert Rev. Mol. Diagn. 2018, 18, 1041–1051. [Google Scholar] [CrossRef]
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [PubMed] [Green Version]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Hamilton, R.L. Molecular diagnostics of gliomas. Arch. Pathol. Lab. Med. 2011, 135, 558–568. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH mutation and MGMT promoter methylation in glioblastoma: Results of a prospective registry. Oncotarget 2015, 6, 40896–40906. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, J.; Chumbalkar, V.; Vaillant, B.; Gururaj, A.E.; Hill, K.S.; Latha, K.; Yao, J.; Priebe, W.; Colman, H.; Elferink, L.A.; et al. Regulation of HGF expression by δEGFR-mediated c-Met activation in glioblastoma cells. Neoplasia 2013, 15, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863. [Google Scholar] [CrossRef] [Green Version]
- Montemurro, N. Glioblastoma Multiforme and Genetic Mutations: The Issue Is Not Over Yet. An Overview of the Current Literature. J. Neurol. Surg. A 2020, 81, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Eoli, M.; Menghi, F.; Bruzzone, M.G.; De Simone, T.; Valletta, L.; Pollo, B.; Bissola, L.; Silvani, A.; Bianchessi, D.; D’Incerti, L.; et al. Methylation of O6-methylguanine DNA methytransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin. Cancer Res. 2007, 13, 2606–2613. [Google Scholar] [CrossRef] [Green Version]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llaguno, S.R.A.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Oliva, R.; Domínguez-García, S.; Carrascal, L.; Abalos-Martínez, J.; Pardillo-Díaz, R.; Verástegui, C.; Castro, C.; Nunez-Abades, P.; Geribaldi-Doldán, N. Evolution of Experimental Models in the Study of Glioblastoma: Toward Finding Efficient Treatments. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I. Genetic secrets of long-term glioblastoma survivors. Bosn. J. Basic Med. Sci. 2019, 19, 116–124. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Steed, T.C.; Treiber, J.M.; Patel, K.; Ramakrishnan, V.; Merk, A.; Smith, A.R.; Carter, B.S.; Dale, A.M.; Chow, L.M.L.; Chen, C.C. Differential localization of glioblastoma subtype: Implications on glioblastoma pathogenesis. Oncotarget 2016, 7, 24899–24907. [Google Scholar] [CrossRef] [Green Version]
- Sidaway, P. CNS cancer: Glioblastoma subtypes revisited. Nat. Rev. Clin. Oncol. 2017, 14, 587. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastomas. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [Green Version]
- Karim, R.; Palazzo, C.; Evrard, B.; Piel, G. Nanocarriers for the treatment of glioblastoma multiforme: Current state-of-the-art. J. Control. Release 2016, 227, 23–37. [Google Scholar] [CrossRef]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.I.; Iwama, T.; Park, D.M. The evidence of glioblastoma heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Sattler, U.G.A.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef]
- Caffery, B.; Lee, J.S.; Alexander-Bryant, A.A. Vectors for glioblastoma gene therapy: Viral & non-viral delivery strategies. Nanomaterials 2019, 9, 105. [Google Scholar]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; Codon Publications: Brisbane, QLD, Australia, 2017; pp. 143–153. ISBN 9780994438126. [Google Scholar]
- Zhang, W.-b.; Wang, Z.; Shu, F.; Jin, Y.H.; Liu, H.Y.; Wang, Q.J.; Yang, Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 2010, 285, 40461–40471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Batista, L.F.Z.; Naumann, S.C.; Wick, W.; Weller, M.; Menck, C.F.M.; Kaina, B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene 2007, 26, 186–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Dico, A.; Martelli, C.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Hypoxia-inducible factor-1α activity as a switch for glioblastoma responsiveness to temozolomide. Front. Oncol. 2018, 8, 249. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, A.L.; Pelloski, C.E.; Gilbert, M.R.; Colman, H.; De La Cruz, C.; Sulman, E.P.; Bekele, B.N.; Aldape, K.D. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro. Oncol. 2010, 12, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Malcolm, F.G.S.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; De Tribolet, N.; Stupp, R. Clinical Trial Substantiates the Predictive Value of O-6-Methylguanine-DNA Methyltransferase Promoter Methylation in Glioblastoma Patients Treated with Temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, C.R.R.; Kajitani, G.S.; Quinet, A.; Fortunato, R.S.; Menck, C.F.M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 2016, 7, 48081–48092. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Du, S.; Du, Y.; Ren, J.; Ying, G.; Yan, Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J. Neurochem. 2018, 144, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynes, J.P.; Nwankwo, A.K.; Sur, H.P.; Sanchez, V.E.; Sarpong, K.A.; Ariyo, O.I.; Dominah, G.A.; Nduom, E.K. Biomarkers for immunotherapy for treatment of glioblastoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Badie, B.; Schartner, J.M. Flow Cytometric Characterization of Tumor-associated Macrophages in Experimental Gliomas. Neurosurgery 2000, 46, 957–962. [Google Scholar]
- Beier, C.P.; Kumar, P.; Meyer, K.; Leukel, P.; Bruttel, V.; Aschenbrenner, I.; Riemenschneider, M.J.; Fragoulis, A.; Rümmele, P.; Lamszus, K.; et al. The cancer stem cell subtype determines immune infiltration of Glioblastoma. Stem Cells Dev. 2012, 21, 2753–2761. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Do, A.S.M.S.; Amano, T.; Edwards, L.A.; Zhang, L.; De Peralta-Venturina, M.; Yu, J.S. CD133 mRNA-Loaded Dendritic Cell Vaccination Abrogates Glioma Stem Cell Propagation in Humanized Glioblastoma Mouse Model. Mol. Ther. Oncolytics 2020, 18, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 1. [Google Scholar]
- Eoli, M.; Corbetta, C.; Anghileri, E.; Di Ianni, N.; Milani, M.; Cuccarini, V.; Musio, S.; Paterra, R.; Frigerio, S.; Nava, S.; et al. Expansion of effector and memory T cells is associated with increased survival in recurrent glioblastomas treated with dendritic cell immunotherapy. Neuro-Oncol. Adv. 2019, 1, vdz022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.; Debinski, W. Challenges to successful implementation of the immune checkpoint inhibitors for treatment of glioblastoma. Int. J. Mol. Sci. 2020, 21, 2759. [Google Scholar] [CrossRef]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do they have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Tudor, T.; Binder, Z.A.; O’Rourke, D.M. CAR T Cells. Neurosurg. Clin. N. Am. 2021, 32, 249–263. [Google Scholar] [CrossRef]
- Majc, B.; Novak, M.; Jerala, N.K.; Jewett, A.; Breznik, B. Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells 2021, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Martínez Bedoya, D.; Dutoit, V.; Migliorini, D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front. Immunol. 2021, 12, 640082. [Google Scholar] [CrossRef] [PubMed]
- Sharifzad, F.; Mardpour, S.S.; Mardpour, S.S.; Fakharian, E.; Taghikhani, A.; Sharifzad, A.; Kiani, S.; Heydarian, Y.; Łos, M.J.; Azizi, Z.; et al. HSP70/IL-2 treated NK cells effectively cross the blood brain barrier and target tumor cells in a rat model of induced glioblastoma multiforme (GBM). Int. J. Mol. Sci. 2020, 21, 2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.T.; Nguyen, N.T.; Choi, Y.J.; Zhang, G.; Nguyen, H.T.N.; Filka, E.; Green, S.; Yong, W.H.; Liau, L.M.; Green, R.M.; et al. Phase 2 Study of Bortezomib Combined with Temozolomide and Regional Radiation Therapy for Upfront Treatment of Patients with Newly Diagnosed Glioblastoma Multiforme: Safety and Efficacy Assessment. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Brekke, J.; Arnesen, V.; Hannisdal, M.H.; Navarro, A.G.; Waha, A.; Herfindal, L.; Rygh, C.B.; Bratland, E.; Brandal, P.; et al. Sequential bortezomib and temozolomide treatment promotes immunological responses in glioblastoma patients with positive clinical outcomes: A phase 1B study. Immun. Inflamm. Dis. 2020, 8, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Milde-Langosch, K. The Fos family of transcription factors and their role in tumourigenesis. Eur. J. Cancer 2005, 41, 2449–2461. [Google Scholar] [CrossRef]
- Motrich, R.D.; Castro, G.M.; Caputto, B.L. Old players with a newly defined function: Fra-1 and c-Fos support growth of human malignant breast tumors by activating membrane biogenesis at the cytoplasm. PLoS ONE 2013, 8, e53211. [Google Scholar] [CrossRef] [Green Version]
- Silvestre, D.C.; Gil, G.A.; Tomasini, N.; Bussolino, D.F.; Caputto, B.L. Growth of peripheral and central nervous system tumors is supported by cytoplasmic c-Fos in humans and mice. PLoS ONE 2010, 5, e9544. [Google Scholar] [CrossRef]
- Rodríguez-Berdini, L.; Ferrero, G.O.; Bustos Plonka, F.; Cardozo Gizzi, A.M.; Prucca, C.G.; Quiroga, S.; Caputto, B.L. The moonlighting protein c-Fos activates lipid synthesis in neurons, an activity that is critical for cellular differentiation and cortical development. J. Biol. Chem. 2020, 295, 8808–8818. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Berdini, L.; Caputto, B.L. Lipid Metabolism in Neurons: A Brief Story of a Novel c-Fos-Dependent Mechanism for the Regulation of Their Synthesis. Front. Cell. Neurosci. 2019, 13, 198. [Google Scholar] [CrossRef] [Green Version]
- Gil, G.A.; Silvestre, D.C.; Tomasini, N.; Bussolino, D.F.; Caputto, B.L. Controlling cytoplasmic c-Fos controls tumor growth in the peripheral and central nervous system. Neurochem. Res. 2012, 37, 1364–1371. [Google Scholar] [CrossRef]
- Alfonso Pecchio, A.R.; Cardozo Gizzi, A.M.; Renner, M.L.; Molina-Calavita, M.; Caputto, B.L. c-Fos activates and physically interacts with specific enzymes of the pathway of synthesis of polyphosphoinositides. Mol. Biol Cell 2011, 22, 4716–4725. [Google Scholar] [CrossRef]
- Racca, A.C.; Prucca, C.G.; Caputto, B.L. Fra-1 and c-Fos N-Terminal Deletion Mutants Impair Breast Tumor Cell Proliferation by Blocking Lipid Synthesis Activation. Front. Oncol. 2019, 9, 544. [Google Scholar] [CrossRef]
- Cardozo Gizzi, A.M.; Caputto, B.L. Mechanistic insights into the nongenomic regulation of phospholipid synthesizing enzymes. IUBMB Life 2013, 65, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Prucca, C.G.; Racca, A.C.; Velazquez, F.N.; Gizzi, A.M.C.; Berdini, L.R.; Caputto, B.L. Impairing activation of phospholipid synthesis by c-fos interferes with glioblastoma cell proliferation. Biochem. J. 2020, 477, 4675–4688. [Google Scholar] [CrossRef]
- Miretti, M.; Prucca, C.G.; Tempesti, T.C.; Baumgartner, M.T. Current phthalocyanines delivery systems in photodynamic therapy: An updated review. Curr. Med. Chem. 2021, 28. [Google Scholar] [CrossRef]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Bechet, D.; Mordon, S.R.; Guillemin, F.; Barberi-Heyob, M.A. Photodynamic therapy of malignant brain tumours: A complementary approach to conventional therapies. Cancer Treat. Rev. 2014, 40, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Quirk, B.J.; Brandal, G.; Donlon, S.; Vera, J.C.; Mang, T.S.; Foy, A.B.; Lew, S.M.; Girotti, A.W.; Jogal, S.; LaViolette, P.S.; et al. Photodynamic therapy (PDT) for malignant brain tumors—Where do we stand? Photodiagnosis Photodyn. Ther. 2015, 12, 530–544. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, L.E.; Vilchez, M.L.; Caverzán, M.D.; Milla Sanabria, L.N. Understanding the glioblastoma tumor biology to optimize photodynamic therapy: From molecular to cellular events. J. Neurosci. Res. 2021, 99, 1024–1047. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, F.N.; Miretti, M.; Baumgartner, M.T.; Caputto, B.L.; Tempesti, T.C.T.; Prucca, C.G. Effectiveness of ZnPc and of an amine derivative to inactivate Glioblastoma cells by Photodynamic Therapy: An in vitro comparative study. Sci. Rep. 2019, 9, 3010. [Google Scholar] [CrossRef] [PubMed]
- Miretti, M.; Tempesti, T.C.; Prucca, C.G.; Baumgartner, M.T. Zn phthalocyanines loaded into liposomes: Characterization and enhanced performance of photodynamic activity on glioblastoma cells. Bioorganic Med. Chem. 2020, 28. [Google Scholar] [CrossRef]
- Rominiyi, O.; Vanderlinden, A.; Clenton, S.J.; Bridgewater, C.; Al-Tamimi, Y.; Collis, S.J. Tumour treating fields therapy for glioblastoma: Current advances and future directions. Br. J. Cancer 2021, 124, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma a randomized clinical trial. JAMA J. Am. Med. Assoc. 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Guzauskas, G.F.; Salzberg, M.; Wang, B.C. Estimated lifetime survival benefit of tumor treating fields and temozolomide for newly diagnosed glioblastoma patients. CNS Oncol. 2018, 7, CNS23. [Google Scholar] [CrossRef] [Green Version]
- Anton, K.; Baehring, J.M.; Mayer, T. Glioblastoma Multiforme: Overview of Current Treatment and Future Perspectives. Hematol. Oncol. Clin. North. Am. 2012, 26, 825–853. [Google Scholar] [CrossRef]
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Golombek, D.A.; Rosenstein, R.E. Physiology of circadian entrainment. Physiol. Rev. 2010, 90, 1063–1102. [Google Scholar] [CrossRef] [Green Version]
- Welsh, D.K.; Takahashi, J.S.; Kay, S.A. Suprachiasmatic nucleus: Cell autonomy and network properties. Annu. Rev. Physiol. 2009, 72, 551–577. [Google Scholar] [CrossRef] [Green Version]
- Guido, M.E.; Goguen, D.; De Guido, L.; Robertson, H.A.; Rusak, B. Circadian and photic regulation of immediate-early gene expression in the hamster suprachiasmatic nucleus. Neuroscience 1999, 90, 555–571. [Google Scholar] [CrossRef]
- Guido, M.E.; Garbarino-Pico, E.; Contin, M.A.; Valdez, D.J.; Nieto, P.S.; Verra, D.M.; Acosta-Rodriguez, V.A.; de Zavalía, N.; Rosenstein, R.E. Inner retinal circadian clocks and non-visual photoreceptors: Novel players in the circadian system. Prog. Neurobiol. 2010, 92, 484–504. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Numano, R.; Abe, M.; Hida, A.; Takahashi, R.I.; Ueda, M.; Block, G.D.; Sakaki, Y.; Menaker, M.; Tei, H. Resetting central and peripheral circadian oscillators in transgenic rats. Science 2000, 288, 682–685. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-L.; Fu, C.-J.; Bu, R.-F. Role of Circadian Clocks in the Development and Therapeutics of Cancer. J. Int. Med. Res. 2011, 39. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Ozasa, K.; Mikami, K.; Wakai, K.; Fujino, Y.; Watanabe, Y.; Miki, T.; Nakao, M.; Hayashi, K.; Suzuki, K.; et al. Prospective cohort study of the risk of prostate cancer among rotating-shift workers: Findings from the Japan Collaborative Cohort Study. Am. J. Epidemiol. 2006, 164, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, S.; Sassone-Corsi, P. The emerging link between cancer, metabolism, and circadian rhythms. Nat. Med. 2018, 24, 1795–1803. [Google Scholar] [CrossRef]
- Green, C.B.; Takahashi, J.S.; Bass, J. The Meter of Metabolism. Cell 2008, 134, 728–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardin, P.E.; Hall, J.C.; Rosbash, M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature 1990, 343, 536–540. [Google Scholar] [CrossRef]
- Buhr, E.D.; Takahashi, J.S. Molecular components of the mammalian circadian clock. Handb. Exp. Pharmacol. 2013, 217, 3–27. [Google Scholar]
- Lee, C.; Etchegaray, J.P.; Cagampang, F.R.A.; Loudon, A.S.I.; Reppert, S.M. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001, 107, 855–867. [Google Scholar] [CrossRef]
- Eide, E.J.; Kang, H.; Crapo, S.; Gallego, M.; Virshup, D.M. Casein kinase I in the mammalian circadian clock. Methods Enzymol. 2005, 393, 408–418. [Google Scholar] [PubMed] [Green Version]
- Godinho, S.I.H.; Maywood, E.S.; Shaw, L.; Tucci, V.; Barnard, A.R.; Busino, L.; Pagano, M.; Kendall, R.; Quwailid, M.M.; Romero, M.R.; et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science 2007, 316, 897–900. [Google Scholar] [CrossRef] [Green Version]
- Shirogane, T.; Jin, J.; Ang, X.L.; Harper, J.W. SCFβ-TRCP controls Clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J. Biol. Chem. 2005, 280, 26863–26872. [Google Scholar] [CrossRef] [Green Version]
- Siepka, S.M.; Yoo, S.H.; Park, J.; Song, W.; Kumar, V.; Hu, Y.; Lee, C.; Takahashi, J.S. Circadian Mutant Overtime Reveals F-box Protein FBXL3 Regulation of Cryptochrome and Period Gene Expression. Cell 2007, 129, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.H.; Mohawk, J.A.; Siepka, S.M.; Shan, Y.; Huh, S.K.; Hong, H.K.; Kornblum, I.; Kumar, V.; Koike, N.; Xu, M.; et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 2013, 152, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Preitner, N.; Damiola, F.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Sato, T.K.; Panda, S.; Miraglia, L.J.; Reyes, T.M.; Rudic, R.D.; McNamara, P.; Naik, K.A.; Fitzgerald, G.A.; Kay, S.A.; Hogenesch, J.B. A functional genomics strategy reveals rora as a component of the mammalian circadian clock. Neuron 2004, 43, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Sancar, G.; Brunner, M. Circadian clocks and energy metabolism. Cell. Mol. Life Sci. 2014, 71, 2667–2680. [Google Scholar] [CrossRef]
- Fu, L.; Kettner, N.M. The circadian clock in cancer development and therapy. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2013; Volume 119, pp. 221–282. ISBN 9780123969712. [Google Scholar]
- Panda, S.; Antoch, M.P.; Miller, B.H.; Su, A.I.; Schook, A.B.; Straume, M.; Schultz, P.G.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.H.; McDearmon, E.L.; Panda, S.; Hayes, K.R.; Zhang, J.; Andrews, J.L.; Antoch, M.P.; Walker, J.R.; Esser, K.A.; Hogenesch, J.B.; et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc. Natl. Acad. Sci. USA 2007, 104, 3342–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arafa, K.; Emara, M. Insights About Circadian Clock and Molecular Pathogenesis in Gliomas. Front. Oncol. 2020, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 2019, 5, 475–494. [Google Scholar] [CrossRef]
- Shafi, A.A.; Knudsen, K.E. Cancer and the circadian clock. Cancer Res. 2019, 79, 3806–3814. [Google Scholar] [CrossRef] [Green Version]
- Soták, M.; Sumová, A.; Pácha, J. Cross-talk between the circadian clock and the cell cycle in cancer. Ann. Med. 2014, 46, 221–232. [Google Scholar] [CrossRef]
- Shostak, A. Circadian clock, cell division, and cancer: From molecules to organism. Int. J. Mol. Sci. 2017, 18, 873. [Google Scholar] [CrossRef]
- Farshadi, E.; Yan, J.; Leclere, P.; Goldbeter, A.; Chaves, I.; van der Horst, G.T.J. The positive circadian regulators CLOCK and BMAL1 control G2/M cell cycle transition through Cyclin B1. Cell Cycle 2019, 18, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, S.; Beaulieu-Laroche, L.; Blum, I.D.; Landgraf, D.; Welsh, D.K.; Storch, K.F.; Labrecque, N.; Cermakian, N. Enhancing circadian clock function in cancer cells inhibits tumor growth. BMC Biol. 2017, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, A.E.; Zheng, T.; Ba, Y.; Stevens, R.G.; Yi, C.H.; Leaderer, D.; Zhu, Y. Phenotypic effects of the circadian gene Cryptochrome 2 on cancer-related pathways. BMC Cancer 2010, 10, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Donehower, L.A.; Herron, A.J.; Moore, D.D.; Fu, L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS ONE 2010, 5, e10995. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C.C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, T.; Vila-Caballer, M.; Santos, C.S.; Liu, J.; Yang, J.; Finkielstein, C.V. The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol. Biol. Cell 2014, 25, 3081–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, T.; Kim, J.K.; Liu, J.; Vila-Caballer, M.; Stauffer, P.E.; Tyson, J.J.; Finkielstein, C.V. Model-driven experimental approach reveals the complex regulatory distribution of p53 by the circadian factor period 2. Proc. Natl. Acad. Sci. USA 2016, 113, 13516–13521. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, T.; Vila-Caballer, M.; Liu, J.; Schiffhauer, S.; Finkielstein, C.V. Association of the circadian factor Period 2 to p53 influences p53’s function in DNA-damage signaling. Mol. Biol. Cell 2015, 26, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Zhanfeng, N.; Chengquan, W.; Hechun, X.; Jun, W.; Lijian, Z.; Dede, M.; Wenbin, L.; Lei, Y. Period2 downregulation inhibits glioma cell apoptosis by activating the MDM2-TP53 pathway. Oncotarget 2016, 7, 27350–27362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relógio, A.; Thomas, P.; Medina-Pérez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schäfer, R.; et al. Ras-Mediated Deregulation of the Circadian Clock in Cancer. PLoS Genet. 2014, 10, e1004338. [Google Scholar] [CrossRef] [PubMed]
- El-Athman, R.; Genov, N.N.; Mazuch, J.; Zhang, K.; Yu, Y.; Fuhr, L.; Abreu, M.; Li, Y.; Wallach, T.; Kramer, A.; et al. The Ink4a/Arf locus operates as a regulator of the circadian clock modulating RAS activity. PLoS Biol. 2017, 15, e2002940. [Google Scholar] [CrossRef] [Green Version]
- Wagner, P.M.; Sosa Alderete, L.G.; Gorné, L.D.; Gaveglio, V.; Salvador, G.; Pasquaré, S.; Guido, M.E. Proliferative Glioblastoma Cancer Cells Exhibit Persisting Temporal Control of Metabolism and Display Differential Temporal Drug Susceptibility in Chemotherapy. Mol. Neurobiol. 2019, 56, 1276–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez, S.; Crespo, P.; Carlini, V.; Garbarino-Pico, E.; Baler, R.; Caputto, B.L.; Guido, M.E. The metabolism of phospholipids oscillates rhythmically in cultures of fibroblasts and is regulated by the clock protein PERIOD 1. FASEB J. 2004, 18, 519–521. [Google Scholar] [CrossRef]
- Verlande, A.; Masri, S. Circadian Clocks and Cancer: Timekeeping Governs Cellular Metabolism. Trends Endocrinol. Metab. 2019, 30, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Méndez, I.; Díaz-Muñoz, M. Circadian and metabolic perspectives in the role played by NADPH in cancer. Front. Endocrinol. 2018, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Dyar, K.A.; Lutter, D.; Artati, A.; Ceglia, N.J.; Liu, Y.; Armenta, D.; Jastroch, M.; Schneider, S.; de Mateo, S.; Cervantes, M.; et al. Atlas of Circadian Metabolism Reveals System-wide Coordination and Communication between Clocks. Cell 2018, 174, 1571.e11–1585.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollmers, C.; Gill, S.; DiTacchio, L.; Pulivarthy, S.R.; Le, H.D.; Panda, S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21453–21458. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Scheiermann, C.; Gibbs, J.; Ince, L.; Loudon, A. Clocking in to immunity. Nat. Rev. Immunol. 2018, 18, 423–437. [Google Scholar] [CrossRef]
- Hadadi, E.; Acloque, H. Role of circadian rhythm disorders on EMT and tumour-immune interactions in endocrine-related cancers. Endocr. Relat. Cancer 2021, 28, R67–R80. [Google Scholar] [CrossRef]
- Matsunaga, N.; Kohno, Y.; Kakimoto, K.; Hayashi, A.; Koyanagi, S.; Ohdo, S. Influence of CLOCK on cytotoxicity induced by diethylnitrosamine in mouse primary hepatocytes. Toxicology 2011, 280, 144–151. [Google Scholar] [CrossRef]
- Wu, Y.; Sato, F.; Bhawal, U.K.; Kawamoto, T.; Fujimoto, K.; Noshiro, M.; Seino, H.; Morohashi, S.; Kato, Y.; Kijima, H. BHLH transcription factor DEC2 regulates pro-apoptotic factor bim in human oral cancer HSC-3 cells. Biomed. Res. 2012, 33, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mauvoisin, D.; Martin, E.; Atger, F.; Galindo, A.N.; Dayon, L.; Sizzano, F.; Palini, A.; Kussmann, M.; Waridel, P.; et al. Nuclear Proteomics Uncovers Diurnal Regulatory Landscapes in Mouse Liver. Cell Metab. 2017, 25, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The Circadian Gene Per1 Plays an Important Role in Cell Growth and DNA Damage Control in Human Cancer Cells. Mol. Cell 2006, 22, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Leem, S.H. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 2014, 42, 4427–4434. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Reardon, J.T.; Kemp, M.; Sancar, A. Orcadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. USA 2009, 106, 2864–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, S.J.; Huber, A.L.; Jordan, S.D.; Kriebs, A.; Nguyen, M.; Moresco, J.J.; Yates, J.R.; Lamia, K.A. DNA damage shifts circadian clock time via Hausp-dependent Cry1 stabilization. eLife 2015, 4, e04883. [Google Scholar] [CrossRef] [Green Version]
- Oklejewicz, M.; Destici, E.; Tamanini, F.; Hut, R.A.; Janssens, R.; van der Horst, G.T.J. Phase Resetting of the Mammalian Circadian Clock by DNA Damage. Curr. Biol. 2008, 18, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Chong, S.T.; Ngeow, J. Molecular connections between circadian rhythm and genome maintenance pathways. Endocr. Relat. Cancer 2021, 28, R55–R66. [Google Scholar] [CrossRef]
- Wendeu-Foyet, M.G.; Menegaux, F. Circadian Disruption and Prostate Cancer Risk: An Updated Review of Epidemiological Evidences. Cancer Epidemiol. Biomark. Prev. 2017, 26, 985–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickerman, B.A.; Markt, S.C.; Koskenvuo, M.; Hublin, C.; Pukkala, E.; Mucci, L.A.; Kaprio, J. Sleep disruption, chronotype, shift work, and prostate cancer risk and mortality: A 30-year prospective cohort study of Finnish twins. Cancer Causes Control. 2016, 27, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Markt, S.C.; Flynn-Evans, E.E.; Valdimarsdottir, U.A.; Sigurdardottir, L.G.; Tamimi, R.M.; Batista, J.L.; Haneuse, S.; Lockley, S.W.; Stampfer, M.; Wilson, K.M.; et al. Sleep duration and disruption and prostate cancer risk: A 23-year prospective study. Cancer Epidemiol. Biomark. Prev. 2016, 25, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Sigurdardottir, L.G.; Valdimarsdottir, U.A.; Fall, K.; Rider, J.R.; Lockley, S.W.; Schernhammer, E.; Mucci, L.A. Circadian disruption, sleep loss, and prostate cancer risk: A systematic review of epidemiologic studies. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamanca-Fernández, E.; Rodríguez-Barranco, M.; Guevara, M.; Ardanaz, E.; Olry de Labry Lima, A.; Sánchez, M.J. Night-shift work and breast and prostate cancer risk: Updating the evidence from epidemiological studies. Anales del Sistema Sanitario de Navarra 2018, 41, 211–226. [Google Scholar] [PubMed] [Green Version]
- Papantoniou, K.; Devore, E.E.; Massa, J.; Strohmaier, S.; Vetter, C.; Yang, L.; Shi, Y.; Giovannucci, E.; Speizer, F.; Schernhammer, E.S. Rotating night shift work and colorectal cancer risk in the nurses’ health studies. Int. J. Cancer 2018, 143, 2709–2717. [Google Scholar] [CrossRef] [Green Version]
- Straif, K.; Baan, R.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Benbrahim-Tallaa, L.; Cogliano, V. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007, 8, 1065–1066. [Google Scholar] [CrossRef]
- Travis, R.C.; Reeves, G.K.; Peto, R.; Key, T.J.; Beral, V. Response. JNCI J. Natl. Cancer Inst. 2017, 109, 8. [Google Scholar] [CrossRef]
- Jones, M.E.; Schoemaker, M.J.; McFadden, E.C.; Wright, L.B.; Johns, L.E.; Swerdlow, A.J. Night shift work and risk of breast cancer in women: The Generations Study cohort. Br. J. Cancer 2019, 121, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Haus, E.L.; Smolensky, M.H. Shift work and cancer risk: Potential mechanistic roles of circadian disruption, light at night, and sleep deprivation. Sleep Med. Rev. 2013, 17, 273–284. [Google Scholar] [CrossRef]
- Zhu, Y.; Stevens, R.G.; Hoffman, A.E.; Tjonneland, A.; Vogel, U.B.; Zheng, T.; Hansen, J. Epigenetic impact of long-term shiftwork: Pilot evidence from circadian genes and whole-genome methylation analysis. Chronobiol. Int. 2011, 28, 852–861. [Google Scholar] [CrossRef] [Green Version]
- Pukkala, E.; Aspholm, R.; Auvinen, A.; Eliasch, H.; Gundestrup, M.; Haldorsen, T.; Hammar, N.; Hrafnkelsson, J.; Kyyrönen, P.; Linnersjö, A.; et al. Cancer incidence among 10,211 airline pilots: A nordic study. Aviat. Sp. Enviorn. Med. 2003, 74, 699–706. [Google Scholar]
- Lie, J.A.S.; Roessink, J.; Kjærheim, K. Breast cancer and night work among Norwegian nurses. Cancer Causes Control. 2006, 17, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Gianotti, T.F.; Burgueno, A.; Pirola, C.J. Gene-gene interaction between serotonin transporter (SLC6A4) and clock modulates the risk of metabolic syndrome in rotating shiftworkers. Chronobiol. Int. 2010, 27, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Kogevinas, M.; Espinosa, A.; Castelló, A.; Gómez-Acebo, I.; Guevara, M.; Martin, V.; Amiano, P.; Alguacil, J.; Peiro, R.; Moreno, V.; et al. Effect of mistimed eating patterns on breast and prostate cancer risk (MCC-Spain Study). Int. J. Cancer 2018, 143, 2380–2389. [Google Scholar] [CrossRef] [Green Version]
- Srour, B.; Plancoulaine, S.; Andreeva, V.A.; Fassier, P.; Julia, C.; Galan, P.; Hercberg, S.; Deschasaux, M.; Latino-Martel, P.; Touvier, M. Circadian nutritional behaviours and cancer risk: New insights from the NutriNet-santé prospective cohort study: Disclaimers. Int. J. Cancer 2018, 143, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Li, X.M.; Delaunay, F.; Dulong, S.; Claustrat, B.; Zampera, S.; Fujii, Y.; Teboul, M.; Beau, J.; Lévi, F. Cancer inhibition through circadian reprogramming of tumor transcriptome with meal timing. Cancer Res. 2010, 70, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xiang, Y.; Ozguc, F.M.; Kim, Y.; Liu, C.J.; Park, P.K.; Hu, Q.; Diao, L.; Lou, Y.; Lin, C.; et al. The Genomic Landscape and Pharmacogenomic Interactions of Clock Genes in Cancer Chronotherapy. Cell Syst. 2018, 6, 314.e2–328.e2. [Google Scholar] [CrossRef] [Green Version]
- Reszka, E.; Zienolddiny, S. Epigenetic basis of circadian rhythm disruption in cancer. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1856, pp. 173–201. [Google Scholar]
- Masri, S.; Kinouchi, K.; Sassone-Corsi, P. Circadian clocks, epigenetics, and cancer. Curr. Opin. Oncol. 2015, 27, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Joska, T.M.; Zaman, R.; Belden, W.J. Regulated DNA methylation and the circadian clock: Implications in cancer. Biology 2014, 3, 560–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipski, E.; King, V.M.; Li, X.M.; Granda, T.G.; Mormont, M.C.; Liu, X.H.; Claustrat, B.; Hastings, M.H.; Lévi, F. Host circadian clock as a control point in tumor progression. J. Natl. Cancer Inst. 2002, 94, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Filipski, E.; Delaunay, F.; King, V.M.; Wu, M.-W.; Claustrat, B.; Gréchez-Cassiau, A.; Guettier, C.; Hastings, M.H.; Francis, L. Effects of Chronic Jet Lag on Tumor Progression in Mice. Cancer Res. 2004, 64, 7879–7885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef] [Green Version]
- Aiello, I.; Mul Fedele, M.L.; Román, F.; Marpegan, L.; Caldart, C.; Chiesa, J.J.; Golombek, D.A.; Finkielstein, C.V.; Paladino, N. Circadian disruption promotes tumor-immune microenvironment remodeling favoring tumor cell proliferation. Sci. Adv. 2020, 6, eaaz4530. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Van Gelder, R.N. Clocks, cancer, and chronochemotherapy. Science 2021, 371. [Google Scholar] [CrossRef]
- Puram, R.V.; Kowalczyk, M.S.; De Boer, C.G.; Schneider, R.K.; Miller, P.G.; McConkey, M.; Tothova, Z.; Tejero, H.; Heckl, D.; Järås, M.; et al. Core Circadian Clock Genes Regulate Leukemia Stem Cells in AML. Cell 2016, 165, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.M.; Huang, S.F.; Zeng, J.M.; Liu, D.B.; Xiao, Q.; Tian, W.J.; Zhu, X.D.; Huang, Z.G.; Feng, W.L. Per2 inhibits K562 leukemia cell growth in vitro and in vivo through cell cycle arrest and apoptosis induction. Pathol. Oncol. Res. 2010, 16, 403–411. [Google Scholar] [CrossRef]
- Antoch, M.P.; Toshkov, I.; Kuropatwinski, K.K.; Jackson, M. Deficiency in PER proteins has no effect on the rate of spontaneous and radiation-induced carcinogenesis. Cell Cycle 2013, 12, 3673–3680. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Ozturk, N.; Lee, J.H.; Gaddameedhi, S. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2841–2846. [Google Scholar]
- Dong, Z.; Zhang, G.; Qu, M.; Gimple, R.C.; Wu, Q.; Qiu, Z.; Prager, B.C.; Wang, X.; Kim, L.J.Y.; Morton, A.R.; et al. Targeting glioblastoma stem cells through disruption of the circadian clock. Cancer Discov. 2019, 9, 1556–1573. [Google Scholar] [CrossRef] [Green Version]
- De, A.; Beligala, D.H.; Sharma, V.P.; Fry, B.R.; Geusz, M.E. Abstract 858: The circadian clock of glioma cells undergoing epithelial-mesenchymal transition. Cancer Res. 2017, 77, 858. [Google Scholar]
- Li, A.; Lin, X.; Tan, X.; Yin, B.; Han, W.; Zhao, J.; Yuan, J.; Qiang, B.; Peng, X. Circadian gene clock contributes to cell proliferation and migration of glioma and is directly regulated by tumor-suppressive miR-124. FEBS Lett. 2013, 587, 2455–2460. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Li, W.; Wang, Q.; Wang, Y.; Lu, F. Circadian regulator NR1D2 regulates glioblastoma cell proliferation and motility. Oncogene 2018, 37, 4838–4853. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Lai, A.G. Timing gone awry: Distinct tumour suppressive and oncogenic roles of the circadian clock and crosstalk with hypoxia signalling in diverse malignancies. J. Transl. Med. 2019, 17, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Antiangiogenic therapies for high-grade glioma. Nat. Rev. Neurol. 2009, 5, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Verhoeff, J.J.C.; Van Tellingen, O.; Claes, A.; Stalpers, L.J.A.; Van Linde, M.E.; Richel, D.J.; Leenders, W.P.J.; Van Furth, W.R. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer 2009, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razorenova, O.V. Brain and muscle ARNT-like protein BMAL1 regulates ROS homeostasis and senescence: A possible link to hypoxia-inducible factor-mediated pathway. Cell Cycle 2012, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, F.; Matsubara, C.; Myung, J.; Yoritaka, T.; Kamimura, N.; Tsutsumi, S.; Kanai, A.; Suzuki, Y.; Sassone-Corsi, P.; Aburatani, H.; et al. Genome-Wide Profiling of the Core Clock Protein BMAL1 Targets Reveals a Strict Relationship with Metabolism. Mol. Cell. Biol. 2010, 30, 5636–5648. [Google Scholar] [CrossRef] [Green Version]
- Khapre, R.V.; Kondratova, A.A.; Susova, O.; Kondratov, R.V. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle 2011, 10, 4162–4169. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Hsu, W.H.; Chang, A.; Tan, Z.; Lan, Z.; Zhou, A.; Spring, D.J.; Lang, F.F.; Wang, Y.A.; Depinho, R.A. Circadian regulator CLOCK recruits immune-suppressive microglia into the GBM tumor microenvironment. Cancer Discov. 2020, 10, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Kim, E.M.; Park, J.K.; Hwang, S.G.; Moon, S.K.; Kim, W.J.; Um, H.D. Bmal1 suppresses cancer cell invasion by blocking the phosphoinositide 3-kinase-Akt-MMP-2 signaling pathway. Oncol. Rep. 2013, 29, 2109–2113. [Google Scholar] [CrossRef] [Green Version]
- Gwon, D.H.; Lee, W.Y.; Shin, N.; Kim, S.I.; Jeong, K.; Lee, W.H.; Kim, D.W.; Hong, J.; Lee, S.Y. BMAL1 suppresses proliferation, migration, and invasion of U87MG cells by downregulating cyclin b1, phospho-AKT, and metalloproteinase-9. Int. J. Mol. Sci. 2020, 21, 2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Liu, Y.; Siddique, R.; Nabi, G.; Xue, M.; Hou, H. Impact of chronically alternating light-dark cycles on circadian clock mediated expression of cancer (Glioma)-related genes in the brain. Int. J. Biol. Sci. 2019, 15, 1816–1834. [Google Scholar] [CrossRef] [Green Version]
- Crespo, I.; Tão, H.; Nieto, A.B.; Rebelo, O.; Domingues, P.; Vital, A.L.; Patino, M.D.C.; Barbosa, M.; Lopes, M.C.; Oliveira, C.R.; et al. Amplified and Homozygously Deleted Genes in Glioblastoma: Impact on Gene Expression Levels. PLoS ONE 2012, 7, e46088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Liu, P.; Li, C.; Luo, Y.; Chen, I.; Liang, W.; Chen, X.; Feng, Y.; Xia, H.; Wang, F. Deregulated expression of the clock genes in gliomas. Technol. Cancer Res. Treat. 2013, 12, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, M.H.; Anic, G.M.; Thompson, R.C.; Nabors, L.B.; Olson, J.J.; Browning, J.E.; Monteiro, A.N.; Egan, K.M. Circadian pathway genes in relation to glioma risk and outcome. Cancer Causes Control 2014, 25, 25–32. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Chen, L. The Circadian Gene Clock Plays an Important Role in Cell Apoptosis and the DNA Damage Response In Vitro. Technol. Cancer Res. Treat. 2016, 15, 480–486. [Google Scholar] [CrossRef]
- Wang, Z.; Su, G.; Dai, Z.; Meng, M.; Zhang, H.; Fan, F.; Liu, Z.; Zhang, L.; Weygant, N.; He, F.; et al. Circadian clock genes promote glioma progression by affecting tumour immune infiltration and tumour cell proliferation. Cell Prolif. 2021, 54, e12988. [Google Scholar] [CrossRef]
- Xia, H.C.; Niu, Z.F.; Ma, H.; Cao, S.Z.; Hao, S.C.; Liu, Z.T.; Wang, F. Deregulated expression of the Per1 and Per2 in human gliomas. Can. J. Neurol. Sci. 2010, 37, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Wang, Q.; Hu, Y.; Wang, F. The circadian gene PER1 plays an important role in radiation-induced apoptosis and DNA damage in glioma. Asian Pac. J. Cancer Prev. 2019, 20, 2195–2201. [Google Scholar] [CrossRef] [Green Version]
- Zhanfeng, N.; Yanhui, L.; Zhou, F.; Shaocai, H.; Guangxing, L.; Hechun, X. Circadian genes Per1 and Per2 increase radiosensitivity of glioma in vivo. Oncotarget 2015, 6, 9951–9958. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wu, Y.; Zhang, N.; Yuan, H.; Wang, F.; Xu, H.; Yu, J.; Ma, J.; Hou, S.; Cao, X. IDH1 gene mutation activates Smad signaling molecules to regulate the expression levels of cell cycle and biological rhythm genes in human glioma U87-MG cells. Mol. Med. Rep. 2021, 23. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Luo, Y.; Li, C.; Chen, L. Correlation between deregulated expression of PER2 gene and degree of glioma malignancy. Tumori 2014, 100, e266–e272. [Google Scholar] [CrossRef]
- Ma, D.; Hou, L.; Xia, H.; Li, H.; Fan, H.; Jia, X.; Niu, Z. PER2 inhibits proliferation and stemness of glioma stem cells via the Wnt/ß-catenin signaling pathway. Oncol. Rep. 2020, 44, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, F.; Chen, L.A.; Chen, X.W.; Chen, Z.J.; Liu, P.F.; Li, F.F.; Li, C.Y.; Liang, W. Deregulated expression of Cry1 and Cry2 in human gliomas. Asian Pac. J. Cancer Prev. 2012, 13, 5725–5728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Caiyan, L.; Ling, Z.; Jiayun, Z. Aberrant rhythmic expression of cryptochrome2 regulates the radiosensitivity of rat gliomas. Oncotarget 2017, 8, 77809–77818. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Zhao, X.; Hatori, M.; Yu, R.T.; Barish, G.D.; Lam, M.T.; Chong, L.W.; Ditacchio, L.; Atkins, A.R.; Glass, C.K.; et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 2012, 485, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Woldt, E.; Sebti, Y.; Solt, L.A.; Duhem, C.; Lancel, S.; Eeckhoute, J.; Hesselink, M.K.C.; Paquet, C.; Delhaye, S.; Shin, Y.; et al. Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat. Med. 2013, 19, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Vieira, E.; Marroquí, L.; Batista, T.M.; Caballero-Garrido, E.; Carneiro, E.M.; Boschero, A.C.; Nadal, A.; Quesada, I. The clock gene Rev-erbα regulates pancreatic β-cell function: Modulation by leptin and high-fat diet. Endocrinology 2012, 153, 592–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiermann, C.; Kunisaki, Y.; Frenette, P.S. Circadian control of the immune system. Nat. Rev. Immunol. 2013, 13, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Wagner, P.M.; Monjes, N.M.; Guido, M.E. Chemotherapeutic Effect of SR9009, a REV-ERB Agonist, on the Human Glioblastoma T98G Cells. ASN Neuro 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, Q.X. The analysis of deregulated expression of the timeless genes in gliomas. J. Cancer Res. Ther. 2018, 14, S708–S712. [Google Scholar]
- Goldsmith, C.S.; Kim, S.M.; Karunarathna, N.; Neuendorff, N.; Gerard Toussaint, L.; Earnest, D.J.; Bell-Pedersen, D. Inhibition of p38 MAPK activity leads to cell type-specific effects on the molecular circadian clock and time-dependent reduction of glioma cell invasiveness. BMC Cancer 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Damato, A.R.; Luo, J.; Katumba, R.G.N.; Talcott, G.R.; Rubin, J.B.; Herzog, E.D.; Campian, J.L. Temozolomide chronotherapy in patients with glioblastoma: A retrospective single institute study. Neuro-Oncol. Adv. 2021, 3, 1–11. [Google Scholar]
- Fischer, D.; Lombardi, D.A.; Marucci-Wellman, H.; Roenneberg, T. Chronotypes in the US—Influence of age and sex. PLoS ONE 2017, 12, e0178782. [Google Scholar] [CrossRef] [Green Version]



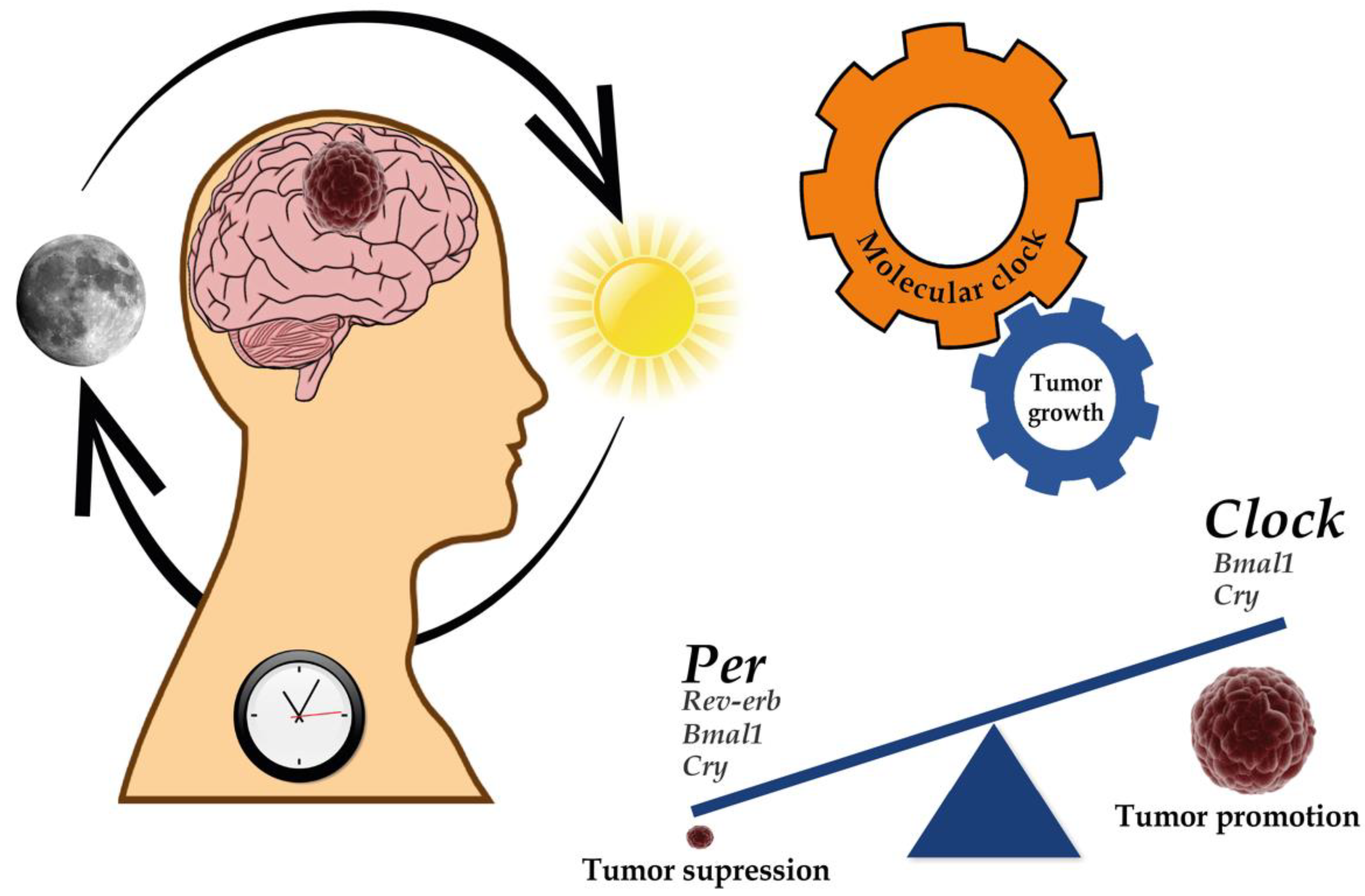{kind=link}
{kind=link}
| Primary GBM [27,43,44,45,46] | Secondary GBM [27,32,45,46] |
|---|---|
| LOH chromosome 10q (70%) | IDH1/2 mutation (73–85%) |
| LOH chromosome 10p (50–70%) | TP53 mutation (65–81%) |
| EGFR amplification or mutation (35–45%) | ATRX mutation (65–71%) |
| TP53 mutation (27–30%) | LOH chromosome 10q (63%) |
| PTEN mutation (25%) | LOH chromosome 19q (~50%) |
| O6-MGMT promoter methylation (42%) | MGMT promoter methylation (79%) |
| TERT promoter mutation (72%) | p16INK4a deletion (~19%) |
| PDGFR amplification (~7%) | EGFR amplification (8%) |
| MDM2 mutation (7–12%) | PTEN mutation (<5%) |
| NF1 mutation/deletion (11%) | |
| GLI1 mutation (5–22%) | |
| IDH1/2 mutation (5%) | |
| PIK3CA mutation (1%) |
| GBM Subtype | Molecular and Genetic Profile | Median Survival (Months) |
|---|---|---|
| Proneural [6,23,47,48,50] |
| 11.3 (9.3–14.7) |
| Neural [23,47,50] |
| 13.1 (9.8–18) |
| Classical [23,47,48,50] |
| 12.2 (11.08–18) |
| Mesenchymal [23,47,48,50] |
| 11.8 (9.57–15.4) |
| Gene | Evidence of Gene Deregulation Associated with Gliomagenesis—Evidence of Potentiality for Therapeutic Targeting |
|---|---|
| Clock | [209,211,213,219,223,224,225,226,227] |
| Bmal1 | [21,22,161,209,219,220,221,222] |
| Per1 | [21,161,213,225,227,228,229,230,231] |
| Per2 | [213,227,228,230,231,232,233] |
| Per3 | [213,225,227,231] |
| Cry1 | [209,222,225,227,231,234] |
| Cry2 | [213,222,225,231,234,235] |
| Npas2 | [225,227] |
| Rev-erb | [209,212,213,219,241,242] |
| RORα | [213] |
| RORβ | [213] |
| Timeless | [227,243] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, P.M.; Prucca, C.G.; Caputto, B.L.; Guido, M.E. Adjusting the Molecular Clock: The Importance of Circadian Rhythms in the Development of Glioblastomas and Its Intervention as a Therapeutic Strategy. Int. J. Mol. Sci. 2021, 22, 8289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158289
Wagner PM, Prucca CG, Caputto BL, Guido ME. Adjusting the Molecular Clock: The Importance of Circadian Rhythms in the Development of Glioblastomas and Its Intervention as a Therapeutic Strategy. International Journal of Molecular Sciences. 2021; 22(15):8289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158289
Chicago/Turabian StyleWagner, Paula M., César G. Prucca, Beatriz L. Caputto, and Mario E. Guido. 2021. "Adjusting the Molecular Clock: The Importance of Circadian Rhythms in the Development of Glioblastomas and Its Intervention as a Therapeutic Strategy" International Journal of Molecular Sciences 22, no. 15: 8289. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158289