
Targeting Lipocalin-2 in Inflammatory Breast Cancer Cells with Small Interference RNA and Small Molecule Inhibitors


 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
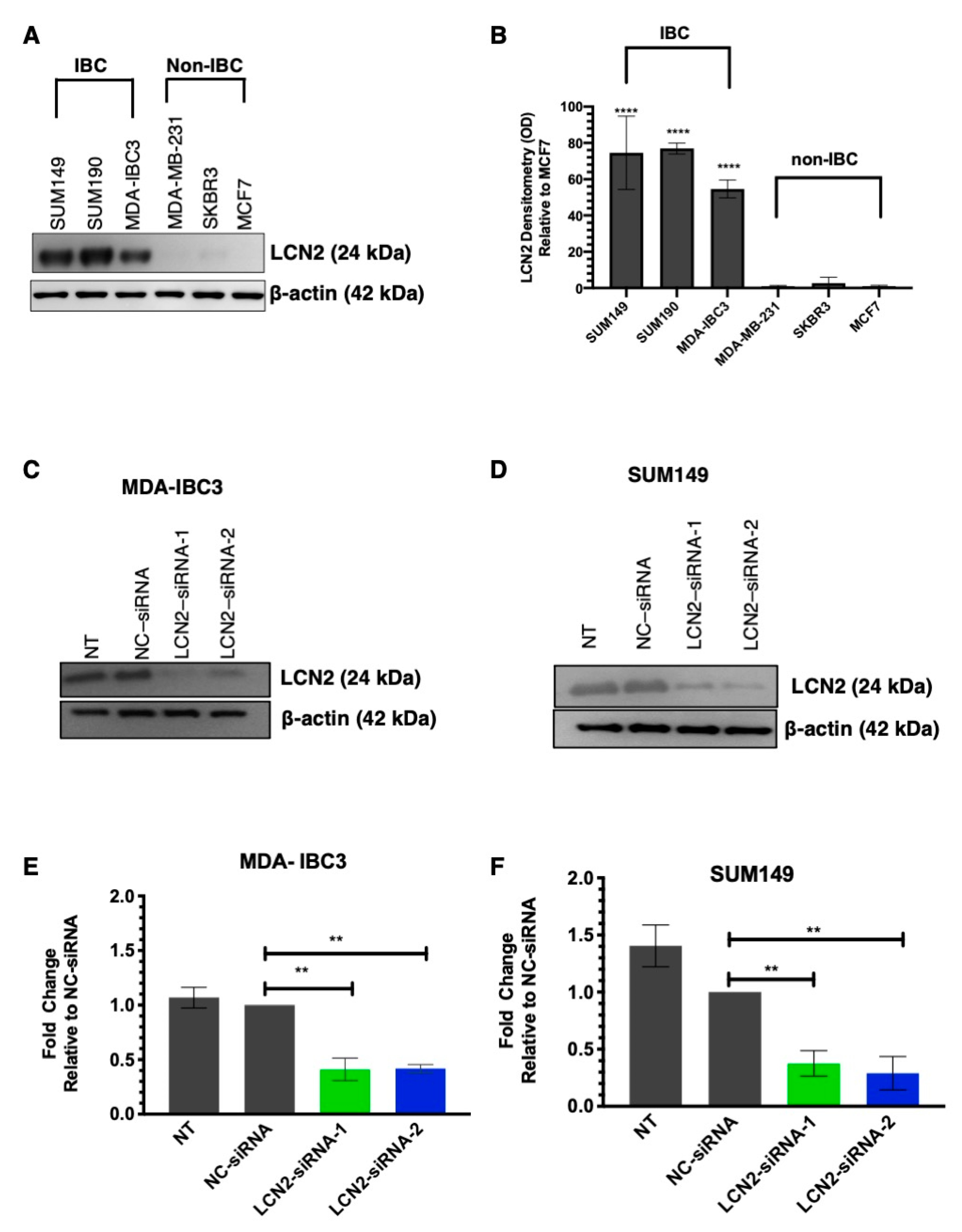
2.1. LCN2 Protein Levels Are Increased in Inflammatory Breast Cancer Cell (IBC) Lines
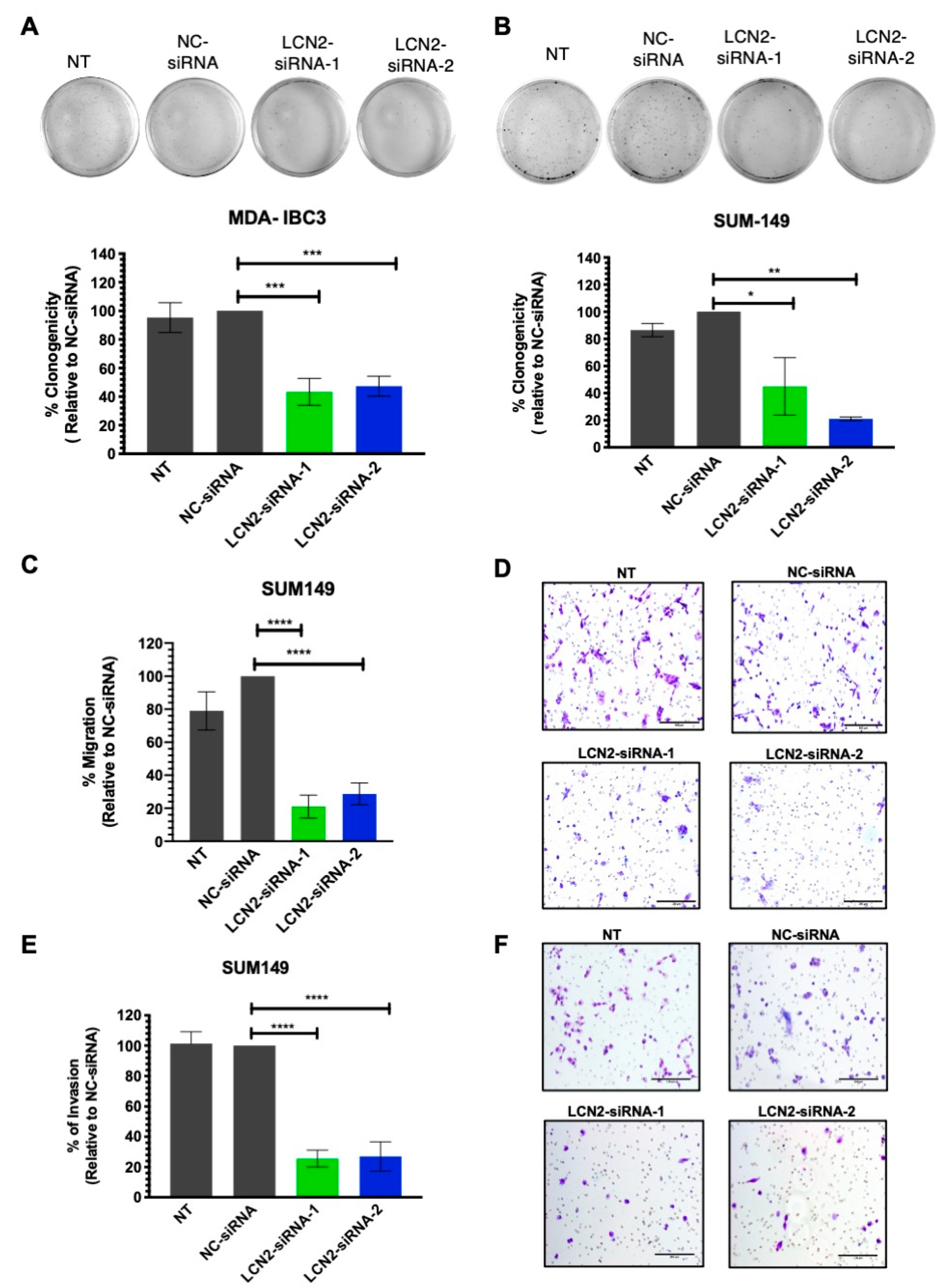
2.2. LCN2-siRNA-Based Silencing Reduces Cell Colony Formation, Cell Invasion and Migration in IBC Cells
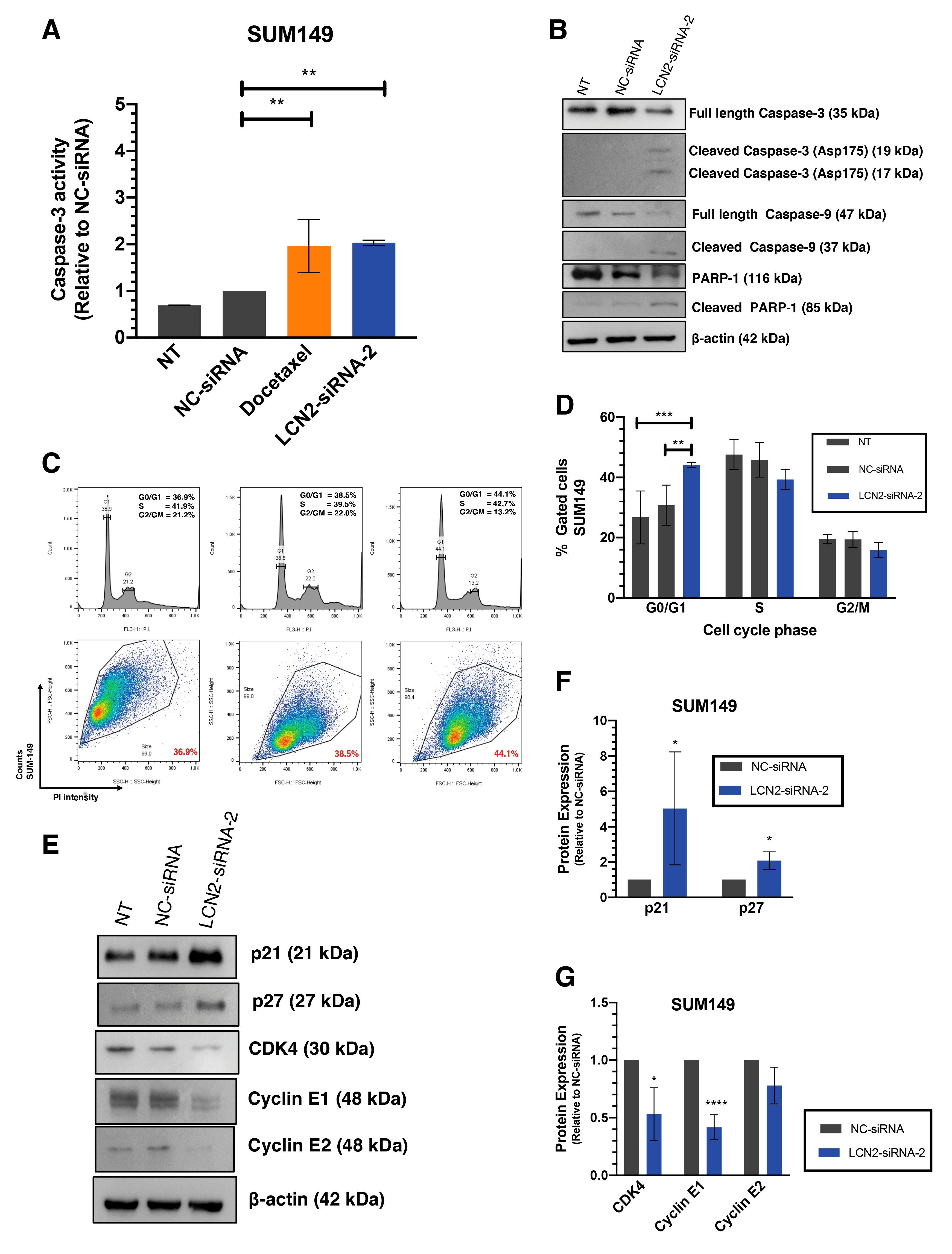
2.3. LCN2 Silencing Induces Apoptosis and Cell Cycle Arrest
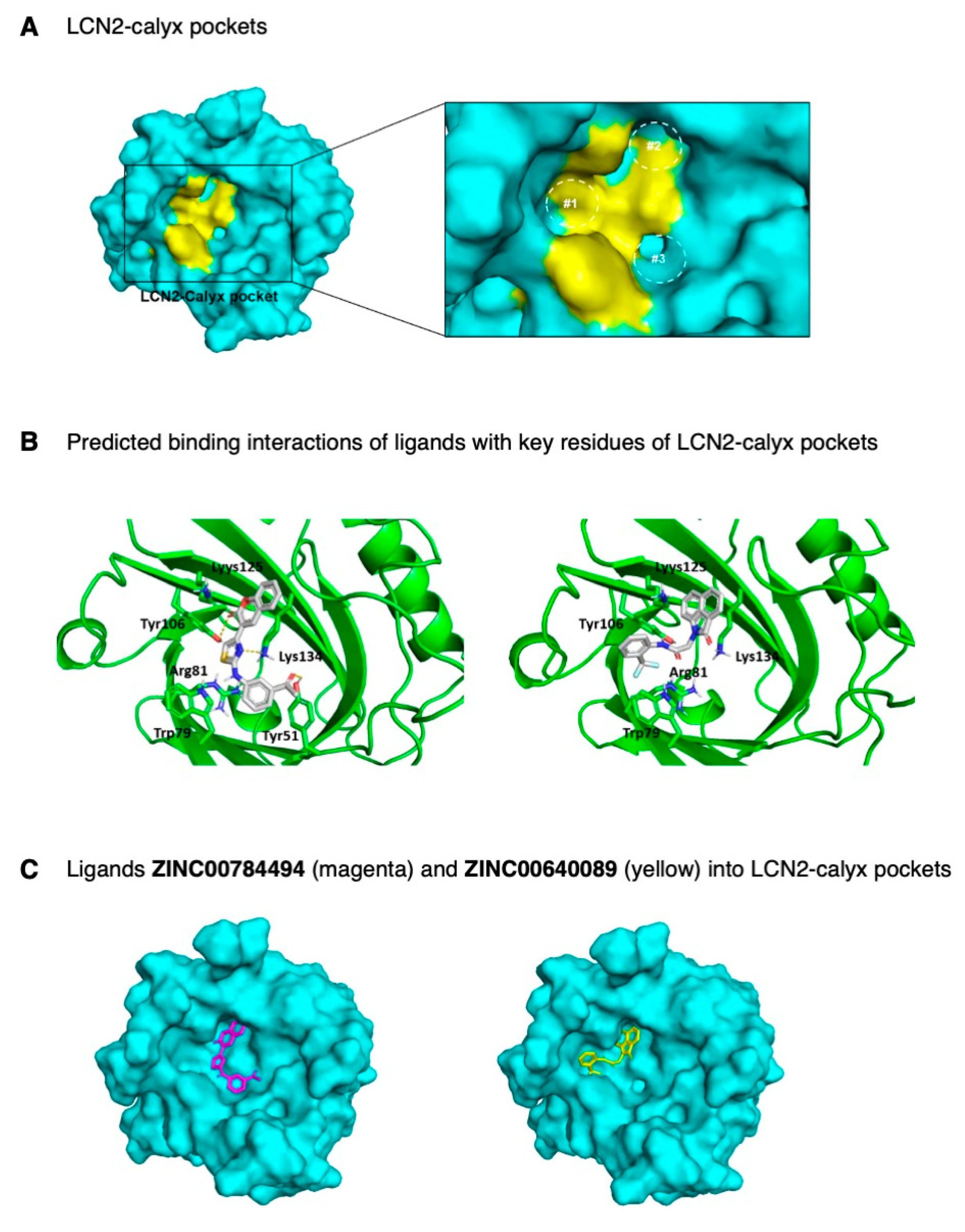
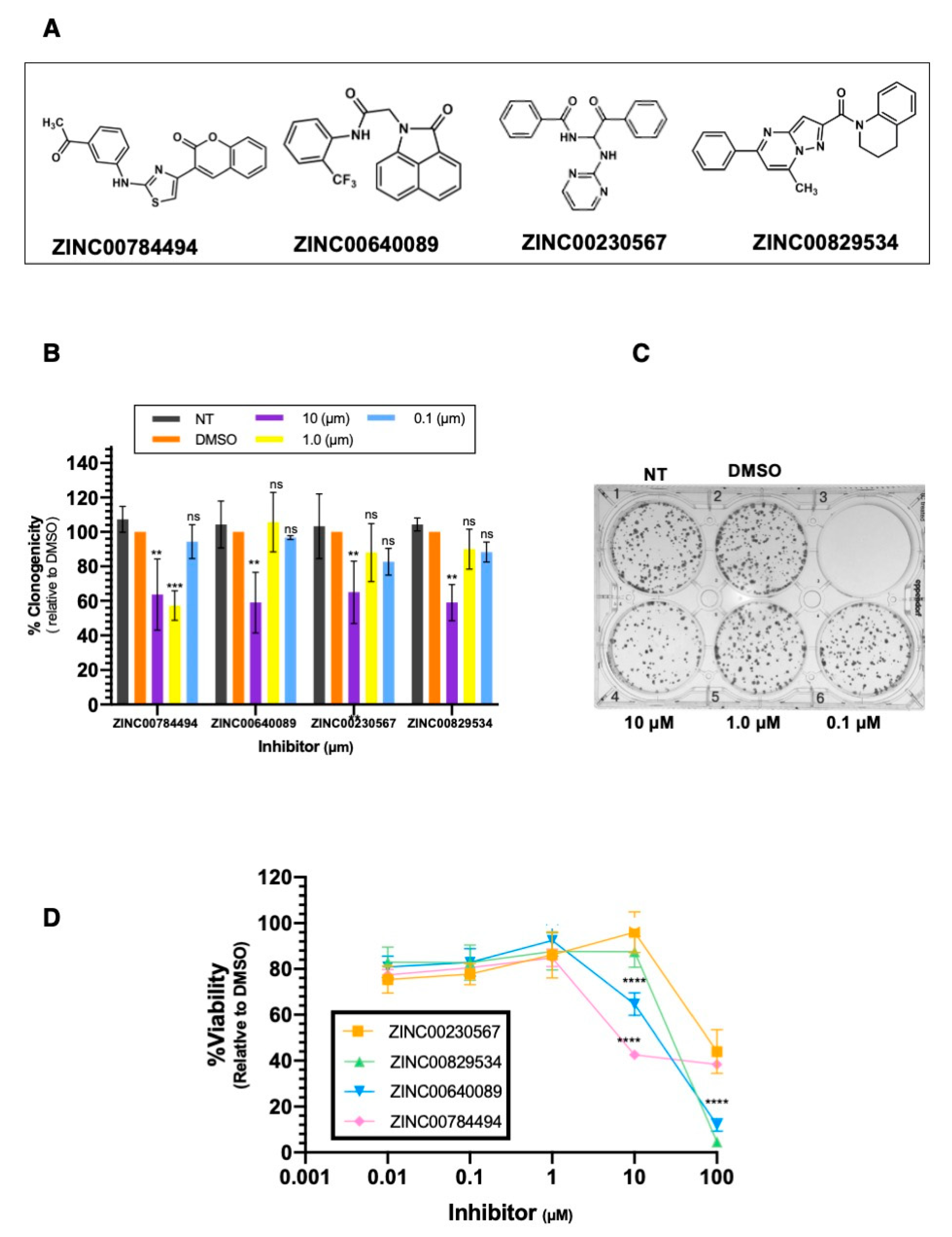
2.4. Identification of LCN2 Small Molecule Inhibitors by In-Silico Analysis
2.5. LCN2-Inhibitors Reduce Colony Formation and Cell Viability in SUM149 Cells
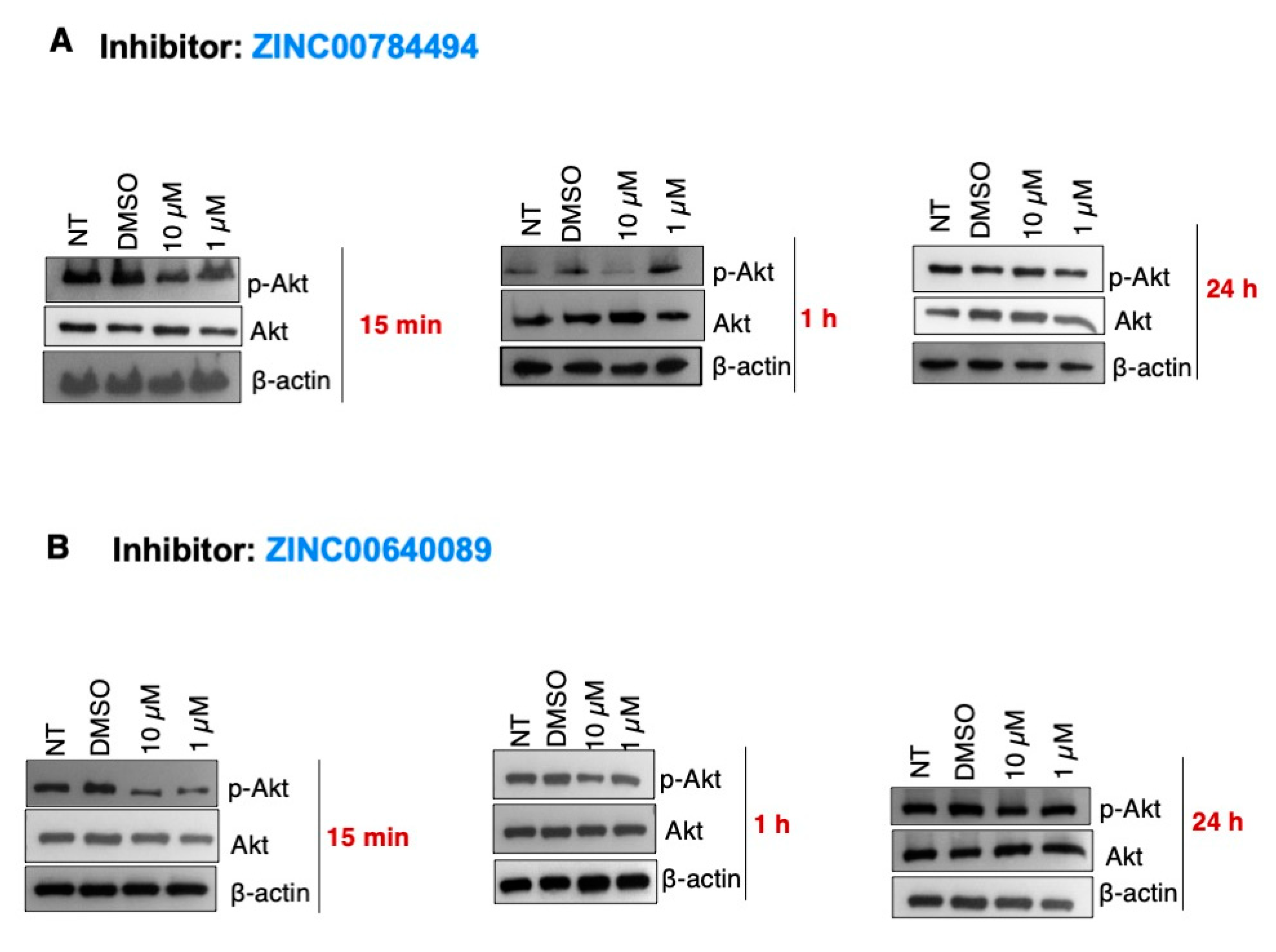
2.6. The LCN2 Inhibitors ZINC00784494 and ZINC00640089 Reduced the p-Akt Levels in SUM149 Cells
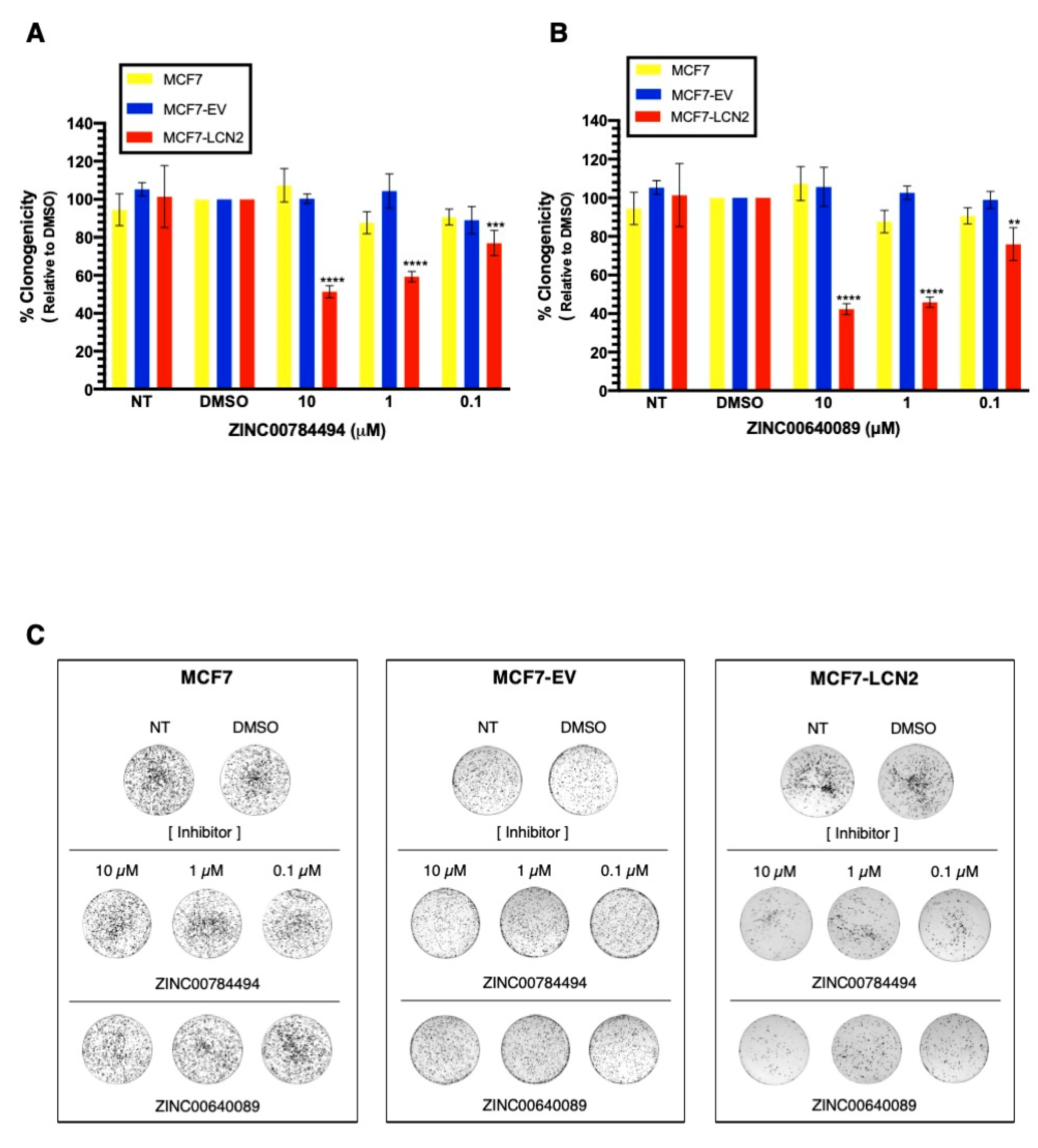
2.7. LCN2-Inhibitors ZINC00784494 and ZINC00640089 Showed Specificity toward LCN2
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Western Blot Analysis
4.3. Transient and Stable Transfections
4.4. Colony Formation Assays
4.5. Cell Viability Assays
4.6. Invasion and Migration Assays
4.7. In Vitro Testing of Small Molecules Inhibitors
4.8. Akt/p-Akt Measurements
4.9. Caspase-3 Fluorometric Assay
4.10. Flow Cytometry Analysis
4.11. Molecular Docking
4.12. Physicochemical and Pharmacokinetic Properties
4.13. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chiang, K.C.; Yeh, T.S.; Wu, R.C.; Pang, J.H.S.; Cheng, C.T.; Wang, S.Y.; Yeh, C.N. Lipocalin 2 (LCN2) is a promising target for cholangiocarcinoma treatment and bile LCN2 level is a potential cholangiocarcinoma diagnostic marker. Sci. Rep. 2016, 6, 36138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Young, H.A.; Levine, P.H. Trends in inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program at the National Cancer Institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, M.; Pan, J.; Wang, X.; Chen, X.-S.; Shen, K.-W. Pattern of distant metastases in inflammatory breast cancer—A large-cohort retrospective study. J. Cancer 2020, 11, 292–300. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Valero, V.; Buzdar, A.U.; Kau, S.W.; Broglio, K.R.; Gonzalez-Angulo, A.M.; Sneige, N.; Islam, R.; Ueno, N.T.; Buchholz, T.A.; et al. Inflammatory breast cancer (IBC) and patterns of recurrence: Understanding the biology of a unique disease. Cancer 2007, 110, 1436–1444. [Google Scholar] [CrossRef]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Iwamoto, T.; Brewer, T.; Pusztai, L.; Kai, K.; Kogawa, T.; Finetti, P.; Birnbaum, D.; et al. Comparison of molecular subtype distribution in triple-negative inflammatory and non-inflammatory breast cancers. Breast Cancer Res. 2013, 15, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fouad, T.M. Survival of patients with metastatic breast cancer with or without locoregional therapy. Lancet Oncol. 2015, 16, e585–e586. [Google Scholar] [CrossRef] [Green Version]
- Robertson, F.M.; Bondy, M.; Yang, W.; Yamauchi, H.; Wiggins, S.; Kamrudin, S.; Krishnamurthy, S.; Le-Petross, H.; Bidaut, L.; Player, A.N.; et al. Inflammatory breast cancer: The disease, the biology, the treatment. CA Cancer J. Clin. 2010, 60, 351–375. [Google Scholar] [CrossRef]
- Saigal, K.; Hurley, J.; Takita, C.; Reis, I.M.; Zhao, W.; Rodgers, S.E.; Wright, J.L. Risk factors for locoregional failure in patients with inflammatory breast cancer treated with trimodality therapy. Clin. Breast Cancer 2013, 13, 335–343. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Kertmen, N.; Babacan, T.; Keskin, O.; Solak, M.; Sarici, F.; Akin, S.; Arik, Z.; Aslan, A.; Ates, O.; Aksoy, S.; et al. Molecular subtypes in patients with inflammatory breast cancer; a single center experience. J. BUON 2015, 20, 35–39. [Google Scholar] [PubMed]
- Parton, M.; Dowsett, M.; Ashley, S.; Hills, M.; Lowe, F.; Smith, I. High incidence of HER-2 positivity in inflammatory breast cancer. Breast 2004, 13, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Sánchez, G.S.; Pita-Grisanti, V.; Quiñones-Díaz, B.; Gumpper, K.; Cruz-Monserrate, Z.; Vivas-Mejía, P.E. Biological Functions and Therapeutic Potential of Lipocalin 2 in Cancer. Int. J. Mol. Sci. 2020, 21, 4365. [Google Scholar] [CrossRef] [PubMed]
- Friedl, A.; Stoesz, S.; Buckley, P.; Gould, M. Neutrophil gelatinase-associated lipocalin in normal and neoplastic human tissues. Cell type-specific pattern of expression. Histochem. J. 1999, 31, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Ashfaq, R.; Maitra, A.; Adsay, N.V.; Shen-Ong, G.L.; Berg, K.; Hollingsworth, M.A.; Cameron, J.L.; Yeo, C.J.; Kern, S.E.; et al. Highly expressed genes in pancreatic ductal adenocarcinomas: A comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003, 63, 8614–8622. [Google Scholar] [PubMed]
- Furutani, M.; Arii, S.; Mizumoto, M.; Kato, M.; Imamura, M. Identification of a neutrophil gelatinase-associated lipocalin mRNA in human pancreatic cancers using a modified signal sequence trap method. Cancer Lett. 1998, 122, 209–214. [Google Scholar] [CrossRef]
- Yang, J.; Bielenberg, D.R.; Rodig, S.J.; Doiron, R.; Clifton, M.C.; Kung, A.L.; Moses, M.A. Lipocalin 2 promotes breast cancer progression. Proc. Natl. Acad. Sci. USA 2009, 106, 3913–3918. [Google Scholar] [CrossRef] [Green Version]
- Grønborg, M.; Bunkenborg, J.; Kristiansen, T.Z.; Jensen, O.N.; Yeo, C.J.; Hruban, R.H.; Maitra, A.; Goggins, M.G.; Pandey, A. Comprehensive proteomic analysis of human pancreatic juice. J. Proteome Res. 2004, 3, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Chou, S.B.; Swidnicka-Siergiejko, A.K.; Badi, N.; Chavez-Tomar, M.; Lesinski, G.B.; Bekaii-Saab, T.; Farren, M.R.; Mace, T.A.; Schmidt, C.; Liu, Y.; et al. Lipocalin-2 Promotes Pancreatic Ductal Adenocarcinoma by Regulating Inflammation in the Tumor Microenvironment. Cancer Res. 2017, 77, 2647–2660. [Google Scholar] [CrossRef] [Green Version]
- Mannelqvist, M.; Stefansson, I.M.; Wik, E.; Kusonmano, K.; Raeder, M.B.; Øyan, A.M.; Kalland, K.H.; Moses, M.A.; Salvesen, H.B.; Akslen, L.A. Lipocalin 2 expression is associated with aggressive features of endometrial cancer. BMC Cancer 2012, 12, 169. [Google Scholar] [CrossRef] [Green Version]
- Villodre, E.; Larson, R.; Hu, X.; Stecklein, S.; Gomez, K.; Finetti, P.; Krishnamurthy, S.; Ivan, C.; Su, X.; Ueno, N.; et al. Lipocalin 2 promotes inflammatory breast cancer tumorigenesis and skin invasion. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bauer, M.; Eickhoff, J.C.; Gould, M.N.; Mundhenke, C.; Maass, N.; Friedl, A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res. Treat. 2008, 108, 389–397. [Google Scholar] [CrossRef]
- Missiaglia, E.; Blaveri, E.; Terris, B.; Wang, Y.-H.; Costello, E.; Neoptolemos, J.; Crnogorac-Jurcevic, T.; Lemoine, N.R. Analysis of gene expression in cancer cell lines identifies candidate markers for pancreatic tumorigenesis and metastasis. Int. J. Cancer 2004, 112, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Tanaka, Y.; Kono, S.; Nishimura, M.; Mukohara, T.; Morinaga, Y.; Hara, S.; Takao, S. Effectiveness of Pertuzumab, Trastuzumab, and Docetaxel Combination Neoadjuvant Chemotherapy for HER2-Positive Inflammatory Breast Cancer: A Case Report. Breast Care 2017, 12, 45–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Parker, S.L.; Pham, T.; Buzdar, A.U.; Hursting, S.D. Inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program of the National Cancer Institute, 1975–1992. Cancer 1998, 82, 2366–2372. [Google Scholar] [CrossRef]
- Clifton, M.C.; Rupert, P.B.; Hoette, T.M.; Raymond, K.N.; Abergel, R.J.; Strong, R.K. Parsing the functional specificity of Siderocalin/Lipocalin 2/NGAL for siderophores and related small-molecule ligands. J. Struct. Biol. X 2019, 2, 100008. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Schiffrin, B.; Radford, S.E.; Brockwell, D.J.; Calabrese, A.N. PyXlinkViewer: A flexible tool for visualization of protein chemical crosslinking data within the PyMOL molecular graphics system. Protein Sci. 2020, 29, 1851–1857. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta 2012, 1826, 129–169. [Google Scholar] [CrossRef] [Green Version]
- Candido, S.; Abrams, S.L.; Steelman, L.S.; Lertpiriyapong, K.; Fitzgerald, T.L.; Martelli, A.M.; Cocco, L.; Montalto, G.; Cervello, M.; Polesel, J.; et al. Roles of NGAL and MMP-9 in the tumor microenvironment and sensitivity to targeted therapy. Biochim. Biophys. Acta 2016, 1863, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Maestro, R.; Polesel, J.; Catania, A.; Maira, F.; Signorelli, S.S.; Libra, M. Roles of neutrophil gelatinase-associated lipocalin (NGAL) in human cancer. Oncotarget 2014, 5, 1576–1594. [Google Scholar] [CrossRef] [Green Version]
- Anderson, W.F.; Chu, K.C.; Chang, S. Inflammatory breast carcinoma and noninflammatory locally advanced breast carcinoma: Distinct clinicopathologic entities? J. Clin. Oncol. 2003, 21, 2254–2259. [Google Scholar] [CrossRef] [PubMed]
- Ohshiro, K.; Schwartz, A.M.; Levine, P.H.; Kumar, R. Alternate estrogen receptors promote invasion of inflammatory breast cancer cells via non-genomic signaling. PLoS ONE 2012, 7, e30725. [Google Scholar]
- Fernandez, S.V.; Robertson, F.M.; Pei, J.; Aburto-Chumpitaz, L.; Mu, Z.; Chu, K.; Alpaugh, R.K.; Huang, Y.; Cao, Y.; Ye, Z.; et al. Inflammatory breast cancer (IBC): Clues for targeted therapies. Breast Cancer Res. Treat. 2013, 140, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, F.J.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z. E-cadherin’s dark side: Possible role in tumor progression. Biochim. Biophys. Acta 2012, 1826, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Bartholomeusz, C.; Mansour, O.; Humphries, J.; Hortobagyi, G.N.; Ordentlich, P.; Ueno, N.T. A class I histone deacetylase inhibitor, entinostat, enhances lapatinib efficacy in HER2-overexpressing breast cancer cells through FOXO3-mediated Bim1 expression. Breast Cancer Res. Treat. 2014, 146, 259–272. [Google Scholar] [CrossRef]
- Hsu, M.Y.; Mina, E.; Roetto, A.; Porporato, P.E. Iron: An Essential Element of Cancer Metabolism. Cells 2020, 9, 2591. [Google Scholar] [CrossRef]
- Correnti, C.; Richardson, V.; Sia, A.K.; Bandaranayake, A.D.; Ruiz, M.; Rahmanto, Y.S.; Kovacevic, Z.; Clifton, M.C.; Holmes, M.A.; Kaiser, B.K.; et al. Siderocalin/Lcn2/NGAL/24p3 does not drive apoptosis through gentisic acid mediated iron withdrawal in hematopoietic cell lines. PLoS ONE 2012, 7, e43696. [Google Scholar] [CrossRef] [Green Version]
- Reyes-González, J.M.; Armaiz-Pena, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarría-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejía, P.E. Targeting c-MYC in Platinum-Resistant Ovarian Cancer. Mol. Cancer Ther. 2015, 14, 2260–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The Design of Leadlike Combinatorial Libraries. Angew. Chem. Int. Ed. Engl. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago-Sánchez, G.S.; Noriega-Rivera, R.; Hernández-O’Farrill, E.; Valiyeva, F.; Quiñones-Diaz, B.; Villodre, E.S.; Debeb, B.G.; Rosado-Albacarys, A.; Vivas-Mejía, P.E. Targeting Lipocalin-2 in Inflammatory Breast Cancer Cells with Small Interference RNA and Small Molecule Inhibitors. Int. J. Mol. Sci. 2021, 22, 8581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168581
Santiago-Sánchez GS, Noriega-Rivera R, Hernández-O’Farrill E, Valiyeva F, Quiñones-Diaz B, Villodre ES, Debeb BG, Rosado-Albacarys A, Vivas-Mejía PE. Targeting Lipocalin-2 in Inflammatory Breast Cancer Cells with Small Interference RNA and Small Molecule Inhibitors. International Journal of Molecular Sciences. 2021; 22(16):8581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168581
Chicago/Turabian StyleSantiago-Sánchez, Ginette S., Ricardo Noriega-Rivera, Eliud Hernández-O’Farrill, Fatma Valiyeva, Blanca Quiñones-Diaz, Emilly S. Villodre, Bisrat G. Debeb, Andrea Rosado-Albacarys, and Pablo E. Vivas-Mejía. 2021. "Targeting Lipocalin-2 in Inflammatory Breast Cancer Cells with Small Interference RNA and Small Molecule Inhibitors" International Journal of Molecular Sciences 22, no. 16: 8581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168581