
Multiomic Approaches to Uncover the Complexities of Dystrophin-Associated Cardiomyopathy

 , , ,
, , ,  ,
,
Abstract
:1. Dystrophin-Associated Cardiomyopathy (DAC)
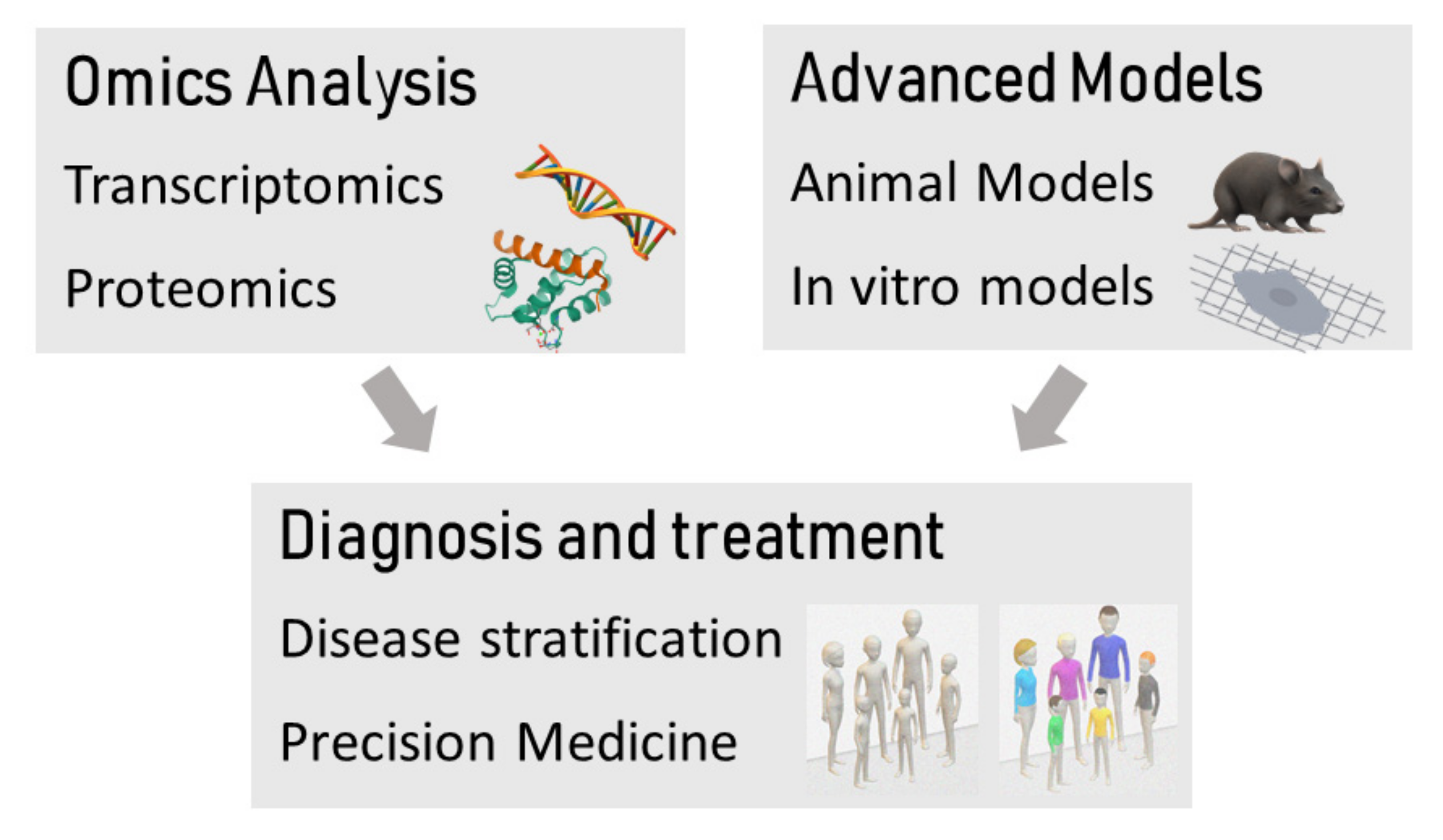
2. Omics Application in DAC Searching for Insight
3. Genome-Editing-Based Models of DAC
4. Proteomic Investigations of Dystrophin Deficiency
5. Transcriptomic Approaches to Uncover DAC Pathological Mechanisms
6. Bioinformatics for Omic Data Integration
7. Relevance of Omics for In Vitro DAC Modelling
8. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Constantin, B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 2014, 1838, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M. The muscular dystrophies. Semin. Neurol. 2012, 32, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Bushby, K.; Connor, E. Clinical outcome measures for trials in Duchenne muscular dystrophy: Report from International Working Group meetings. Clin. Investig. 2011, 1, 1217–1235. [Google Scholar] [CrossRef]
- Szabo, S.M.; Salhany, R.M.; Deighton, A.; Harwood, M.; Mah, J.; Gooch, K.L. The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: A systematic literature review. Orphanet J. Rare Dis. 2021, 16, 237. [Google Scholar] [CrossRef]
- Finsterer, J.; Cripe, L. Treatment of dystrophin cardiomyopathies. Nat. Rev. Cardiol. 2014, 11, 168–179. [Google Scholar] [CrossRef]
- Verhaert, D.; Richards, K.; Rafael-Fortney, J.A.; Raman, S.V. Cardiac involvement in patients with muscular dystrophies: Magnetic resonance imaging phenotype and genotypic considerations. Circ. Cardiovasc. Imaging 2011, 4, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Melacini, P.; Vianello, A.; Villanova, C.; Fanin, M.; Miorin, M.; Angelini, C.; Dalla Volta, S. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul. Disord. 1996, 6, 367–376. [Google Scholar] [CrossRef]
- Tsuda, T.; Fitzgerald, K.K. Dystrophic cardiomyopathy: Complex pathobiological processes to generate clinical phenotype. J. Cardiovasc. Dev. Dis. 2017, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Vicente, A.M.; Ballensiefen, W.; Jonsson, J.I. How personalised medicine will transform healthcare by 2030: The ICPerMed vision. J. Transl. Med. 2020, 18, 180. [Google Scholar] [CrossRef]
- Gaina, G.; Popa Gruianu, A. Muscular dystrophy: Experimental animal models and therapeutic approaches (Review). Exp. Ther. Med. 2021, 21, 610. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.A.; Morgan, J.E. Duchenne’s muscular dystrophy: Animal models used to investigate pathogenesis and develop therapeutic strategies. Int. J. Exp. Pathol. 2003, 84, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Rota, M.; Nurzynska, D.; Misao, Y.; Tillmanns, J.; Ojaimi, C.; Padin-Iruegas, M.E.; Muller, P.; Esposito, G.; Bearzi, C.; et al. Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan. Circ. Res. 2008, 102, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Young, C.S.; Mokhonova, E.; Quinonez, M.; Pyle, A.D.; Spencer, M.J. Creation of a novel humanized dystrophic mouse model of duchenne muscular dystrophy and application of a CRISPR/Cas9 gene editing therapy. J. Neuromuscul. Dis. 2017, 4, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorova, T.V.; Zotova, E.D.; Reshetov, D.A.; Polikarpova, A.V.; Vassilieva, S.G.; Vlodavets, D.V.; Gavrilov, A.A.; Ulianov, S.V.; Buchman, V.L.; Deykin, A.V. CRISPR/Cas9-generated mouse model of Duchenne muscular dystrophy recapitulating a newly identified large 430 kb deletion in the human DMD gene. Dis. Model. Mech. 2019, 12, dmm037655. [Google Scholar] [CrossRef] [Green Version]
- Amoasii, L.; Li, H.; Zhang, Y.; Min, Y.L.; Sanchez-Ortiz, E.; Shelton, J.M.; Long, C.; Mireault, A.A.; Bhattacharyya, S.; McAnally, J.R.; et al. In vivo non-invasive monitoring of dystrophin correction in a new Duchenne muscular dystrophy reporter mouse. Nat. Commun. 2019, 10, 4537. [Google Scholar] [CrossRef]
- Koo, T.; Lu-Nguyen, N.B.; Malerba, A.; Kim, E.; Kim, D.; Cappellari, O.; Cho, H.Y.; Dickson, G.; Popplewell, L.; Kim, J.S. Functional rescue of dystrophin deficiency in mice caused by frameshift mutations using campylobacter jejuni Cas9. Mol. Ther. 2018, 26, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.M.; Koo, T.; Kim, K.; Lim, K.; Baek, G.; Kim, S.T.; Kim, H.S.; Kim, D.E.; Lee, H.; Chung, E.; et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018, 36, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Veltrop, M.; van Vliet, L.; Hulsker, M.; Claassens, J.; Brouwers, C.; Breukel, C.; van der Kaa, J.; Linssen, M.M.; den Dunnen, J.T.; Verbeek, S.; et al. A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLoS ONE 2018, 13, e0193289. [Google Scholar] [CrossRef]
- Nakamura, K.; Fujii, W.; Tsuboi, M.; Tanihata, J.; Teramoto, N.; Takeuchi, S.; Naito, K.; Yamanouchi, K.; Nishihara, M. Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci. Rep. 2014, 4, 5635. [Google Scholar] [CrossRef]
- Larcher, T.; Lafoux, A.; Tesson, L.; Remy, S.; Thepenier, V.; Francois, V.; Le Guiner, C.; Goubin, H.; Dutilleul, M.; Guigand, L.; et al. Characterization of dystrophin deficient rats: A new model for Duchenne muscular dystrophy. PLoS ONE 2014, 9, e110371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, M.; Tochinai, R.; Sekizawa, S.I.; Shiga, T.; Uchida, K.; Tsuru, Y.; Kuwahara, M. Cardiac lesions in Duchenne muscular dystrophy model rats with out-of-frame Dmd gene mutation mediated by CRISPR/Cas9 system. J. Toxicol. Pathol. 2020, 33, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, H.; Kimura, K.; Yamanouchi, K.; Teramoto, N.; Okano, T.; Daimon, M.; Morita, H.; Takenaka, K.; Shiga, T.; Tanihata, J.; et al. Age-dependent echocardiographic and pathologic findings in a rat model with duchenne muscular dystrophy generated by CRISPR/Cas9 genome editing. Int. Heart J. 2020, 61, 1279–1284. [Google Scholar] [CrossRef]
- Sui, T.; Lau, Y.S.; Liu, D.; Liu, T.; Xu, L.; Gao, Y.; Lai, L.; Li, Z.; Han, R. A novel rabbit model of Duchenne muscular dystrophy generated by CRISPR/Cas9. Dis. Model. Mech. 2018, 11, dmm032201. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zheng, Y.; Kang, Y.; Yang, W.; Niu, Y.; Guo, X.; Tu, Z.; Si, C.; Wang, H.; Xing, R.; et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 2015, 24, 3764–3774. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ren, S.; Bai, R.; Xiao, P.; Zhou, Q.; Zhou, Y.; Zhou, Z.; Niu, Y.; Ji, W.; Chen, Y. No off-target mutations in functional genome regions of a CRISPR/Cas9-generated monkey model of muscular dystrophy. J. Biol. Chem. 2018, 293, 11654–11658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. CRISPR-mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.C.; Wu, W.S. CRISPR/Cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Moretti, A.; Fonteyne, L.; Giesert, F.; Hoppmann, P.; Meier, A.B.; Bozoglu, T.; Baehr, A.; Schneider, C.M.; Sinnecker, D.; Klett, K.; et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020, 26, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Banfi, C.; Brioschi, M.; Tremoli, E.; Straino, S.; Spallotta, F.; Mai, A.; Rotili, D.; Capogrossi, M.C.; Gaetano, C. Proteomic profile of differentially expressed plasma proteins from dystrophic mice and following suberoylanilide hydroxamic acid treatment. Proteom. Clin. Appl. 2010, 4, 71–83. [Google Scholar] [CrossRef]
- Gulston, M.K.; Rubtsov, D.V.; Atherton, H.J.; Clarke, K.; Davies, K.E.; Lilley, K.S.; Griffin, J.L. A combined metabolomic and proteomic investigation of the effects of a failure to express dystrophin in the mouse heart. J. Proteome Res. 2008, 7, 2069–2077. [Google Scholar] [CrossRef]
- Lewis, C.; Jockusch, H.; Ohlendieck, K. Proteomic profiling of the dystrophin-deficient MDX heart reveals drastically altered levels of key metabolic and contractile proteins. J. Biomed. Biotechnol. 2010, 2010, 648501. [Google Scholar] [CrossRef]
- Carr, S.J.; Zahedi, R.P.; Lochmuller, H.; Roos, A. Mass spectrometry-based protein analysis to unravel the tissue pathophysiology in Duchenne muscular dystrophy. Proteom. Clin. Appl. 2018, 12, 1700071. [Google Scholar] [CrossRef] [PubMed]
- Rouillon, J.; Zocevic, A.; Leger, T.; Garcia, C.; Camadro, J.M.; Udd, B.; Wong, B.; Servais, L.; Voit, T.; Svinartchouk, F. Proteomics profiling of urine reveals specific titin fragments as biomarkers of Duchenne muscular dystrophy. Neuromuscul. Disord. 2014, 24, 563–573. [Google Scholar] [CrossRef]
- Awano, H.; Matsumoto, M.; Nagai, M.; Shirakawa, T.; Maruyama, N.; Iijima, K.; Nabeshima, Y.I.; Matsuo, M. Diagnostic and clinical significance of the titin fragment in urine of Duchenne muscular dystrophy patients. Clin. Chim. Acta 2018, 476, 111–116. [Google Scholar] [CrossRef]
- Robertson, A.S.; Majchrzak, M.J.; Smith, C.M.; Gagnon, R.C.; Devidze, N.; Banks, G.B.; Little, S.C.; Nabbie, F.; Bounous, D.I.; DiPiero, J.; et al. Dramatic elevation in urinary amino terminal titin fragment excretion quantified by immunoassay in Duchenne muscular dystrophy patients and in dystrophin deficient rodents. Neuromuscul. Disord. 2017, 27, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Awano, H.; Maruyama, N.; Nishio, H. Titin fragment in urine: A noninvasive biomarker of muscle degradation. Adv. Clin. Chem. 2019, 90, 1–23. [Google Scholar] [CrossRef]
- Rouillon, J.; Poupiot, J.; Zocevic, A.; Amor, F.; Leger, T.; Garcia, C.; Camadro, J.M.; Wong, B.; Pinilla, R.; Cosette, J.; et al. Serum proteomic profiling reveals fragments of MYOM3 as potential biomarkers for monitoring the outcome of therapeutic interventions in muscular dystrophies. Hum. Mol. Genet. 2015, 24, 4916–4932. [Google Scholar] [CrossRef] [Green Version]
- Brinkmeier, H.; Ohlendieck, K. Chaperoning heat shock proteins: Proteomic analysis and relevance for normal and dystrophin-deficient muscle. Proteom. Clin. Appl. 2014, 8, 875–895. [Google Scholar] [CrossRef]
- Holland, A.; Dowling, P.; Ohlendieck, K. New pathobiochemical insights into dystrophinopathy from the proteomics of senescent mdx mouse muscle. Front. Aging Neurosci. 2014, 6, 109. [Google Scholar] [CrossRef]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the dystrophin-deficient mdx phenocopy of dystrophinopathy-associated cardiomyopathy. Biomed. Res. Int. 2014, 2014, 246195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef]
- Johnson, E.K.; Zhang, L.; Adams, M.E.; Phillips, A.; Freitas, M.A.; Froehner, S.C.; Green-Church, K.B.; Montanaro, F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE 2012, 7, e43515. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yue, Y.; Lai, Y.; Hakim, C.H.; Duan, D. Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin-null mice. J. Pathol. 2011, 223, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.S.; Kim, G.E.; Holewinski, R.J.; Venkatraman, V.; Zhu, G.; Bedja, D.; Kass, D.A.; Van Eyk, J.E. Transient receptor potential channel 6 regulates abnormal cardiac S-nitrosylation in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E10763–E10771. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Kohr, M.; Sun, J.; Nguyen, T.; Steenbergen, C. S-nitrosylation: A radical way to protect the heart. J. Mol. Cell. Cardiol. 2012, 52, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The relationship between protein S-nitrosylation and human diseases: A review. Neurochem. Res. 2020, 45, 2815–2827. [Google Scholar] [CrossRef]
- Tamiyakul, H.; Kemter, E.; Kosters, M.; Ebner, S.; Blutke, A.; Klymiuk, N.; Flenkenthaler, F.; Wolf, E.; Arnold, G.J.; Frohlich, T. Progressive proteome changes in the myocardium of a pig model for duchenne muscular dystrophy. iScience 2020, 23, 101516. [Google Scholar] [CrossRef]
- Judge, D.P.; Kass, D.A.; Thompson, W.R.; Wagner, K.R. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am. J. Cardiovasc. Drugs 2011, 11, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Diegoli, M.; Grasso, M.; Favalli, V.; Serio, A.; Gambarin, F.I.; Klersy, C.; Pasotti, M.; Agozzino, E.; Scelsi, L.; Ferlini, A.; et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J. Am. Coll. Cardiol. 2011, 58, 925–934. [Google Scholar] [CrossRef] [Green Version]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, T.; Straw, S.; Cheng, C.; Garnham, J.O.; Scragg, J.L.; Smith, J.; Koshy, A.O.; Levelt, E.; Sukumar, P.; Gierula, J.; et al. Unique transcriptome signature distinguishes patients with heart failure with myopathy. J. Am. Heart Assoc. 2020, 9, e017091. [Google Scholar] [CrossRef]
- Chiesa, M.; Piacentini, L.; Bono, E.; Milazzo, V.; Campodonico, J.; Marenzi, G.; Colombo, G.I. Whole blood transcriptome profile at hospital admission discriminates between patients with ST-segment elevation and non-ST-segment elevation acute myocardial infarction. Sci. Rep. 2020, 10, 8731. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, L.; Werba, J.P.; Bono, E.; Saccu, C.; Tremoli, E.; Spirito, R.; Colombo, G.I. Genome-wide expression profiling unveils autoimmune response signatures in the perivascular adipose tissue of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef]
- Haslett, J.N.; Sanoudou, D.; Kho, A.T.; Bennett, R.R.; Greenberg, S.A.; Kohane, I.S.; Beggs, A.H.; Kunkel, L.M. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 15000–15005. [Google Scholar] [CrossRef] [Green Version]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’Amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Marotta, M.; Ruiz-Roig, C.; Sarria, Y.; Peiro, J.L.; Nunez, F.; Ceron, J.; Munell, F.; Roig-Quilis, M. Muscle genome-wide expression profiling during disease evolution in mdx mice. Physiol. Genom. 2009, 37, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Guo, W.; Andrade, F.H. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2002, 11, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Porter, J.D.; Merriam, A.P.; Leahy, P.; Gong, B.; Khanna, S. Dissection of temporal gene expression signatures of affected and spared muscle groups in dystrophin-deficient (mdx) mice. Hum. Mol. Genet. 2003, 12, 1813–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, D.; Davies, K.E. Microarray analysis of mdx mice expressing high levels of utrophin: Therapeutic implications for dystrophin deficiency. Neuromuscul. Disord. 2008, 18, 239–247. [Google Scholar] [CrossRef]
- Almeida, C.F.; Martins, P.C.; Vainzof, M. Comparative transcriptome analysis of muscular dystrophy models Large(myd), Dmd(mdx)/Large(myd) and Dmd(mdx): What makes them different? Eur. J. Hum. Genet. 2016, 24, 1301–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haslett, J.N.; Kang, P.B.; Han, M.; Kho, A.T.; Sanoudou, D.; Volinski, J.M.; Beggs, A.H.; Kohane, I.S.; Kunkel, L.M. The influence of muscle type and dystrophin deficiency on murine expression profiles. Mamm. Genome 2005, 16, 739–748. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Van Pelt, D.W.; Kharaz, Y.A.; Sarver, D.C.; Eckhardt, L.R.; Dzierzawski, J.T.; Disser, N.P.; Piacentini, A.N.; Comerford, E.; McDonagh, B.; Mendias, C.L. Multiomics analysis of the mdx/mTR mouse model of Duchenne muscular dystrophy. Connect. Tissue Res. 2021, 62, 24–39. [Google Scholar] [CrossRef]
- Paik, D.T.; Cho, S.; Tian, L.; Chang, H.Y.; Wu, J.C. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat. Rev. Cardiol. 2020, 17, 457–473. [Google Scholar] [CrossRef]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Lu, X.; Shao, X.; Zhu, L.; Fan, X. Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 2021, 39, 43–58. [Google Scholar] [CrossRef]
- Chemello, F.; Wang, Z.; Li, H.; McAnally, J.R.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Degenerative and regenerative pathways underlying Duchenne muscular dystrophy revealed by single-nucleus RNA sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 29691–29701. [Google Scholar] [CrossRef]
- Gonorazky, H.; Liang, M.; Cummings, B.; Lek, M.; Micallef, J.; Hawkins, C.; Basran, R.; Cohn, R.; Wilson, M.D.; MacArthur, D.; et al. RNAseq analysis for the diagnosis of muscular dystrophy. Ann. Clin. Transl. Neurol. 2016, 3, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Kamdar, F.; Das, S.; Gong, W.; Klaassen Kamdar, A.; Meyers, T.A.; Shah, P.; Ervasti, J.M.; Townsend, D.; Kamp, T.J.; Wu, J.C.; et al. Stem cell-derived cardiomyocytes and beta-adrenergic receptor blockade in duchenne muscular dystrophy cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 1159–1174. [Google Scholar] [CrossRef]
- Signorelli, M.; Ebrahimpoor, M.; Veth, O.; Hettne, K.; Verwey, N.; Garcia-Rodriguez, R.; Tanganyika-deWinter, C.L.; Lopez Hernandez, L.B.; Escobar Cedillo, R.; Gomez Diaz, B.; et al. Peripheral blood transcriptome profiling enables monitoring disease progression in dystrophic mice and patients. EMBO Mol. Med. 2021, 13, e13328. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Shameer, K.; Johnson, K.W.; Glicksberg, B.S.; Dudley, J.T.; Sengupta, P.P. Machine learning in cardiovascular medicine: Are we there yet? Heart 2018, 104, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics factor analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- Pai, S.; Hui, S.; Isserlin, R.; Shah, M.A.; Kaka, H.; Bader, G.D. netDx: Interpretable patient classification using integrated patient similarity networks. Mol. Syst. Biol. 2019, 15, e8497. [Google Scholar] [CrossRef]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine learning and integrative analysis of biomedical big data. Genes 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, M.; Colombo, G.I.; Piacentini, L. DaMiRseq-an R/Bioconductor package for data mining of RNA-Seq data: Normalization, feature selection and classification. Bioinformatics 2018, 34, 1416–1418. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, M.; Maioli, G.; Colombo, G.I.; Piacentini, L. GARS: Genetic Algorithm for the identification of a robust subset of features in high-dimensional datasets. BMC Bioinform. 2020, 21, 54. [Google Scholar] [CrossRef] [Green Version]
- Heydemann, A.; Wheeler, M.T.; McNally, E.M. Cardiomyopathy in animal models of muscular dystrophy. Curr. Opin. Cardiol. 2001, 16, 211–217. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A brief review of current maturation methods for human induced pluripotent stem cells-derived cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Pu, W.T. Cardiomyocyte maturation: New phase in development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Maroli, G.; Braun, T. The long and winding road of cardiomyocyte maturation. Cardiovasc. Res. 2021, 117, 712–726. [Google Scholar] [CrossRef]
- Smith, A.S.T.; Davis, J.; Lee, G.; Mack, D.L.; Kim, D.H. Muscular dystrophy in a dish: Engineered human skeletal muscle mimetics for disease modeling and drug discovery. Drug Discov. Today 2016, 21, 1387–1398. [Google Scholar] [CrossRef] [Green Version]
- Caputo, L.; Granados, A.; Lenzi, J.; Rosa, A.; Ait-Si-Ali, S.; Puri, P.L.; Albini, S. Acute conversion of patient-derived Duchenne muscular dystrophy iPSC into myotubes reveals constitutive and inducible over-activation of TGFbeta-dependent pro-fibrotic signaling. Skelet. Muscle 2020, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Model Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef] [Green Version]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C.; American Heart Association Council on Functional, G.; Translational, B.; et al. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: A scientific statement from the american heart association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [Green Version]
- Biendarra-Tiegs, S.M.; Secreto, F.J.; Nelson, T.J. Addressing variability and heterogeneity of induced pluripotent stem cell-derived cardiomyocytes. Adv. Exp. Med. Biol. 2020, 1212, 1–29. [Google Scholar] [CrossRef]
- Mesquita, F.C.P.; Morrissey, J.; Lee, P.F.; Monnerat, G.; Xi, Y.; Andersson, H.; Nogueira, F.C.S.; Domont, G.B.; Sampaio, L.C.; Hochman-Mendez, C.; et al. Cues from human atrial extracellular matrix enrich the atrial differentiation of human induced pluripotent stem cell-derived cardiomyocytes. Biomater. Sci. 2021, 9, 3737–3749. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, R.; Campbell, K.F.; Carvalho, J.L.; Ruiz, E.C.; Kim, G.C.; Schmuck, E.G.; Raval, A.N.; da Rocha, A.M.; Herron, T.J.; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 2019, 10, 2238. [Google Scholar] [CrossRef] [Green Version]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.J.; Osteil, P.; et al. Single-cell transcriptomic analysis of cardiac differentiation from human PSCs reveals HOPX-dependent cardiomyocyte maturation. Cell Stem Cell 2018, 23, 586–598.e588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, H.; Liao, Y.; Ren, Z.; Mao, L.; Yao, F.; Yu, P.; Ye, Y.; Zhang, Z.; Li, S.; Xu, H.; et al. Single-cell reconstruction of differentiation trajectory reveals a critical role of ETS1 in human cardiac lineage commitment. BMC Biol. 2019, 17, 89. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.J.; Krotenberg Garcia, A.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell 2020, 26, 862–879.e811. [Google Scholar] [CrossRef]
- Beauchamp, P.; Jackson, C.B.; Ozhathil, L.C.; Agarkova, I.; Galindo, C.L.; Sawyer, D.B.; Suter, T.M.; Zuppinger, C. 3D Co-culture of hiPSC-derived cardiomyocytes with cardiac fibroblasts improves tissue-like features of cardiac spheroids. Front. Mol. Biosci. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tian, L.; Shen, M.; Tu, C.; Wu, H.; Gu, M.; Paik, D.T.; Wu, J.C. Generation of quiescent cardiac fibroblasts from human induced pluripotent stem cells for in vitro modeling of cardiac fibrosis. Circ. Res. 2019, 125, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Dressen, M.; Geyer, P.E.; Itzhak, D.N.; Braun, C.; Doppler, S.A.; Meier, F.; Deutsch, M.A.; Lahm, H.; Lange, R.; et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017, 8, 1469. [Google Scholar] [CrossRef]
- Sun, C.; Choi, I.Y.; Rovira Gonzalez, Y.I.; Andersen, P.; Talbot, C.C., Jr.; Iyer, S.R.; Lovering, R.M.; Wagner, K.R.; Lee, G. Duchenne muscular dystrophy hiPSC-derived myoblast drug screen identifies compounds that ameliorate disease in mdx mice. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.H.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Malatras, A.; Duguez, S.; Duddy, W. Muscle gene sets: A versatile methodological aid to functional genomics in the neuromuscular field. Skelet. Muscle 2019, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Balabanov, P.; Bushby, K.; Ensini, M.; Goemans, N.; De Luca, A.; Pereda, A.; Hemmings, R.; Campion, G.; Kaye, E.; et al. Stakeholder cooperation to overcome challenges in orphan medicine development: The example of Duchenne muscular dystrophy. Lancet Neurol. 2016, 15, 882–890. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Model | Age | Methods | Findings | Reference |
|---|---|---|---|---|
| mdx mice | 1,3,5,7,9 months | 2D-DIGE and NMR | Altered mitochondrial and glycolytic enzymes | Gulston 2008 |
| mdx mice | 9 months | 2D-DIGE | Altered energy metabolism, contraction and cytoskeletal proteins | Lewis 2010 |
| mdx mice | 7 weeks and 20 months | label free MS-based method | Reduced laminin, nidogen and annexin at 20 months | Holland 2013 |
| mdx-4cv mice | 20 months | label-free MS-based method | Altered basal lamina components, Ca2+-binding proteins, ECM proteins, cardiac myosin light chain kinase, stress response proteins and many mitochondrial and glycolytic enzymes | Murphy 2016 |
| mdx mice | 20 weeks | MS analysis of dystrophin interactors | Reduced association of dystrophin with nNOS and beta-1-syntrophin in the heart; altered cardiac ion flux and sarcomeric contraction | Johnson 2012 |
| mdx mice | 50 weeks | MS analysis of nitrosylated peptides | Increased mitochondrial and sarcomeric protein nitrosylation | Chung 2017 |
| Pig | 2 days and 3 months | Offgel prefractionation and iTRAQ-based MS analysis | Impaired mitochondrial energy production; increased inflammation; decreased DGC components | Tamiyakul 2020 |
| Model | Tissue | Method | Findings | Reference |
|---|---|---|---|---|
| human, DMD and alpha-sarcoglycan deficiency | muscle biopsy | microarray | developmental genes (e.g., alpha-actinin) overexpression; energy metabolism dysregulation; Ca2+ signalling; inflammation | Chen 2000 |
| C57BL/10ScSn-Dmdmdx/J mouse, mdx | gastrocnemius and soleus muscle | microarray | upregulation of secreted phosphoprotein 1 (minopontin, osteopontin) | Porter 2002 |
| human, DMD | skeletal muscle | microarray | overexpression of immune response signals and ECM genes | Haslett 2002 |
| C57BL/10ScSn-Dmdmdx/J mouse, mdx | gastrocnemius and soleus muscle | microarray | ECM and inflammation transcript dysregulation | Porter 2003 |
| C57BL/10ScSn-Dmdmdx/J B6Ros.Cg-Dmdmdx−5cv mouse, mdx | diaphragm, extensor digitorum longus, gastrocnemius, quadriceps, soleus and tibialis anterior muscles | microarray | muscle specific molecular signatures explain fragility of muscle tissues | Haslett 2005 |
| human, DMD | skeletal muscle | microarray | dystrophinopathy molecular signature characterised by genes involved in inflammation, ECM, muscle regeneration and energy metabolism | Pescatori 2007 |
| C57BL/10ScSn-Dmdmdx/J and Fiona transgenic line mouse, mdx | Tibialis anterior muscle | microarray | utrophin modulates gene expression profile, thus is likely to be beneficial in dystrophin deficiency | Baban 2008 |
| C57BL/10ScSn-mdx mouse, mdx | medial gastrocnemius | microarray | upregulated genes related to inflammation, ECM, muscle regeneration; identified candidate genes declining muscle necrosis, possible therapeutic targets; | Marotta 2009 |
| human, DMD | skeletal muscle | RNA-seq | clinical genetic diagnostics, identification of intronic mutation | Gonorazky 2015 |
| C57BL/10ScSn-Dmdmdx/J; B6C3Fe a/a-Largemyd/J; and double mutant Dmdmdx/J/Largemyd/J mouse, mdx | calf muscle | microarray | model signatures differ in genes regulating immune system, muscle degeneration/regeneration and ECM remodelling | Almeida 2016 |
| C57/BL6N-DmdΔEx51 mouse | tibialis anterior muscle | RNA-seq; and snRNA-seq | myonuclei subtypes marker identification | Chemello 2020 |
| human, DMD; and C57BL/10ScSn-Dmdmdx/J mouse, mdx | DMD patient-specific iPSC-derived cardiomyocytes; and mouse left ventricle | RNA-seq and scRNA-seq | assessment for DAC modelling, comparing profiling of DMD patients, with animal and in vitro models. | Kamdar 2020 |
| mouse, mdx/mTRG2 | plantaris and TA muscles | RNA-seq; Proteomics, metablomics lipidomics (MS) | DMD transcriptome and proteome signatures are different in protein balance, contractile elements, ECM and metabolism | Van Pelt 2021 |
| human, DMD; and C57BL/10ScSn-Dmdmdx/J mouse, mdx | human and mouse blood | RNA-seq | DMD progression and therapy response evaluation in surrogate tissue | Signorelli 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gowran, A.; Brioschi, M.; Rovina, D.; Chiesa, M.; Piacentini, L.; Mallia, S.; Banfi, C.; Pompilio, G.; Santoro, R. Multiomic Approaches to Uncover the Complexities of Dystrophin-Associated Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 8954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168954
Gowran A, Brioschi M, Rovina D, Chiesa M, Piacentini L, Mallia S, Banfi C, Pompilio G, Santoro R. Multiomic Approaches to Uncover the Complexities of Dystrophin-Associated Cardiomyopathy. International Journal of Molecular Sciences. 2021; 22(16):8954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168954
Chicago/Turabian StyleGowran, Aoife, Maura Brioschi, Davide Rovina, Mattia Chiesa, Luca Piacentini, Sara Mallia, Cristina Banfi, Giulio Pompilio, and Rosaria Santoro. 2021. "Multiomic Approaches to Uncover the Complexities of Dystrophin-Associated Cardiomyopathy" International Journal of Molecular Sciences 22, no. 16: 8954. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168954