
Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARα


 ,
,  and
and
Abstract
:1. Introduction
2. Peroxisomal β-Oxidation Systems
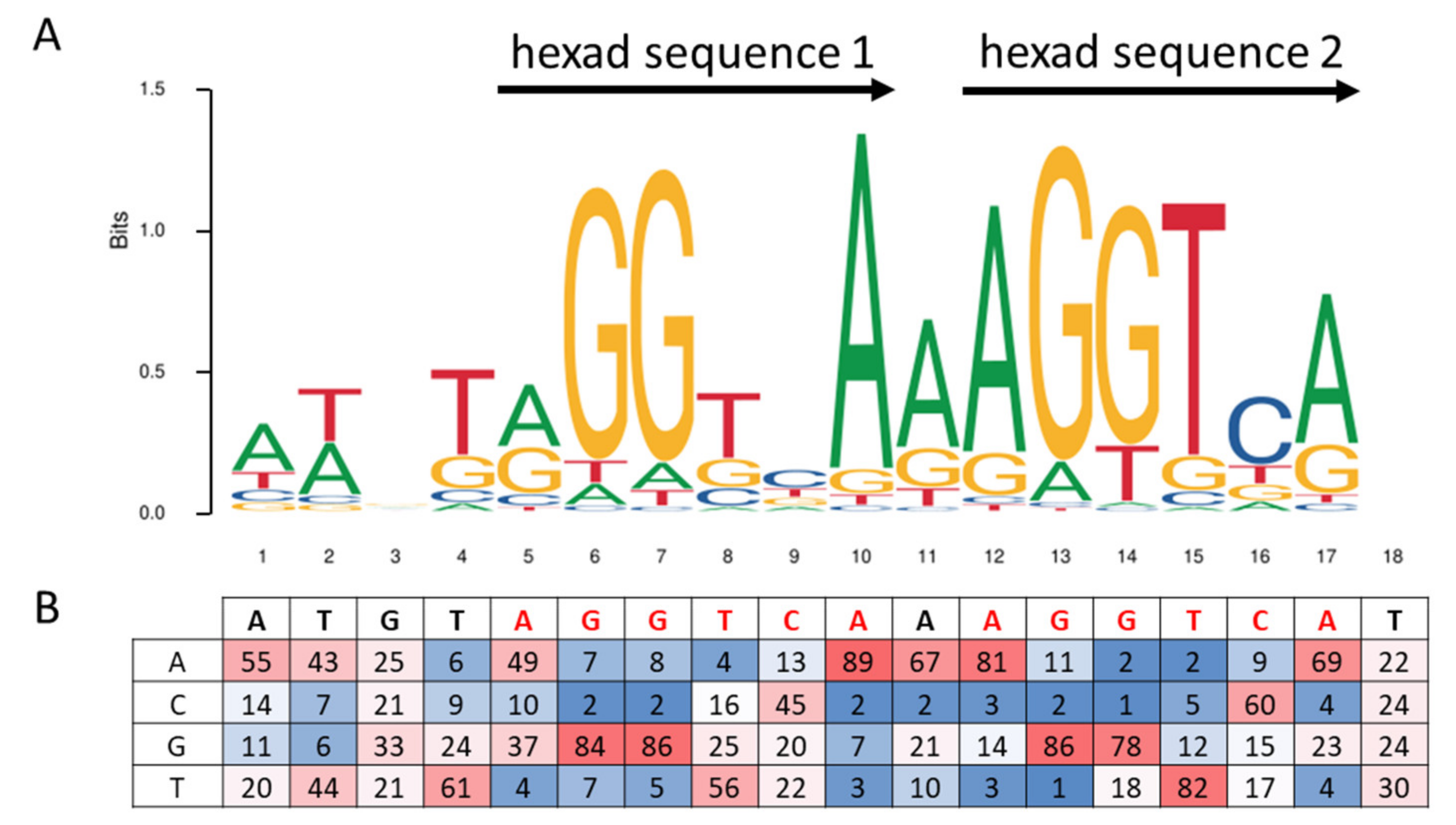
3. Peroxisome Proliferator Response Element, PPRE
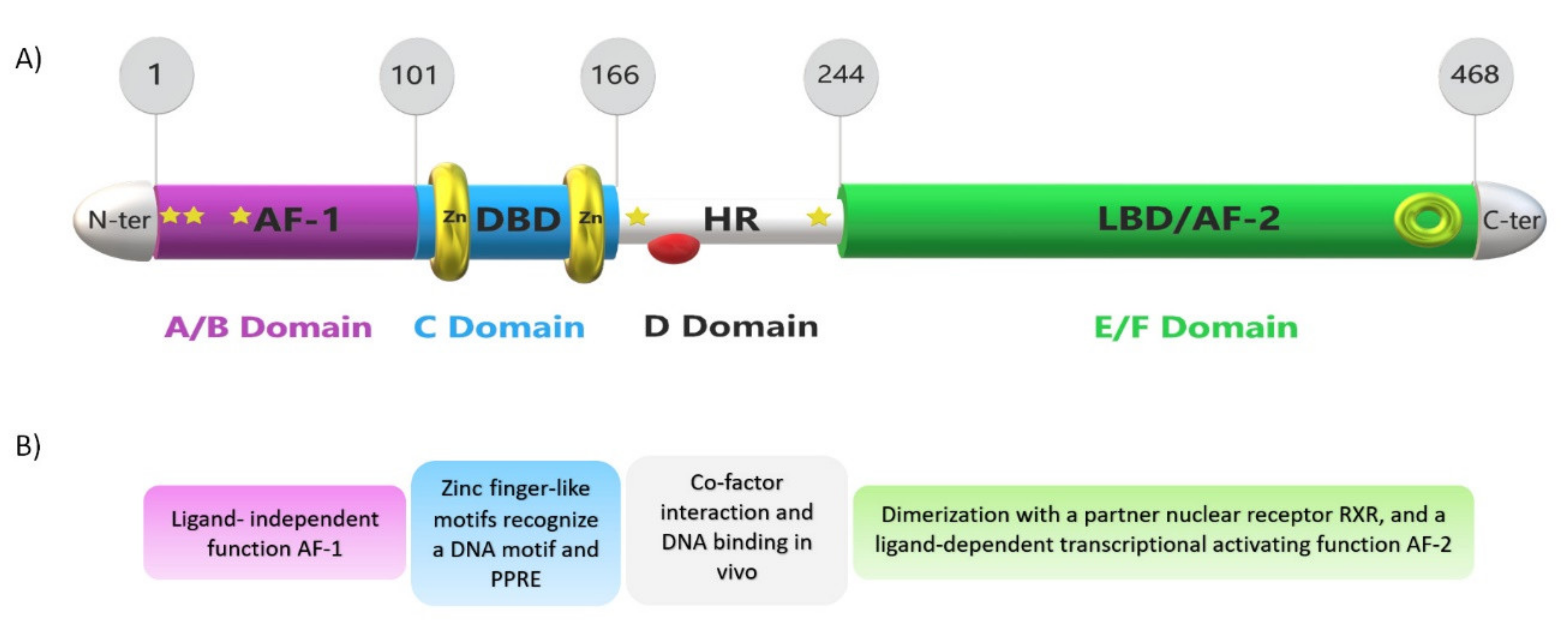
4. PPARs and PPARα Structure and Function
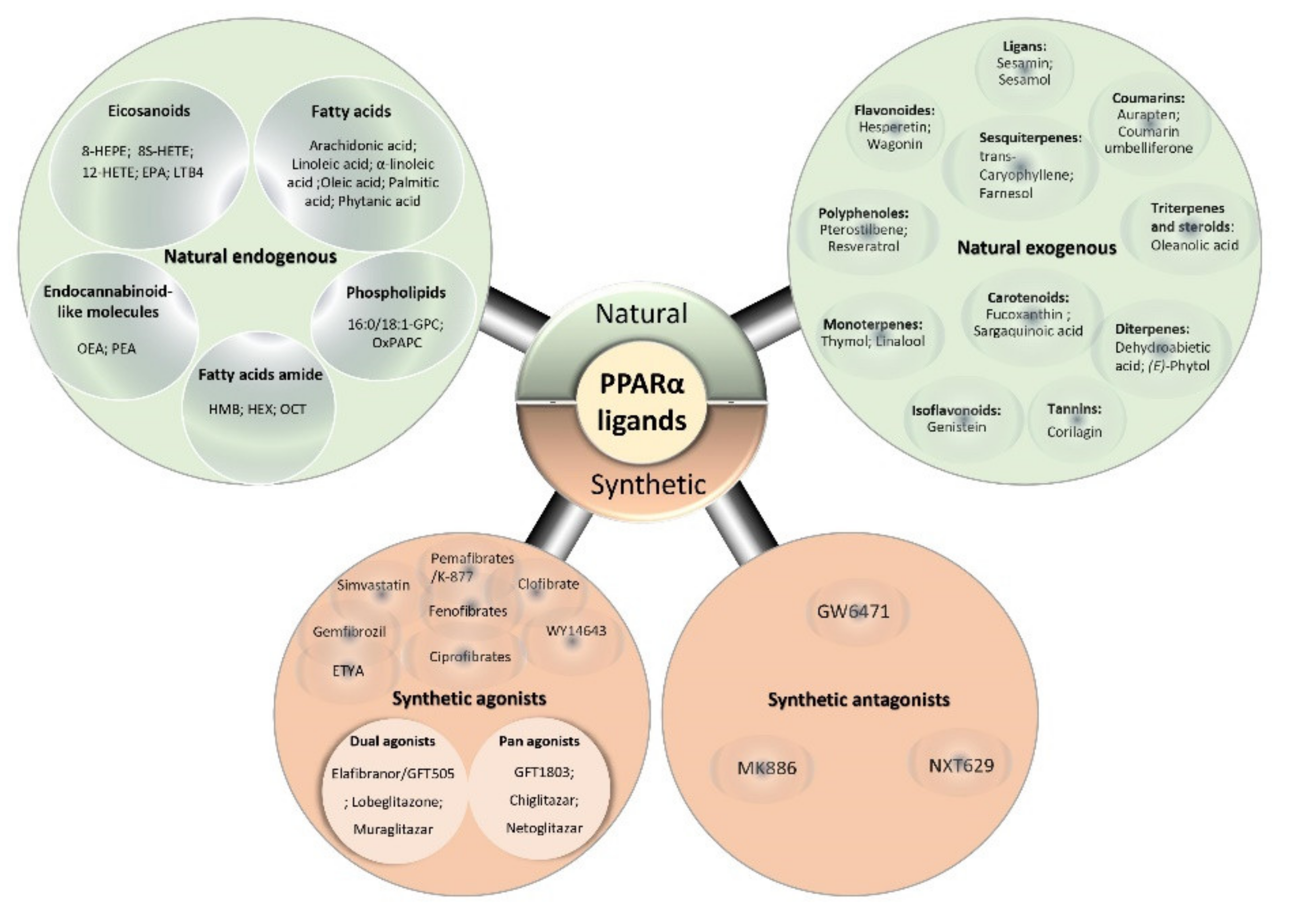
5. PPARα Ligands
5.1. PPARα Natural Ligands
5.2. PPARα Synthetic Ligands
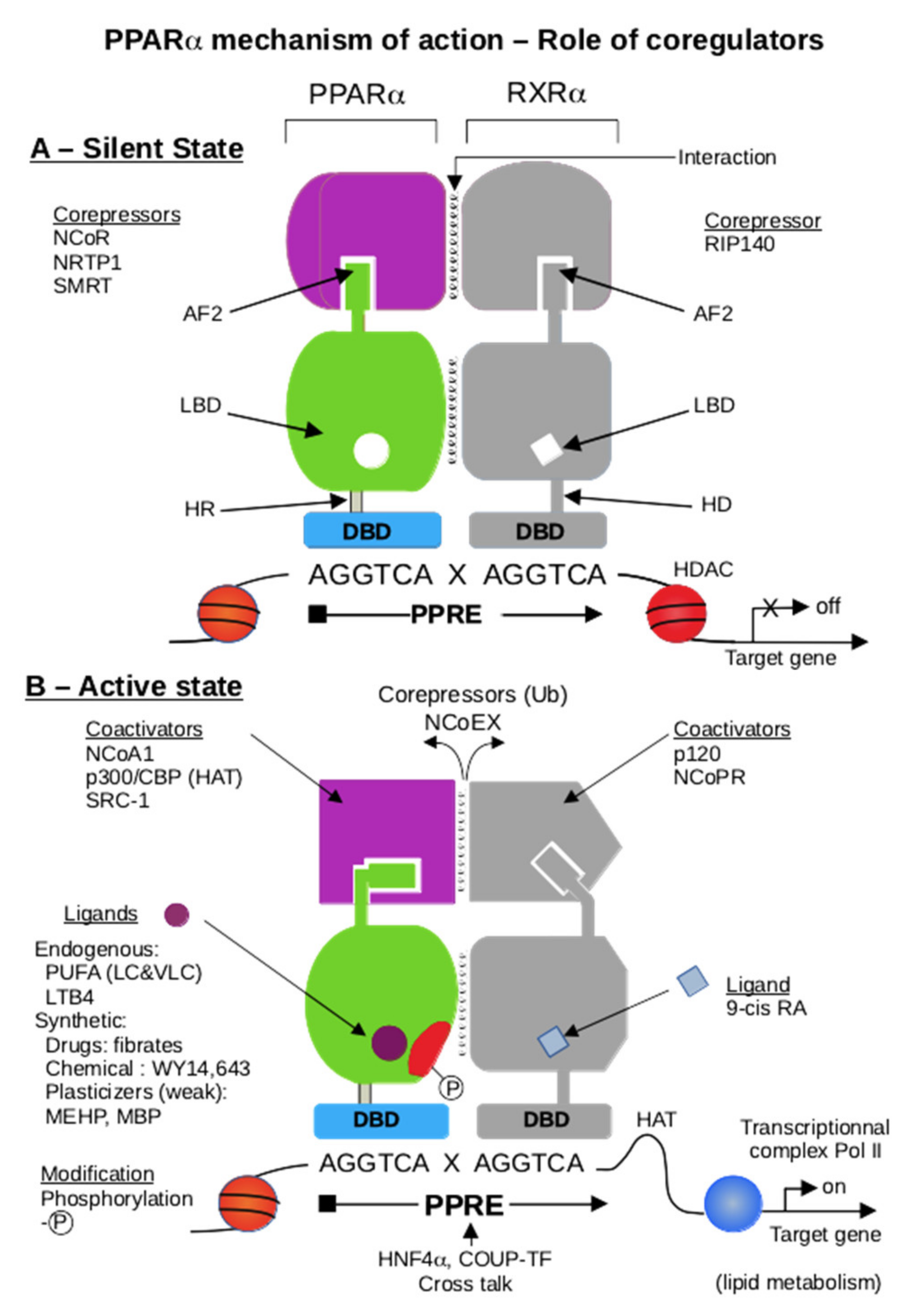
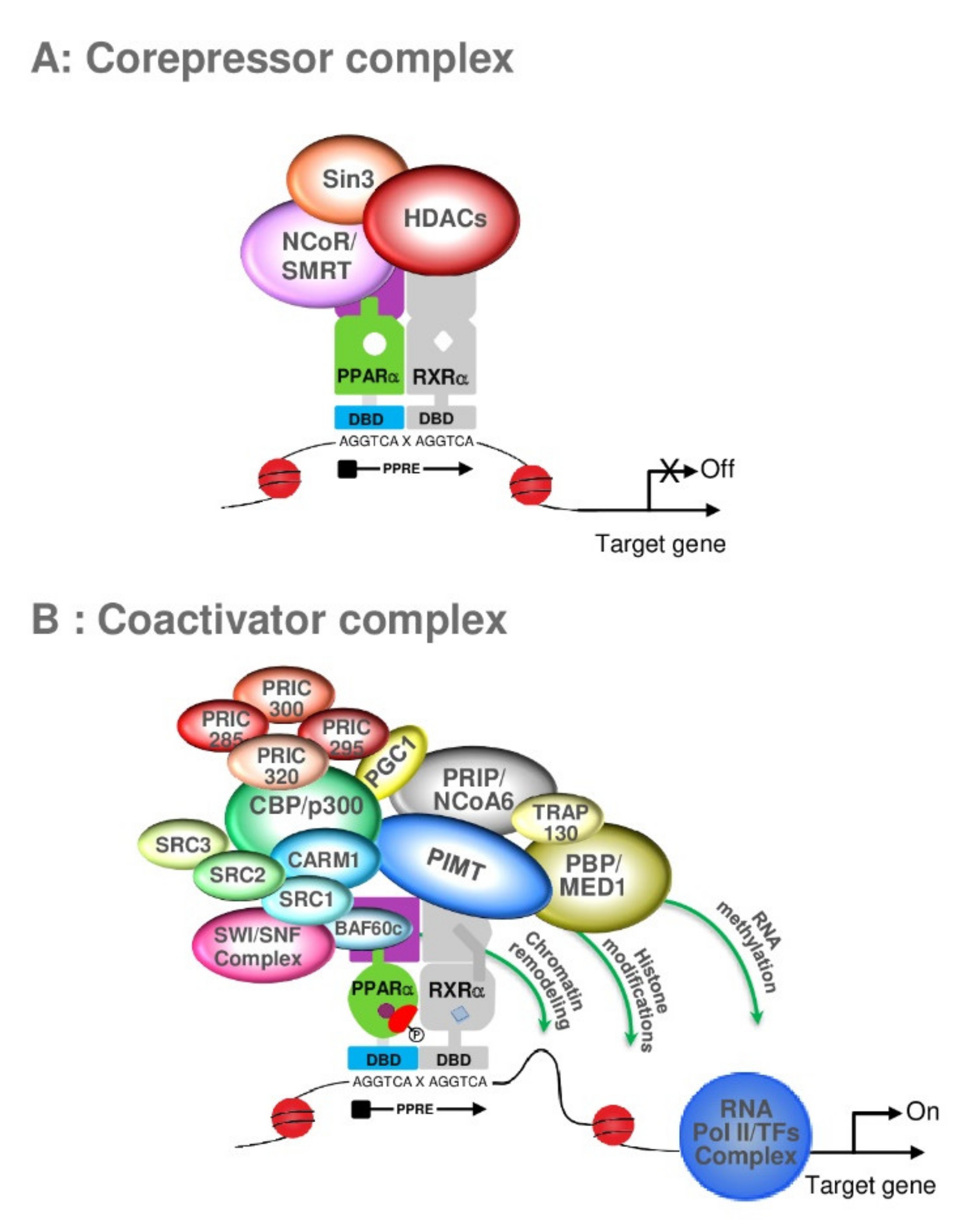
6. PPARα and Coregulators
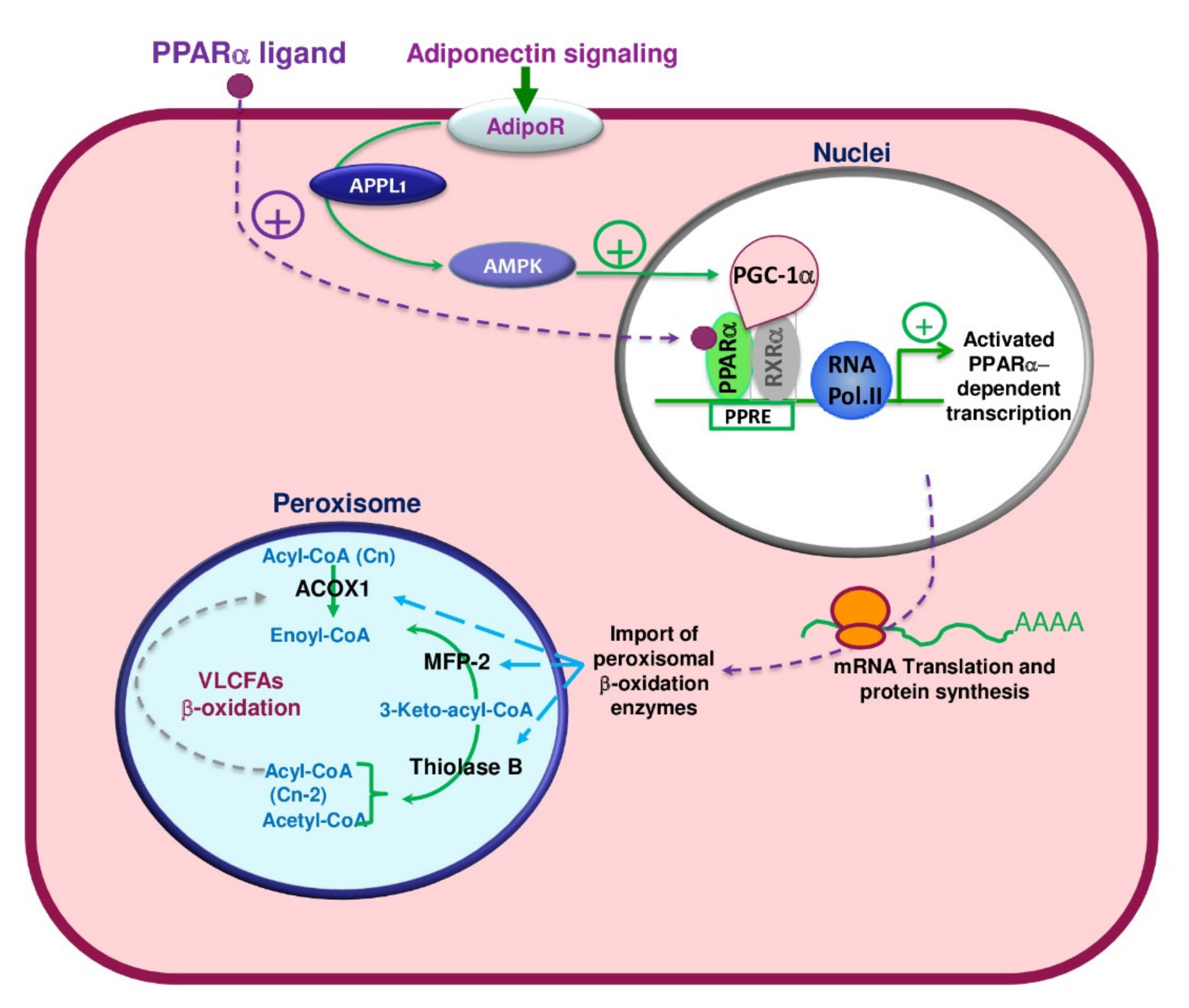
7. Metabolic Regulation of the Peroxisomal β-Oxidation Pathways
8. PPARα Expression in Species and Tissue Distribution
8.1. PPARα Expression in Different Species
8.2. PPARα Tissue Distribution
8.3. Lessons from Pparα Knockout
8.4. Lessons from Pparα-KO in the Liver
8.5. Lessons from Ppara-KO in the Heart
9. PPARα and Micronutrients
10. Effect of Polyphenols, Known as Antioxidants and Anti-Aging Compounds
10.1. Resveratrol
10.2. Quercetin
10.3. EGCG (Epigallocatechin-3-Gallate)
10.4. Curcumin
10.5. Anthocyanins
10.6. Coffee
10.7. Edible Oil Products
11. Conclusions and Future Directions
- The shuttling of substrates and cofactors from and into peroxisome.
- What is the exact role of peroxisomal β-oxidation in lipid metabolism and cell signaling?
- How can peroxisome be a mediator and responder of metabolic and environmental stresses?
- What are the molecular events that are required at the metabolic level?
- (a)
- Does heterodimerization of PPAR/RXR control the regulation? Is it controlled by coregulators?
- (b)
- What is the nature of ligands?
- (c)
- What is the nature of micronutrients? Are they natural agonists or antagonists or their balance?
- (d)
- Is PPARα the only nuclear receptor governing peroxisomal β-oxidation-related genes?
- (e)
- How do coregulators play in concert to fine-tune metabolically peroxisomal β-oxidation pathway?
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Latruffe, N.; Vamecq, J. Evolutionary Aspects of Peroxisomes as Cell Organelles, and of Genes Encoding Peroxisomal Proteins. Biol. Cell 2000, 92, 389–395. [Google Scholar] [CrossRef]
- Hess, R.; Stäubli, W.; Riess, W. Nature of the Hepatomegalic Effect Produced by Ethyl-Chlorophenoxy-Isobutyrate in the Rat. Nature 1965, 208, 856–858. [Google Scholar] [CrossRef]
- Lalwani, N.D.; Reddy, M.K.; Qureshi, S.A.; Sirtori, C.R.; Abiko, Y.; Reddy, J.K. Evaluation of Selected Hypolipidemic Agents for the Induction of Peroxisomal Enzymes and Peroxisome Proliferation in the Rat Liver. Hum. Toxicol. 1983, 2, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a Member of the Steroid Hormone Receptor Superfamily by Peroxisome Proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Zhou, T.; Yan, X.; Wang, G.; Liu, H.; Gan, X.; Zhang, T.; Wang, J.; Li, L. Evolutionary Pattern and Regulation Analysis to Support Why Diversity Functions Existed within PPAR Gene Family Members. BioMed Res. Int. 2015, 2015, e613910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarow, P.B.; De Duve, C. A Fatty Acyl-CoA Oxidizing System in Rat Liver Peroxisomes; Enhancement by Clofibrate, a Hypolipidemic Drug. Proc. Natl. Acad. Sci. USA 1976, 73, 2043–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkaoui-Malki, M.; Surapureddi, S.; El-Hajj, H.I.; Vamecq, J.; Andreoletti, P. Hepatic Steatosis and Peroxisomal Fatty Acid Beta-Oxidation. Curr. Drug Metab. 2012, 13, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, P.; Raas, Q.; Gondcaille, C.; Cherkaoui-Malki, M.; Trompier, D.; Savary, S. Predictive Structure and Topology of Peroxisomal ATP-Binding Cassette (ABC) Transporters. Int. J. Mol. Sci. 2017, 18, 1593. [Google Scholar] [CrossRef] [Green Version]
- Watkins, P.A.; Ellis, J.M. Peroxisomal Acyl-CoA Synthetases. Biochim. Biophys. Acta 2012, 1822, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caira, F.; Clémencet, M.C.; Cherkaoui-Malki, M.; Dieuaide-Noubhani, M.; Pacot, C.; Van Veldhoven, P.P.; Latruffe, N. Differential Regulation by a Peroxisome Proliferator of the Different Multifunctional Proteins in Guinea Pig: CDNA Cloning of the Guinea Pig D-Specific Multifunctional Protein 2. Biochem. J. 1998, 330 Pt 3, 1361–1368. [Google Scholar] [CrossRef]
- Osumi, T.; Ishii, N.; Hijikata, M.; Kamijo, K.; Ozasa, H.; Furuta, S.; Miyazawa, S.; Kondo, K.; Inoue, K.; Kagamiyama, H.; et al. Molecular Cloning and Nucleotide Sequence of the CDNA for Rat Peroxisomal Enoyl-CoA: Hydratase-3-Hydroxyacyl-CoA Dehydrogenase Bifunctional Enzyme. J. Biol. Chem. 1985, 260, 8905–8910. [Google Scholar] [CrossRef]
- Latruffe, N. Human Peroxisomal 3-Ketoacyl-CoA Thiolase: Tissue Expression and Metabolic Regulation: Human Peroxisomal Thiolase. Adv. Exp. Med. Biol. 2020, 1299, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Baes, M.; Van Veldhoven, P.P. Hepatic Dysfunction in Peroxisomal Disorders. Biochim. Biophys. Acta 2016, 1863, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P.; De Schryver, E.; Young, S.G.; Zwijsen, A.; Fransen, M.; Espeel, M.; Baes, M.; Van Ael, E. Slc25a17 Gene Trapped Mice: PMP34 Plays a Role in the Peroxisomal Degradation of Phytanic and Pristanic Acid. Front. Cell Dev. Biol. 2020, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Seedorf, U.; Brysch, P.; Engel, T.; Schrage, K.; Assmann, G. Sterol Carrier Protein X Is Peroxisomal 3-Oxoacyl Coenzyme A Thiolase with Intrinsic Sterol Carrier and Lipid Transfer Activity. J. Biol. Chem. 1994, 269, 21277–21283. [Google Scholar] [CrossRef]
- Ranea-Robles, P.; Violante, S.; Argmann, C.; Dodatko, T.; Bhattacharya, D.; Chen, H.; Yu, C.; Friedman, S.L.; Puchowicz, M.; Houten, S.M. Murine Deficiency of Peroxisomal L-Bifunctional Protein (EHHADH) Causes Medium-Chain 3-Hydroxydicarboxylic Aciduria and Perturbs Hepatic Cholesterol Homeostasis. Cell. Mol. Life Sci. 2021. [Google Scholar] [CrossRef]
- Wang, H.; Lu, J.; Chen, X.; Schwalbe, M.; Gorka, J.E.; Mandel, J.A.; Wang, J.; Goetzman, E.S.; Ranganathan, S.; Dobrowolski, S.F.; et al. Acquired Deficiency of Peroxisomal Dicarboxylic Acid Catabolism Is a Metabolic Vulnerability in Hepatoblastoma. J. Biol. Chem. 2021, 100283. [Google Scholar] [CrossRef]
- Tillander, V.; Alexson, S.E.H.; Cohen, D.E. Deactivating Fatty Acids: Acyl-CoA Thioesterase-Mediated Control of Lipid Metabolism. Trends Endocrinol. Metab. 2017, 28, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Bowen, K.J.; Kris-Etherton, P.M.; Shearer, G.C.; West, S.G.; Reddivari, L.; Jones, P.J.H. Oleic Acid-Derived Oleoylethanolamide: A Nutritional Science Perspective. Prog. Lipid Res. 2017, 67, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional Regulation of Metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs Are a Unique Set of Fatty Acid Regulated Transcription Factors Controlling Both Lipid Metabolism and Inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef] [PubMed]
- More, V.R.; Campos, C.R.; Evans, R.A.; Oliver, K.D.; Chan, G.N.; Miller, D.S.; Cannon, R.E. PPAR-α, a Lipid-Sensing Transcription Factor, Regulates Blood-Brain Barrier Efflux Transporter Expression. J. Cereb. Blood Flow Metab. 2017, 37, 1199–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, L.-J.; He, A.-Y.; Lu, D.-L.; Li, J.-M.; Qiao, F.; Li, D.-L.; Zhang, M.-L.; Chen, L.-Q.; Du, Z.-Y. Nutritional Background Changes the Hypolipidemic Effects of Fenofibrate in Nile Tilapia (Oreochromis Niloticus). Sci. Rep. 2017, 7, 41706. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Schoonjans, K.; Lefebvre, A.M.; Staels, B.; Auwerx, J. Coordinate Regulation of the Expression of the Fatty Acid Transport Protein and Acyl-CoA Synthetase Genes by PPARalpha and PPARgamma Activators. J. Biol. Chem. 1997, 272, 28210–28217. [Google Scholar] [CrossRef] [Green Version]
- Motojima, K.; Passilly, P.; Peters, J.M.; Gonzalez, F.J.; Latruffe, N. Expression of Putative Fatty Acid Transporter Genes Are Regulated by Peroxisome Proliferator-Activated Receptor Alpha and Gamma Activators in a Tissue- and Inducer-Specific Manner. J. Biol. Chem. 1998, 273, 16710–16714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of Metabolism and as Therapeutic Targets in Cardiovascular Disease. Part I: PPAR-α. Future Cardiol. 2017, 13, 259–278. [Google Scholar] [CrossRef]
- Kandel, B.A.; Thomas, M.; Winter, S.; Damm, G.; Seehofer, D.; Burk, O.; Schwab, M.; Zanger, U.M. Genomewide Comparison of the Inducible Transcriptomes of Nuclear Receptors CAR, PXR and PPARα in Primary Human Hepatocytes. Biochim. Biophys. Acta 2016, 1859, 1218–1227. [Google Scholar] [CrossRef]
- Lefebvre, P.; Benomar, Y.; Staels, B. Retinoid X Receptors: Common Heterodimerization Partners with Distinct Functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972. [Google Scholar] [CrossRef] [Green Version]
- Corrales, P.; Vidal-Puig, A.; Medina-Gómez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the Peroxisomal Beta-Oxidation Pathway by a Novel Family of Nuclear Hormone Receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Tudor, C.; Engelborghs, Y.; Wahli, W.; Desvergne, B. Fluorescence Imaging Reveals the Nuclear Behavior of Peroxisome Proliferator-Activated Receptor/Retinoid X Receptor Heterodimers in the Absence and Presence of Ligand. J. Biol. Chem. 2005, 280, 17880–17890. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-Cis Retinoic Acid and Peroxisome Proliferator Signalling Pathways through Heterodimer Formation of Their Receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Tugwood, J.D.; Issemann, I.; Anderson, R.G.; Bundell, K.R.; McPheat, W.L.; Green, S. The Mouse Peroxisome Proliferator Activated Receptor Recognizes a Response Element in the 5′ Flanking Sequence of the Rat Acyl CoA Oxidase Gene. EMBO J. 1992, 11, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, J.; Byun, J.; Park, J.Y.; Yamamoto, T.; Schesing, K.; Tian, B.; Sadoshima, J.; Oka, S. An Ideal PPAR Response Element Bound to and Activated by PPARα. PLoS ONE 2015, 10, e0134996. [Google Scholar] [CrossRef] [Green Version]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; Van Der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Pranjic, K.; Huber, A.; Ellinger, A.; Hartig, A.; Kragler, F.; Brocard, C. PEX11 Family Members Are Membrane Elongation Factors That Coordinate Peroxisome Proliferation and Maintenance. J. Cell Sci. 2010, 123, 3389–3400. [Google Scholar] [CrossRef] [Green Version]
- Hansmannel, F.; Clémencet, M.-C.; Le Jossic-Corcos, C.; Osumi, T.; Latruffe, N.; Nicolas-Francés, V. Functional Characterization of a Peroxisome Proliferator Response-Element Located in the Intron 3 of Rat Peroxisomal Thiolase B Gene. Biochem. Biophys. Res. Commun. 2003, 311, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Woodyatt, N.J.; Lambe, K.G.; Myers, K.A.; Tugwood, J.D.; Roberts, R.A. The Peroxisome Proliferator (PP) Response Element Upstream of the Human Acyl CoA Oxidase Gene Is Inactive among a Sample Human Population: Significance for Species Differences in Response to PPs. Carcinogenesis 1999, 20, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Ashibe, B.; Motojima, K. Fatty Aldehyde Dehydrogenase Is Up-Regulated by Polyunsaturated Fatty Acid via Peroxisome Proliferator-Activated Receptor Alpha and Suppresses Polyunsaturated Fatty Acid-Induced Endoplasmic Reticulum Stress. FEBS J. 2009, 276, 6956–6970. [Google Scholar] [CrossRef]
- Girnun, G.D.; Domann, F.E.; Moore, S.A.; Robbins, M.E.C. Identification of a Functional Peroxisome Proliferator-Activated Receptor Response Element in the Rat Catalase Promoter. Mol. Endocrinol. 2002, 16, 2793–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardot, O.; Aldridge, T.C.; Latruffe, N.; Green, S. PPAR-RXR Heterodimer Activates a Peroxisome Proliferator Response Element Upstream of the Bifunctional Enzyme Gene. Biochem. Biophys. Res. Commun. 1993, 192, 37–45. [Google Scholar] [CrossRef]
- Lee, G.Y.; Kim, N.H.; Zhao, Z.-S.; Cha, B.S.; Kim, Y.S. Peroxisomal-Proliferator-Activated Receptor Alpha Activates Transcription of the Rat Hepatic Malonyl-CoA Decarboxylase Gene: A Key Regulation of Malonyl-CoA Level. Biochem. J. 2004, 378, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Yamashita, D.; Yamaguchi, T.; Hirose, F.; Osumi, T. Aspects of the Regulatory Mechanisms of PPAR Functions: Analysis of a Bidirectional Response Element and Regulation by Sumoylation. Mol. Cell. Biochem. 2006, 286, 33–42. [Google Scholar] [CrossRef]
- Lopez, D.; Irby, R.B.; McLean, M.P. Peroxisome Proliferator-Activated Receptor Alpha Induces Rat Sterol Carrier Protein x Promoter Activity through Two Peroxisome Proliferator-Response Elements. Mol. Cell. Endocrinol. 2003, 205, 169–184. [Google Scholar] [CrossRef]
- Evans, R.M.; Barish, G.D.; Wang, Y.-X. PPARs and the Complex Journey to Obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular Mediators of Mitochondrial Metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.; Wahli, W. Peroxisome Proliferator-Activated Receptors: Finding the Orphan a Home. Mol. Cell. Endocrinol. 1994, 100, 149–153. [Google Scholar] [CrossRef]
- Zhu, Y.; Qi, C.; Korenberg, J.R.; Chen, X.N.; Noya, D.; Rao, M.S.; Reddy, J.K. Structural Organization of Mouse Peroxisome Proliferator-Activated Receptor Gamma (MPPAR Gamma) Gene: Alternative Promoter Use and Different Splicing Yield Two MPPAR Gamma Isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 7921–7925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göttlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty Acids Activate a Chimera of the Clofibric Acid-Activated Receptor and the Glucocorticoid Receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sher, T.; Yi, H.F.; McBride, O.W.; Gonzalez, F.J. CDNA Cloning, Chromosomal Mapping, and Functional Characterization of the Human Peroxisome Proliferator Activated Receptor. Biochemistry 1993, 32, 5598–5604. [Google Scholar] [CrossRef] [PubMed]
- Vamecq, J.; Latruffe, N. Medical Significance of Peroxisome Proliferator-Activated Receptors. Lancet 1999, 354, 141–148. [Google Scholar] [CrossRef]
- Brown, J.D.; Plutzky, J. Peroxisome Proliferator-Activated Receptors as Transcriptional Nodal Points and Therapeutic Targets. Circulation 2007, 115, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [Green Version]
- Lamichane, S.; Dahal Lamichane, B.; Kwon, S.-M. Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis. Int. J. Mol. Sci. 2018, 19, 949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.T.; Collins, J.L.; Pearce, K.H. The Nuclear Receptor Superfamily and Drug Discovery. ChemMedChem 2006, 1, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Floyd, Z.E.; Stephens, J.M. Controlling a Master Switch of Adipocyte Development and Insulin Sensitivity: Covalent Modifications of PPARγ. Biochim. Biophys. Acta 2012, 1822, 1090–1095. [Google Scholar] [CrossRef] [Green Version]
- Wadosky, K.M.; Willis, M.S. The Story so Far: Post-Translational Regulation of Peroxisome Proliferator-Activated Receptors by Ubiquitination and SUMOylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H515–H526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.-H.; Kim, M.-Y.; Jo, S.-H.; Park, J.-M.; Ahn, Y.-H. Modulation of the Transcriptional Activity of Peroxisome Proliferator-Activated Receptor Gamma by Protein-Protein Interactions and Post-Translational Modifications. Yonsei Med. J. 2013, 54, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Tufano, M.; Pinna, G. Is There a Future for PPARs in the Treatment of Neuropsychiatric Disorders? Molecules 2020, 25, 1062. [Google Scholar] [CrossRef] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARα in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. General Molecular Biology and Architecture of Nuclear Receptors. Curr. Top. Med. Chem. 2012, 12, 486–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas Bervejillo, M.; Ferreira, A.M. Understanding Peroxisome Proliferator-Activated Receptors: From the Structure to the Regulatory Actions on Metabolism. Adv. Exp. Med. Biol. 2019, 1127, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Oyama, T.; Toyota, K.; Waku, T.; Hirakawa, Y.; Nagasawa, N.; Kasuga, J.I.; Hashimoto, Y.; Miyachi, H.; Morikawa, K. Adaptability and Selectivity of Human Peroxisome Proliferator-Activated Receptor (PPAR) Pan Agonists Revealed from Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Kambe, A.; Yamamoto, Y.; Arulmozhiraja, S.; Ito, S.; Nakagawa, Y.; Tokiwa, H.; Nakano, S.; Shimano, H. Elucidation of Molecular Mechanism of a Selective PPARα Modulator, Pemafibrate, through Combinational Approaches of X-Ray Crystallography, Thermodynamic Analysis, and First-Principle Calculations. Int. J. Mol. Sci. 2020, 21, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T.; McKee, D.D.; et al. Structural Determinants of Ligand Binding Selectivity between the Peroxisome Proliferator-Activated Receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα Ligand-Binding Domain Structures with Endogenous Fatty Acids and Fibrates. iScience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic Drugs, Polyunsaturated Fatty Acids, and Eicosanoids Are Ligands for Peroxisome Proliferator-Activated Receptors Alpha and Delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty Acids and Eicosanoids Regulate Gene Expression through Direct Interactions with Peroxisome Proliferator-Activated Receptors Alpha and Gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [Green Version]
- Takada, I.; Makishima, M. Peroxisome Proliferator-Activated Receptor Agonists and Antagonists: A Patent Review (2014-Present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef]
- Elholm, M.; Dam, I.; Jorgensen, C.; Krogsdam, A.M.; Holst, D.; Kratchmarova, I.; Gottlicher, M.; Gustafsson, J.A.; Berge, R.; Flatmark, T.; et al. Acyl-CoA Esters Antagonize the Effects of Ligands on Peroxisome Proliferator-Activated Receptor Alpha Conformation, DNA Binding, and Interaction with Co-Factors. J. Biol. Chem. 2001, 276, 21410–21416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostetler, H.A.; Kier, A.B.; Schroeder, F. Very-Long-Chain and Branched-Chain Fatty Acyl-CoAs Are High Affinity Ligands for the Peroxisome Proliferator-Activated Receptor Alpha (PPARalpha). Biochemistry 2006, 45, 7669–7681. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, M.V.; Lodhi, I.J.; Yin, L.; Malapaka, R.R.V.; Xu, H.E.; Turk, J.; Semenkovich, C.F. Identification of a Physiologically Relevant Endogenous Ligand for PPARalpha in Liver. Cell 2009, 138, 476–488. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.D.; Karimian Azari, E.; Ayala, J.E. Oleoylethanolamide: A Fat Ally in the Fight against Obesity. Physiol. Behav. 2017, 176, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Campolongo, P.; Roozendaal, B.; Trezza, V.; Cuomo, V.; Astarita, G.; Fu, J.; McGaugh, J.L.; Piomelli, D. Fat-Induced Satiety Factor Oleoylethanolamide Enhances Memory Consolidation. Proc. Natl. Acad. Sci. USA 2009, 106, 8027–8031. [Google Scholar] [CrossRef] [Green Version]
- Azhar, S. Peroxisome Proliferator-Activated Receptors, Metabolic Syndrome and Cardiovascular Disease. Future Cardiol. 2010, 6, 657–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigano, D.; Sirignano, C.; Taglialatela-Scafati, O. The Potential of Natural Products for Targeting PPARα. Acta Pharm. Sin. B 2017, 7, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Green, S. PPAR: A Mediator of Peroxisome Proliferator Action. Mutat. Res. 1995, 333, 101–109. [Google Scholar] [CrossRef]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential Activation of Peroxisome Proliferator-Activated Receptors by Eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [Green Version]
- Goto, T.; Takahashi, N.; Kato, S.; Egawa, K.; Ebisu, S.; Moriyama, T.; Fushiki, T.; Kawada, T. Phytol Directly Activates Peroxisome Proliferator-Activated Receptor Alpha (PPARalpha) and Regulates Gene Expression Involved in Lipid Metabolism in PPARalpha-Expressing HepG2 Hepatocytes. Biochem. Biophys. Res. Commun. 2005, 337, 440–445. [Google Scholar] [CrossRef]
- Wahli, W.; Michalik, L. PPARs at the Crossroads of Lipid Signaling and Inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Narala, V.R.; Adapala, R.K.; Suresh, M.V.; Brock, T.G.; Peters-Golden, M.; Reddy, R.C. Leukotriene B4 Is a Physiologically Relevant Endogenous Peroxisome Proliferator-Activated Receptor-Alpha Agonist. J. Biol. Chem. 2010, 285, 22067–22074. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Ruuska, S.E.; Shaw, N.S.; Dong, D.; Noy, N. Ligand Selectivity of the Peroxisome Proliferator-Activated Receptor Alpha. Biochemistry 1999, 38, 185–190. [Google Scholar] [CrossRef]
- Delerive, P.; Furman, C.; Teissier, E.; Fruchart, J.; Duriez, P.; Staels, B. Oxidized Phospholipids Activate PPARalpha in a Phospholipase A2-Dependent Manner. FEBS Lett. 2000, 471, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kundu, M.; Jana, M.; Mishra, R.K.; Yung, Y.; Luan, C.-H.; Gonzalez, F.J.; Pahan, K. Identification and Characterization of PPARα Ligands in the Hippocampus. Nat. Chem. Biol. 2016, 12, 1075–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardes, A.; Souza, P.C.T.; Muniz, J.R.C.; Ricci, C.G.; Ayers, S.D.; Parekh, N.M.; Godoy, A.S.; Trivella, D.B.B.; Reinach, P.; Webb, P.; et al. Molecular Mechanism of Peroxisome Proliferator-Activated Receptor α Activation by WY14643: A New Mode of Ligand Recognition and Receptor Stabilization. J. Mol. Biol. 2013, 425, 2878–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-T.; Liao, C.-K.; Chiu, W.-T.; Tzeng, S.-F. Ligands of Peroxisome Proliferator-Activated Receptor-Alpha Promote Glutamate Transporter-1 Endocytosis in Astrocytes. Int. J. Biochem. Cell Biol. 2017, 86, 42–53. [Google Scholar] [CrossRef]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome Proliferator-Activated Receptors and Inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR Receptor in Different Diseases and Their Ligands: Physiological Importance and Clinical Implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and Pan-Peroxisome Proliferator-Activated Receptors (PPAR) Co-Agonism: The Bezafibrate Lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogue, A.; Anthérieu, S.; Vluggens, A.; Umbdenstock, T.; Claude, N.; de la Moureyre-Spire, C.; Weaver, R.J.; Guillouzo, A. PPAR Agonists Reduce Steatosis in Oleic Acid-Overloaded HepaRG Cells. Toxicol. Appl. Pharmacol. 2014, 276, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.-R.; Park, S.-H.; Ko, J.-W.; Cho, Y.-K.; Lee, I.-C.; Kim, J.-C.; Shin, I.-S.; Kim, J.-S. Lobeglitazone Attenuates Airway Inflammation and Mucus Hypersecretion in a Murine Model of Ovalbumin-Induced Asthma. Front. Pharmacol. 2018, 9, 906. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, K.J.; Broadhead, A.R.; Cabrera, G.; Correa, L.D.; Messmer, D.; Bundey, R.; Baccei, C.; Bravo, Y.; Chen, A.; Stock, N.S.; et al. In Vitro and in Vivo Pharmacology of NXT629, a Novel and Selective PPARα Antagonist. Eur. J. Pharmacol. 2017, 809, 130–140. [Google Scholar] [CrossRef]
- Duszka, K.; Gregor, A.; Guillou, H.; König, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction-Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708. [Google Scholar] [CrossRef]
- Kosgei, V.J.; Coelho, D.; Gueant-Rodriguez, R.M.; Gueant, J.L. Sirt1-PPARS Cross-Talk in Complex Metabolic Diseases and Inherited Disorders of the One Carbon Metabolism. Cells 2020, 9, 1882. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The Role and Regulation of the Peroxisome Proliferator Activated Receptor Alpha in Human Liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Laleh, P.; Yaser, K.; Alireza, O. Oleoylethanolamide: A Novel Pharmaceutical Agent in the Management of Obesity-an Updated Review. J. Cell. Physiol. 2019, 234, 7893–7902. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.; Jump, D.B. Unsaturated Fatty Acid Regulation of Peroxisome Proliferator-Activated Receptor Alpha Activity in Rat Primary Hepatocytes. J. Biol. Chem. 2003, 278, 35931–35939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinghaus, P.; Wolfrum, C.; Assmann, G.; Spener, F.; Seedorf, U. Phytanic Acid Activates the Peroxisome Proliferator-Activated Receptor Alpha (PPARalpha) in Sterol Carrier Protein 2-/Sterol Carrier Protein x-Deficient Mice. J. Biol. Chem. 1999, 274, 2766–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, A.W.; van Der Burg, B.; Jansen, G.A.; Wanders, R.J.; Poll-The, B.T.; van Der Saag, P.T. Pristanic Acid and Phytanic Acid: Naturally Occurring Ligands for the Nuclear Receptor Peroxisome Proliferator-Activated Receptor Alpha. J. Lipid Res. 2000, 41, 1801–1807. [Google Scholar] [CrossRef]
- Hostetler, H.A.; Petrescu, A.D.; Kier, A.B.; Schroeder, F. Peroxisome Proliferator-Activated Receptor Alpha Interacts with High Affinity and Is Conformationally Responsive to Endogenous Ligands. J. Biol. Chem. 2005, 280, 18667–18682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, P.S.; Marine, K.A.; Brady, L.J.; Ramsay, R.R. Co-Ordinate Induction of Hepatic Mitochondrial and Peroxisomal Carnitine Acyltransferase Synthesis by Diet and Drugs. Biochem. J. 1989, 260, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, S.L.; Miyata, K.S.; Zhang, B.; Subramani, S.; Rachubinski, R.A.; Capone, J.P. Diverse Peroxisome Proliferator-Activated Receptors Bind to the Peroxisome Proliferator-Responsive Elements of the Rat Hydratase/Dehydrogenase and Fatty Acyl-CoA Oxidase Genes but Differentially Induce Expression. Proc. Natl. Acad. Sci. USA 1993, 90, 5723–5727. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.K.; Mannaerts, G.P. Peroxisomal Lipid Metabolism. Annu. Rev. Nutr. 1994, 14, 343–370. [Google Scholar] [CrossRef]
- Zhang, B.; Marcus, S.L.; Miyata, K.S.; Subramani, S.; Capone, J.P.; Rachubinski, R.A. Characterization of Protein-DNA Interactions within the Peroxisome Proliferator-Responsive Element of the Rat Hydratase-Dehydrogenase Gene. J. Biol. Chem. 1993, 268, 12939–12945. [Google Scholar] [CrossRef]
- Chen, X.; Shang, L.; Deng, S.; Li, P.; Chen, K.; Gao, T.; Zhang, X.; Chen, Z.; Zeng, J. Peroxisomal Oxidation of Erucic Acid Suppresses Mitochondrial Fatty Acid Oxidation by Stimulating Malonyl-CoA Formation in the Rat Liver. J. Biol. Chem. 2020, 295, 10168–10179. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, G.; Ringseis, R.; Wen, G.; Gessner, D.K.; Rost, J.; Fraatz, M.A.; Zorn, H.; Eder, K. Branched-Chain Fatty Acids as Mediators of the Activation of Hepatic Peroxisome Proliferator-Activated Receptor Alpha by a Fungal Lipid Extract. Biomolecules 2020, 10, 1259. [Google Scholar] [CrossRef] [PubMed]
- Latruffe, N.; Cherkaoui Malki, M.; Nicolas-Frances, V.; Clemencet, M.C.; Jannin, B.; Berlot, J.P. Regulation of the Peroxisomal Beta-Oxidation-Dependent Pathway by Peroxisome Proliferator-Activated Receptor Alpha and Kinases. Biochem. Pharmacol. 2000, 60, 1027–1032. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Babich, M.A.; Baetcke, K.P.; Cook, J.C.; Corton, J.C.; David, R.M.; DeLuca, J.G.; Lai, D.Y.; McKee, R.H.; Peters, J.M.; et al. PPARalpha Agonist-Induced Rodent Tumors: Modes of Action and Human Relevance. Crit. Rev. Toxicol. 2003, 33, 655–780. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Lalwai, N.D. Carcinogenesis by Hepatic Peroxisome Proliferators: Evaluation of the Risk of Hypolipidemic Drugs and Industrial Plasticizers to Humans. Crit. Rev. Toxicol. 1983, 12, 1–58. [Google Scholar] [CrossRef]
- Gonzalez, F.J.; Peters, J.M.; Cattley, R.C. Mechanism of Action of the Nongenotoxic Peroxisome Proliferators: Role of the Peroxisome Proliferator-Activator Receptor Alpha. J. Natl. Cancer Inst. 1998, 90, 1702–1709. [Google Scholar] [CrossRef]
- Maloney, E.K.; Waxman, D.J. Trans-Activation of PPARalpha and PPARgamma by Structurally Diverse Environmental Chemicals. Toxicol. Appl. Pharmacol. 1999, 161, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbiyik, F.; Cinar, K.; Demirpence, E.; Ozsullu, T.; Tunca, R.; Haziroglu, R.; Yurdaydin, C.; Uzunalimoglu, O.; Bozkaya, H. Ligand-Induced Expression of Peroxisome Proliferator-Activated Receptor Alpha and Activation of Fatty Acid Oxidation Enzymes in Fatty Liver. Eur. J. Clin. Investig. 2004, 34, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Preiss, D.; Tikkanen, M.J.; Welsh, P.; Ford, I.; Lovato, L.C.; Elam, M.B.; LaRosa, J.C.; DeMicco, D.A.; Colhoun, H.M.; Goldenberg, I.; et al. Lipid-Modifying Therapies and Risk of Pancreatitis: A Meta-Analysis. JAMA 2012, 308, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Estrela, G.R.; Arruda, A.C.; Torquato, H.F.V.; Freitas-Lima, L.C.; Perilhão, M.S.; Wasinski, F.; Budu, A.; Fock, R.A.; Paredes-Gamero, E.J.; Araujo, R.C. Gemfibrozil Induces Anemia, Leukopenia and Reduces Hematopoietic Stem Cells via PPAR-α in Mice. Int. J. Mol. Sci. 2020, 21, 5050. [Google Scholar] [CrossRef] [PubMed]
- Oswal, D.P.; Balanarasimha, M.; Loyer, J.K.; Bedi, S.; Soman, F.L.; Rider, S.D.; Hostetler, H.A. Divergence between Human and Murine Peroxisome Proliferator-Activated Receptor Alpha Ligand Specificities. J. Lipid Res. 2013, 54, 2354–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswal, D.P.; Alter, G.M.; Rider, S.D.; Hostetler, H.A. A Single Amino Acid Change Humanizes Long-Chain Fatty Acid Binding and Activation of Mouse Peroxisome Proliferator-Activated Receptor α. J. Mol. Graph. Model. 2014, 51, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty Acids, Eicosanoids, and Hypolipidemic Agents Identified as Ligands of Peroxisome Proliferator-Activated Receptors by Coactivator-Dependent Receptor Ligand Assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear Receptors and Nonalcoholic Fatty Liver Disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic Steatohepatitis: The Role of Peroxisome Proliferator-Activated Receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Rajak, S.; Singh, B.K.; Yen, P.M. Hepatic Lipid Catabolism via PPARalpha-Lysosomal Crosstalk. Int. J. Mol. Sci. 2020, 21, 2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, N.; Wagner, K.-D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted Disruption of the Alpha Isoform of the Peroxisome Proliferator-Activated Receptor Gene in Mice Results in Abolishment of the Pleiotropic Effects of Peroxisome Proliferators. Mol. Cell. Biol. 1995, 15, 3012–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amber-Vitos, O.; Chaturvedi, N.; Nachliel, E.; Gutman, M.; Tsfadia, Y. The Effect of Regulating Molecules on the Structure of the PPAR-RXR Complex. Biochim. Biophys. Acta 2016, 1861, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Surapureddi, S.; Yu, S.; Bu, H.; Hashimoto, T.; Yeldandi, A.V.; Kashireddy, P.; Cherkaoui-Malki, M.; Qi, C.; Zhu, Y.J.; Rao, M.S.; et al. Identification of a Transcriptionally Active Peroxisome Proliferator-Activated Receptor Alpha -Interacting Cofactor Complex in Rat Liver and Characterization of PRIC285 as a Coactivator. Proc. Natl. Acad. Sci. USA 2002, 99, 11836–11841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skowron, K.J.; Booker, K.; Cheng, C.; Creed, S.; David, B.P.; Lazzara, P.R.; Lian, A.; Siddiqui, Z.; Speltz, T.E.; Moore, T.W. Steroid Receptor/Coactivator Binding Inhibitors: An Update. Mol. Cell. Endocrinol. 2019, 493, 110471. [Google Scholar] [CrossRef] [PubMed]
- Surapureddi, S.; Rana, R.; Reddy, J.K.; Goldstein, J.A. Nuclear Receptor Coactivator 6 Mediates the Synergistic Activation of Human Cytochrome P-450 2C9 by the Constitutive Androstane Receptor and Hepatic Nuclear Factor-4alpha. Mol. Pharmacol. 2008, 74, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Misra, P.; Reddy, J.K. Peroxisome Proliferator-Activated Receptor-α Activation and Excess Energy Burning in Hepatocarcinogenesis. Biochimie 2014, 98, 63–74. [Google Scholar] [CrossRef]
- Rana, R.; Surapureddi, S.; Kam, W.; Ferguson, S.; Goldstein, J.A. Med25 Is Required for RNA Polymerase II Recruitment to Specific Promoters, Thus Regulating Xenobiotic and Lipid Metabolism in Human Liver. Mol. Cell. Biol. 2011, 31, 466–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitler, K.M.; Ponce, J.M.; Oudit, G.Y.; Hall, D.D.; Grueter, C.E. Cardiac Med1 Deletion Promotes Early Lethality, Cardiac Remodeling, and Transcriptional Reprogramming. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H768–H780. [Google Scholar] [CrossRef] [PubMed]
- De Vera, I.M.S.; Zheng, J.; Novick, S.; Shang, J.; Hughes, T.S.; Brust, R.; Munoz-Tello, P.; Gardner, W.J., Jr.; Marciano, D.P.; Kong, X.; et al. Synergistic Regulation of Coregulator/Nuclear Receptor Interaction by Ligand and DNA. Structure 2017, 25, 1506–1518.e4. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-H.; Choudhary, K.; Cloutier, S.C.; Xing, Z.; Aviran, S.; Tran, E.J. Genome-Wide Discovery of DEAD-Box RNA Helicase Targets Reveals RNA Structural Remodeling in Transcription Termination. Genetics 2019, 212, 153–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Hotz-Wagenblatt, A.; Voit, R.; Grummt, I. SIRT7 and the DEAD-Box Helicase DDX21 Cooperate to Resolve Genomic R Loops and Safeguard Genome Stability. Genes Dev. 2017, 31, 1370–1381. [Google Scholar] [CrossRef]
- Taschuk, F.; Cherry, S. DEAD-Box Helicases: Sensors, Regulators, and Effectors for Antiviral Defense. Viruses 2020, 12, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arconzo, M.; Piccinin, E.; Moschetta, A. Increased Risk of Acute Liver Failure by Pain Killer Drugs in NAFLD: Focus on Nuclear Receptors and Their Coactivators. Dig. Liver Dis. 2021, 53, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Fornes, D.; Gomez Ribot, D.; Heinecke, F.; Roberti, S.L.; Capobianco, E.; Jawerbaum, A. Maternal Diets Enriched in Olive Oil Regulate Lipid Metabolism and Levels of PPARs and Their Coactivators in the Fetal Liver in a Rat Model of Gestational Diabetes Mellitus. J. Nutr. Biochem. 2020, 78, 108334. [Google Scholar] [CrossRef]
- Kalliora, C.; Kyriazis, I.D.; Oka, S.-I.; Lieu, M.J.; Yue, Y.; Area-Gomez, E.; Pol, C.J.; Tian, Y.; Mizushima, W.; Chin, A.; et al. Dual Peroxisome-Proliferator-Activated-Receptor-α/γ Activation Inhibits SIRT1-PGC1α Axis and Causes Cardiac Dysfunction. JCI Insight 2019, 5, 129556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Widlund, H.R.; Puigserver, P. PGC-1 Coactivators: Shepherding the Mitochondrial Biogenesis of Tumors. Trends Cancer 2016, 2, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallcup, M.R.; Poulard, C. Gene-Specific Actions of Transcriptional Coregulators Facilitate Physiological Plasticity: Evidence for a Physiological Coregulator Code. Trends Biochem. Sci. 2020, 45, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Emmett, M.J.; Lazar, M.A. Integrative Regulation of Physiology by Histone Deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef]
- Jaiswal, B.; Gupta, A. Modulation of Nuclear Receptor Function by Chromatin Modifying Factor TIP60. Endocrinology 2018, 159, 2199–2215. [Google Scholar] [CrossRef]
- Jankowsky, E.; Guenther, U.-P. A Helicase Links Upstream ORFs and RNA Structure. Curr. Genet. 2019, 65, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Surapureddi, S.; Viswakarma, N.; Yu, S.; Guo, D.; Rao, M.S.; Reddy, J.K. PRIC320, a Transcription Coactivator, Isolated from Peroxisome Proliferator-Binding Protein Complex. Biochem. Biophys. Res. Commun. 2006, 343, 535–543. [Google Scholar] [CrossRef]
- Jia, Y.; Liu, N.; Viswakarma, N.; Sun, R.; Schipma, M.J.; Shang, M.; Thorp, E.B.; Kanwar, Y.S.; Thimmapaya, B.; Reddy, J.K. PIMT/NCOA6IP Deletion in the Mouse Heart Causes Delayed Cardiomyopathy Attributable to Perturbation in Energy Metabolism. Int. J. Mol. Sci. 2018, 19, 1485. [Google Scholar] [CrossRef] [Green Version]
- Jeronimo, C.; Robert, F. The Mediator Complex: At the Nexus of RNA Polymerase II Transcription. Trends Cell Biol. 2017, 27, 765–783. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription Regulation by the Mediator Complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Paiano, A.; Margiotta, A.; De Luca, M.; Bucci, C. Yeast Two-Hybrid Assay to Identify Interacting Proteins. Curr. Protoc. Protein Sci. 2019, 95, e70. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, B.W. Origins of the Field of Molecular Endocrinology: A Personal Perspective. Mol. Endocrinol. 2016, 30, 1015–1018. [Google Scholar] [CrossRef] [Green Version]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A CBP Integrator Complex Mediates Transcriptional Activation and AP-1 Inhibition by Nuclear Receptors. Cell 1996, 85, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.W.S.; Anjum, B.; Shen, H.-M.; Ghosh, S.; Yen, P.M.; Sinha, R.A. Lysosomal Inhibition Attenuates Peroxisomal Gene Transcription via Suppression of PPARA and PPARGC1A Levels. Autophagy 2019, 15, 1455–1459. [Google Scholar] [CrossRef]
- Dumesic, P.A.; Egan, D.F.; Gut, P.; Tran, M.T.; Parisi, A.; Chatterjee, N.; Jedrychowski, M.; Paschini, M.; Kazak, L.; Wilensky, S.E.; et al. An Evolutionarily Conserved UORF Regulates PGC1alpha and Oxidative Metabolism in Mice, Flies, and Bluefin Tuna. Cell Metab. 2019, 30, 190–200.e6. [Google Scholar] [CrossRef] [PubMed]
- Petr, M.; Stastny, P.; Zajac, A.; Tufano, J.J.; Maciejewska-Skrendo, A. The Role of Peroxisome Proliferator-Activated Receptors and Their Transcriptional Coactivators Gene Variations in Human Trainability: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behera, A.K.; Bhattacharya, A.; Vasudevan, M.; Kundu, T.K. P53 Mediated Regulation of Coactivator Associated Arginine Methyltransferase 1 (CARM1) Expression Is Critical for Suppression of Adipogenesis. FEBS J. 2018, 285, 1730–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Chen, H.; Du, K.; Asahara, H.; Tini, M.; Emerson, B.M.; Montminy, M.; Evans, R.M. A Transcriptional Switch Mediated by Cofactor Methylation. Science 2001, 294, 2507–2511. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Fan, R. PPARα and NCOR/SMRT Corepressor Network in Liver Metabolic Regulation. FASEB J. 2020, 34, 8796–8809. [Google Scholar] [CrossRef]
- Ghisletti, S.; Huang, W.; Jepsen, K.; Benner, C.; Hardiman, G.; Rosenfeld, M.G.; Glass, C.K. Cooperative NCoR/SMRT Interactions Establish a Corepressor-Based Strategy for Integration of Inflammatory and Anti-Inflammatory Signaling Pathways. Genes Dev. 2009, 23, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, K.; Gleiberman, A.S.; Shi, C.; Simon, D.I.; Rosenfeld, M.G. Cooperative Regulation in Development by SMRT and FOXP1. Genes Dev. 2008, 22, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Cunningham, T.J.; Duester, G. Nuclear Receptor Corepressors Ncor1 and Ncor2 (Smrt) Are Required for Retinoic Acid-Dependent Repression of Fgf8 during Somitogenesis. Dev. Biol. 2016, 418, 204–215. [Google Scholar] [CrossRef]
- Duong, V.; Augereau, P.; Badia, E.; Jalaguier, S.; Cavailles, V. Regulation of Hormone Signaling by Nuclear Receptor Interacting Proteins. Adv. Exp. Med. Biol. 2008, 617, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Yagi, T.; Guo, T.; Takeda, K.; Ohguchi, H.; Koyama, H.; Aotani, D.; Imaeda, K.; Kataoka, H.; Tanaka, T. Pemafibrate, a Selective PPARα Modulator, and Fenofibrate Suppress Microglial Activation through Distinct PPARα and SIRT1-Dependent Pathways. Biochem. Biophys. Res. Commun. 2020, 524, 385–391. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 Is Regulated by a PPAR{gamma}-SIRT1 Negative Feedback Loop Associated with Senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naiman, S.; Huynh, F.K.; Gil, R.; Glick, Y.; Shahar, Y.; Touitou, N.; Nahum, L.; Avivi, M.Y.; Roichman, A.; Kanfi, Y.; et al. SIRT6 Promotes Hepatic Beta-Oxidation via Activation of PPARalpha. Cell Rep. 2019, 29, 4127–4143.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.K.; Rosenfeld, M.G. The Coregulator Exchange in Transcriptional Functions of Nuclear Receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [PubMed]
- Fritah, A.; Christian, M.; Parker, M.G. The Metabolic Coregulator RIP140: An Update. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E335–E340. [Google Scholar] [CrossRef] [PubMed]
- Venkata, N.G.; Robinson, J.A.; Cabot, P.J.; Davis, B.; Monteith, G.R.; Roberts-Thomson, S.J. Mono(2-Ethylhexyl)Phthalate and Mono-n-Butyl Phthalate Activation of Peroxisome Proliferator Activated-Receptors Alpha and Gamma in Breast. Toxicol. Lett. 2006, 163, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.I.; Xia, Z. The Retinoid X Receptors and Their Ligands. Biochim. Biophys. Acta 2012, 1821, 21–56. [Google Scholar] [CrossRef] [Green Version]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iershov, A.; Nemazanyy, I.; Alkhoury, C.; Girard, M.; Barth, E.; Cagnard, N.; Montagner, A.; Chretien, D.; Rugarli, E.I.; Guillou, H.; et al. The Class 3 PI3K Coordinates Autophagy and Mitochondrial Lipid Catabolism by Controlling Nuclear Receptor PPARα. Nat. Commun. 2019, 10, 1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalev, A.; Siegrist-Kaiser, C.A.; Yen, P.M.; Wahli, W.; Burger, A.G.; Chin, W.W.; Meier, C.A. The Peroxisome Proliferator-Activated Receptor Alpha Is a Phosphoprotein: Regulation by Insulin. Endocrinology 1996, 137, 4499–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamouton, J.; Latruffe, N. PPARα/HNF4α Interplay on Diversified Responsive Elements. Relevance in the Regulation of Liver Peroxisomal Fatty Acid Catabolism. Curr. Drug Metab. 2012, 13, 1436–1453. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic Control of Mitochondrial Biogenesis through the PGC-1 Family Regulatory Network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T. Individual Peroxisomal Beta-Oxidation Enzymes. Ann. N. Y. Acad. Sci. 1982, 386, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K. Peroxisome Proliferators and Peroxisome Proliferator-Activated Receptor Alpha: Biotic and Xenobiotic Sensing. Am. J. Pathol. 2004, 164, 2305–2321. [Google Scholar] [CrossRef]
- Raas, Q.; Gondcaille, C.; Hamon, Y.; Leoni, V.; Caccia, C.; Menetrier, F.; Lizard, G.; Trompier, D.; Savary, S. CRISPR/Cas9-Mediated Knockout of Abcd1 and Abcd2 Genes in BV-2 Cells: Novel Microglial Models for X-Linked Adrenoleukodystrophy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Dixon, E.D.; Nardo, A.D.; Claudel, T.; Trauner, M. The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes 2021, 12, 645. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- Haro, D.; Marrero, P.F.; Relat, J. Nutritional Regulation of Gene Expression: Carbohydrate-, Fat- and Amino Acid-Dependent Modulation of Transcriptional Activity. Int. J. Mol. Sci. 2019, 20, 1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, R.B.; Kelly, D.P. Cardiac Nuclear Receptors: Architects of Mitochondrial Structure and Function. J. Clin. Investig. 2017, 127, 1155–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Jia, Y.; Yang, G.; Zhang, X.; Boddu, P.C.; Petersen, B.; Narsingam, S.; Zhu, Y.-J.; Thimmapaya, B.; Kanwar, Y.S.; et al. PPARα-Deficient Ob/Ob Obese Mice Become More Obese and Manifest Severe Hepatic Steatosis Due to Decreased Fatty Acid Oxidation. Am. J. Pathol. 2015, 185, 1396–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonsgard, J.H.; Getz, G.S. Effect of Reye’s Syndrome Serum on Isolated Chinchilla Liver Mitochondria. J. Clin. Investig. 1985, 76, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-Mediated Autophagy and Fatty Acid Oxidation Prevent Hepatosteatosis and Tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.S.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Hong, Y.A.; Choi, S.R.; Chung, S.; Kim, H.W.; Choi, B.S.; Kim, Y.S.; et al. Resveratrol Increases AdipoR1 and AdipoR2 Expression in Type 2 Diabetic Nephropathy. J. Transl. Med. 2016, 14, 176. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J.A.; Ferdinandusse, S.; Brites, P.; Kemp, S. Peroxisomes, Lipid Metabolism and Lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Pontis, S.; Ribeiro, A.; Sasso, O.; Piomelli, D. Macrophage-Derived Lipid Agonists of PPAR-α as Intrinsic Controllers of Inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Vluggens, A.; Andreoletti, P.; Viswakarma, N.; Jia, Y.; Matsumoto, K.; Kulik, W.; Khan, M.; Huang, J.; Guo, D.; Yu, S.; et al. Reversal of Mouse Acyl-CoA Oxidase 1 (ACOX1) Null Phenotype by Human ACOX1b Isoform [Corrected]. Lab. Investig. 2010, 90, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Gutierrez, G.E.; Gao, X.; Dixon, H.; McDonough, J.A.; Marini, A.M.; Fisher, A.L. The ω-3 Fatty Acid α-Linolenic Acid Extends Caenorhabditis Elegans Lifespan via NHR-49/PPARα and Oxidation to Oxylipins. Aging Cell 2017, 16, 1125–1135. [Google Scholar] [CrossRef] [Green Version]
- Fock, E.; Lavrova, E.; Bachteeva, V.; Nikolaeva, S.; Parnova, R. Suppression of Fatty Acid β-Oxidation and Energy Deficiency as a Cause of Inhibitory Effect of E. Coli Lipopolysaccharide on Osmotic Water Transport in the Frog Urinary Bladder. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2019, 218, 81–87. [Google Scholar] [CrossRef]
- Ling, Y.; Shi, Z.; Yang, X.; Cai, Z.; Wang, L.; Wu, X.; Ye, A.; Jiang, J. Hypolipidemic Effect of Pure Total Flavonoids from Peel of Citrus (PTFC) on Hamsters of Hyperlipidemia and Its Potential Mechanism. Exp. Gerontol. 2020, 130, 110786. [Google Scholar] [CrossRef]
- Li, L.-Y.; Lv, H.-B.; Jiang, Z.-Y.; Qiao, F.; Chen, L.-Q.; Zhang, M.-L.; Du, Z.-Y. Peroxisomal Proliferator-Activated Receptor α-b Deficiency Induces the Reprogramming of Nutrient Metabolism in Zebrafish. J. Physiol. 2020, 598, 4537–4553. [Google Scholar] [CrossRef]
- El Kebbaj, Z.; Andreoletti, P.; Mountassif, D.; Kabine, M.; Schohn, H.; Dauca, M.; Latruffe, N.; El Kebbaj, M.S.; Cherkaoui-Malki, M. Differential Regulation of Peroxisome Proliferator-Activated Receptor (PPAR)-Alpha 1 and Truncated PPAR Alpha 2 as an Adaptive Response to Fasting in the Control of Hepatic Peroxisomal Fatty Acid Beta-Oxidation in the Hibernating Mammal. Endocrinology 2008, 150, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential Expression of Peroxisome Proliferator-Activated Receptors (PPARs): Tissue Distribution of PPAR-Alpha, -Beta, and -Gamma in the Adult Rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR Isotypes in the Adult Mouse and Human Brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef] [PubMed]
- Mariani, M.M.; Malm, T.; Lamb, R.; Jay, T.R.; Neilson, L.; Casali, B.; Medarametla, L.; Landreth, G.E. Neuronally-Directed Effects of RXR Activation in a Mouse Model of Alzheimer’s Disease. Sci. Rep. 2017, 7, 42270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, G.M.; Esposito, E.; Vitiello, S.; Iacono, A.; Santoro, A.; D’Agostino, G.; Sasso, O.; Russo, R.; Piazza, P.V.; Calignano, A.; et al. Palmitoylethanolamide Stimulation Induces Allopregnanolone Synthesis in C6 Cells and Primary Astrocytes: Involvement of Peroxisome-Proliferator Activated Receptor-α. J. Neuroendocrinol. 2011, 23, 591–600. [Google Scholar] [CrossRef]
- Roy, A.; Jana, M.; Corbett, G.T.; Ramaswamy, S.; Kordower, J.H.; Gonzalez, F.J.; Pahan, K. Regulation of Cyclic AMP Response Element Binding and Hippocampal Plasticity-Related Genes by Peroxisome Proliferator-Activated Receptor α. Cell Rep. 2013, 4, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Marx, N.; Duez, H.; Fruchart, J.-C.; Staels, B. Peroxisome Proliferator-Activated Receptors and Atherogenesis: Regulators of Gene Expression in Vascular Cells. Circ. Res. 2004, 94, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Morinishi, T.; Tokuhara, Y.; Ohsaki, H.; Ibuki, E.; Kadota, K.; Hirakawa, E. Activation and Expression of Peroxisome Proliferator-Activated Receptor Alpha Are Associated with Tumorigenesis in Colorectal Carcinoma. PPAR Res. 2019, 2019, 7486727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential Expression and Activation of a Family of Murine Peroxisome Proliferator-Activated Receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costet, P.; Legendre, C.; Moré, J.; Edgar, A.; Galtier, P.; Pineau, T. Peroxisome Proliferator-Activated Receptor Alpha-Isoform Deficiency Leads to Progressive Dyslipidemia with Sexually Dimorphic Obesity and Steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D. Loss of Hepatic PPARα Promotes Inflammation and Serum Hyperlipidemia in Diet-Induced Obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ward, S.C.; Cederbaum, A.I.; Xiong, H.; Lu, Y. Alcoholic Fatty Liver Is Enhanced in CYP2A5 Knockout Mice: The Role of the PPARα-FGF21 Axis. Toxicology 2017, 379, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.-R.; Van Hul, W.; Mertens, I.; et al. PPARα Gene Expression Correlates with Severity and Histological Treatment Response in Patients with Non-Alcoholic Steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central Role of PPARalpha-Dependent Hepatic Lipid Turnover in Dietary Steatohepatitis in Mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef]
- Patsouris, D.; Reddy, J.K.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Mediates the Effects of High-Fat Diet on Hepatic Gene Expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Müller, M. Peroxisome Proliferator-Activated Receptor Alpha Protects against Obesity-Induced Hepatic Inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Rando, G.; Tan, C.K.; Khaled, N.; Montagner, A.; Leuenberger, N.; Bertrand-Michel, J.; Paramalingam, E.; Guillou, H.; Wahli, W. Glucocorticoid Receptor-PPARα Axis in Fetal Mouse Liver Prepares Neonates for Milk Lipid Catabolism. Elife 2016, 5, e11853. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα Is Crucial for Whole-Body Fatty Acid Homeostasis and Is Protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polizzi, A.; Fouché, E.; Ducheix, S.; Lasserre, F.; Marmugi, A.P.; Mselli-Lakhal, L.; Loiseau, N.; Wahli, W.; Guillou, H.; Montagner, A. Hepatic Fasting-Induced PPARα Activity Does Not Depend on Essential Fatty Acids. Int. J. Mol. Sci. 2016, 17, 1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Régnier, M.; Polizzi, A.; Lippi, Y.; Fouché, E.; Michel, G.; Lukowicz, C.; Smati, S.; Marrot, A.; Lasserre, F.; Naylies, C.; et al. Insights into the Role of Hepatocyte PPARα Activity in Response to Fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.N.; Patel, D.P.; Velenosi, T.J.; Kim, D.; Yan, T.; Yue, J.; Li, G.; Krausz, K.W.; Gonzalez, F.J. Extrahepatic PPARα Modulates Fatty Acid Oxidation and Attenuates Fasting-Induced Hepatosteatosis in Mice. J. Lipid Res. 2018, 59, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell 2019, 178, 1102–1114.e17. [Google Scholar] [CrossRef] [PubMed]
- Régnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouché, E.; Lasserre, F.; Naylies, C.; Bétoulières, C.; et al. Hepatocyte-Specific Deletion of Pparα Promotes NAFLD in the Context of Obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar] [CrossRef] [PubMed]
- Batatinha, H.A.P.; Lima, E.A.; Teixeira, A.A.S.; Souza, C.O.; Biondo, L.A.; Silveira, L.S.; Lira, F.S.; Rosa Neto, J.C. Association Between Aerobic Exercise and Rosiglitazone Avoided the NAFLD and Liver Inflammation Exacerbated in PPAR-α Knockout Mice. J. Cell. Physiol. 2017, 232, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.N.; Yue, J.; Kim, D.; Qu, A.; Bonzo, J.A.; Gonzalez, F.J. Hepatocyte-Specific PPARA Expression Exclusively Promotes Agonist-Induced Cell Proliferation without Influence from Nonparenchymal Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G283–G299. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Kaimoto, S.; Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Tateishi, S.; Ono, K.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Activation of PPAR-α in the Early Stage of Heart Failure Maintained Myocardial Function and Energetics in Pressure-Overload Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H305–H313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, R.; Cordaro, M.; Crupi, R.; Siracusa, R.; Campolo, M.; Bruschetta, G.; Fusco, R.; Pugliatti, P.; Esposito, E.; Cuzzocrea, S. Protective Effects of Ultramicronized Palmitoylethanolamide (PEA-Um) in Myocardial Ischaemia and Reperfusion Injury in VIVO. Shock 2016, 46, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Standage, S.W.; Waworuntu, R.L.; Delaney, M.A.; Maskal, S.M.; Bennion, B.G.; Duffield, J.S.; Parks, W.C.; Liles, W.C.; McGuire, J.K. Nonhematopoietic Peroxisome Proliferator-Activated Receptor-α Protects Against Cardiac Injury and Enhances Survival in Experimental Polymicrobial Sepsis. Crit. Care Med. 2016, 44, e594–e603. [Google Scholar] [CrossRef]
- Yammine, A.; Namsi, A.; Vervandier-Fasseur, D.; Mackrill, J.J.; Lizard, G.; Latruffe, N. Polyphenols of the Mediterranean Diet and Their Metabolites in the Prevention of Colorectal Cancer. Molecules 2021, 26, 3483. [Google Scholar] [CrossRef] [PubMed]
- Castrejón-Tellez, V.; Rodríguez-Pérez, J.M.; Pérez-Torres, I.; Pérez-Hernández, N.; Cruz-Lagunas, A.; Guarner-Lans, V.; Vargas-Alarcón, G.; Rubio-Ruiz, M.E. The Effect of Resveratrol and Quercetin Treatment on PPAR Mediated Uncoupling Protein (UCP-) 1, 2, and 3 Expression in Visceral White Adipose Tissue from Metabolic Syndrome Rats. Int. J. Mol. Sci. 2016, 17, 1069. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lin, S.; Zhang, L.; Li, Y. Resveratrol Prevents Renal Lipotoxicity in High-Fat Diet-Treated Mouse Model through Regulating PPAR-α Pathway. Mol. Cell. Biochem. 2016, 411, 143–150. [Google Scholar] [CrossRef]
- Barone, R.; Rizzo, R.; Tabbì, G.; Malaguarnera, M.; Frye, R.E.; Bastin, J. Nuclear Peroxisome Proliferator-Activated Receptors (PPARs) as Therapeutic Targets of Resveratrol for Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantacuzzi, M.; De Filippis, B.; Amoroso, R.; Giampietro, L. PPAR Ligands Containing Stilbene Scaffold. Mini Rev. Med. Chem. 2019, 19, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Bastin, J.; Djouadi, F. Resveratrol and Myopathy. Nutrients 2016, 8, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Yamasaki, M.; Katsube, T.; Shiwaku, K. Effects of Quercetin Derivatives from Mulberry Leaves: Improved Gene Expression Related Hepatic Lipid and Glucose Metabolism in Short-Term High-Fat Fed Mice. Nutr. Res. Pract. 2015, 9, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.L.; Zhang, Z.C.; Hassan, W.; Li, Y.; Liu, J.; Shang, J. Amelioration of Free Fatty Acid-Induced Fatty Liver by Quercetin-3-O-β-D-Glucuronide through Modulation of Peroxisome Proliferator-Activated Receptor-Alpha/Sterol Regulatory Element-Binding Protein-1c Signaling. Hepatol. Res. 2016, 46, 225–238. [Google Scholar] [CrossRef]
- Chen, D.; Daniel, K.G.; Kuhn, D.J.; Kazi, A.; Bhuiyan, M.; Li, L.; Wang, Z.; Wan, S.B.; Lam, W.H.; Chan, T.H.; et al. Green Tea and Tea Polyphenols in Cancer Prevention. Front. Biosci. 2004, 9, 2618–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yang, X.; Luo, J.; Ge, X.; Sun, W.; Zhu, H.; Zhang, W.; Cao, J.; Hou, Y. PPARα Activation Sensitizes Cancer Cells to Epigallocatechin-3-Gallate (EGCG) Treatment via Suppressing Heme Oxygenase-1. Nutr. Cancer 2014, 66, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zuo, X.Z.; Tian, C.; He, D.L.; Yi, W.J.; Chen, Z.; Zhang, P.W.; Ding, S.B.; Ying, C.J. Green Tea Polyphenols Attenuate High-Fat Diet-Induced Renal Oxidative Stress through SIRT3-Dependent Deacetylation. Biomed. Environ. Sci. 2015, 28, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.; Kong, Z.-L.; Tsai, M.-L.; Lo, C.-Y.; Ho, C.-T.; Lai, C.-S. Tetrahydrocurcumin Ameliorates Free Fatty Acid-Induced Hepatic Steatosis and Improves Insulin Resistance in HepG2 Cells. J. Food Drug Anal. 2018, 26, 1075–1085. [Google Scholar] [CrossRef]
- Rimando, A.M.; Khan, S.I.; Mizuno, C.S.; Ren, G.; Mathews, S.T.; Kim, H.; Yokoyama, W. Evaluation of PPARα Activation by Known Blueberry Constituents. J. Sci. Food Agric. 2016, 96, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Vitaglione, P.; Mazzone, G.; Lembo, V.; D’Argenio, G.; Rossi, A.; Guido, M.; Savoia, M.; Salomone, F.; Mennella, I.; De Filippis, F.; et al. Coffee Prevents Fatty Liver Disease Induced by a High-Fat Diet by Modulating Pathways of the Gut-Liver Axis. J. Nutr. Sci. 2019, 8, e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigagli, E.; Toti, S.; Lodovici, M.; Giovannelli, L.; Cinci, L.; D’Ambrosio, M.; Luceri, C. Dietary Extra-Virgin Olive Oil Polyphenols Do Not Attenuate Colon Inflammation in Transgenic HLAB-27 Rats but Exert Hypocholesterolemic Effects through the Modulation of HMGCR and PPAR-α Gene Expression in the Liver. Lifestyle Genom. 2018, 11, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, C.; Lama, A.; Simeoli, R.; Paciello, O.; Pagano, T.B.; Mollica, M.P.; Di Guida, F.; Russo, R.; Magliocca, S.; Canani, R.B.; et al. Hydroxytyrosol Prevents Metabolic Impairment Reducing Hepatic Inflammation and Restoring Duodenal Integrity in a Rat Model of NAFLD. J. Nutr. Biochem. 2016, 30, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.; Illesca, P.; Echeverría, F.; Espinosa, A.; Rincón-Cervera, M.Á.; Ortiz, M.; Hernandez-Rodas, M.C.; Valenzuela, A.; Videla, L.A. Molecular Adaptations Underlying the Beneficial Effects of Hydroxytyrosol in the Pathogenic Alterations Induced by a High-Fat Diet in Mouse Liver: PPAR-α and Nrf2 Activation, and NF-ΚB down-Regulation. Food Funct. 2017, 8, 1526–1537. [Google Scholar] [CrossRef]
- El Kebbaj, R.; Andreoletti, P.; El Hajj, H.I.; El Kharrassi, Y.; Vamecq, J.; Mandard, S.; Saih, F.-E.; Latruffe, N.; El Kebbaj, M.S.; Lizard, G.; et al. Argan Oil Prevents Down-Regulation Induced by Endotoxin on Liver Fatty Acid Oxidation and Gluconeogenesis and on Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1α, (PGC-1α), Peroxisome Proliferator-Activated Receptor α (PPARα) and Estrogen Related Receptor α (ERRα). Biochim. Open 2015, 1, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Q.; Shao, M.; Ma, L.; Guo, D.; Wu, Y.; Gao, P.; Wang, X.; Li, W.; Li, C.; et al. Ginsenoside Rb3 Regulates Energy Metabolism and Apoptosis in Cardiomyocytes via Activating PPARα Pathway. Biomed. Pharmacother. 2019, 120, 109487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Deng, J.; Liu, D.; Tuo, X.; Xiao, L.; Lai, B.; Yao, Q.; Liu, J.; Yang, H.; Wang, N. Nuciferine Ameliorates Hepatic Steatosis in High-Fat Diet/Streptozocin-Induced Diabetic Mice through a PPARα/PPARγ Coactivator-1α Pathway. Br. J. Pharmacol. 2018, 175, 4218–4228. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, C.; Yang, J.; Zhang, T.; Zhou, Q. Berberine Is a Potent Agonist of Peroxisome Proliferator Activated Receptor Alpha. Front. Biosci. 2016, 21, 1052–1060. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Species | PPRE a | PPRE Sequence b | Reference |
|---|---|---|---|---|---|
| Acaaa1b | acetyl-coenzyme A acyltransferase 1B | rat | PPRE2 | AGGTCAAAAGTCA | [40] |
| ACOX1 | acyl-CoA oxidase 1 | human | PPRE | AGGTCAGCTGTCA | [41] |
| Acox1 | acyl-CoA oxidase 1 | rat | PPRE | TGACCTTTGTCCT | [36] |
| Aldh3a2 | aldehyde dehydrogenase family 3, subfamily A2 | mouse | PPRE | nd | [42] |
| Cat | catalase | rat | PPRE | AGGTGAAAGTTGA | [43] |
| Ehhadh | enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase | rat | PPRE | TGACCTATTGAAC | [44] |
| Mlycd | malonyl-CoA decarboxylase | rat | PPRE-2 | AGGCAAGAGGCTG | [45] |
| Mlycd | malonyl-CoA decarboxylase | rat | PPRE-3 | GAACCTTTGGCTG | [45] |
| Pex11a | peroxisomal biogenesis factor 11 alpha | mouse | PPRE | TCACCTTTCACCC | [46] |
| Scp2 | sterol carrier protein 2 | rat | PPRE-A | TCCTGTAACTCCG | [47] |
| Scp2 | sterol carrier protein 2 | rat | PPRE-B | GTGGATTACAGGA | [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahri-Joutey, M.; Andreoletti, P.; Surapureddi, S.; Nasser, B.; Cherkaoui-Malki, M.; Latruffe, N. Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARα. Int. J. Mol. Sci. 2021, 22, 8969. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168969
Tahri-Joutey M, Andreoletti P, Surapureddi S, Nasser B, Cherkaoui-Malki M, Latruffe N. Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARα. International Journal of Molecular Sciences. 2021; 22(16):8969. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168969
Chicago/Turabian StyleTahri-Joutey, Mounia, Pierre Andreoletti, Sailesh Surapureddi, Boubker Nasser, Mustapha Cherkaoui-Malki, and Norbert Latruffe. 2021. "Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARα" International Journal of Molecular Sciences 22, no. 16: 8969. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168969