
Unraveling the Relevance of ARL GTPases in Cutaneous Melanoma Prognosis through Integrated Bioinformatics Analysis


Abstract
:1. Introduction
2. Results
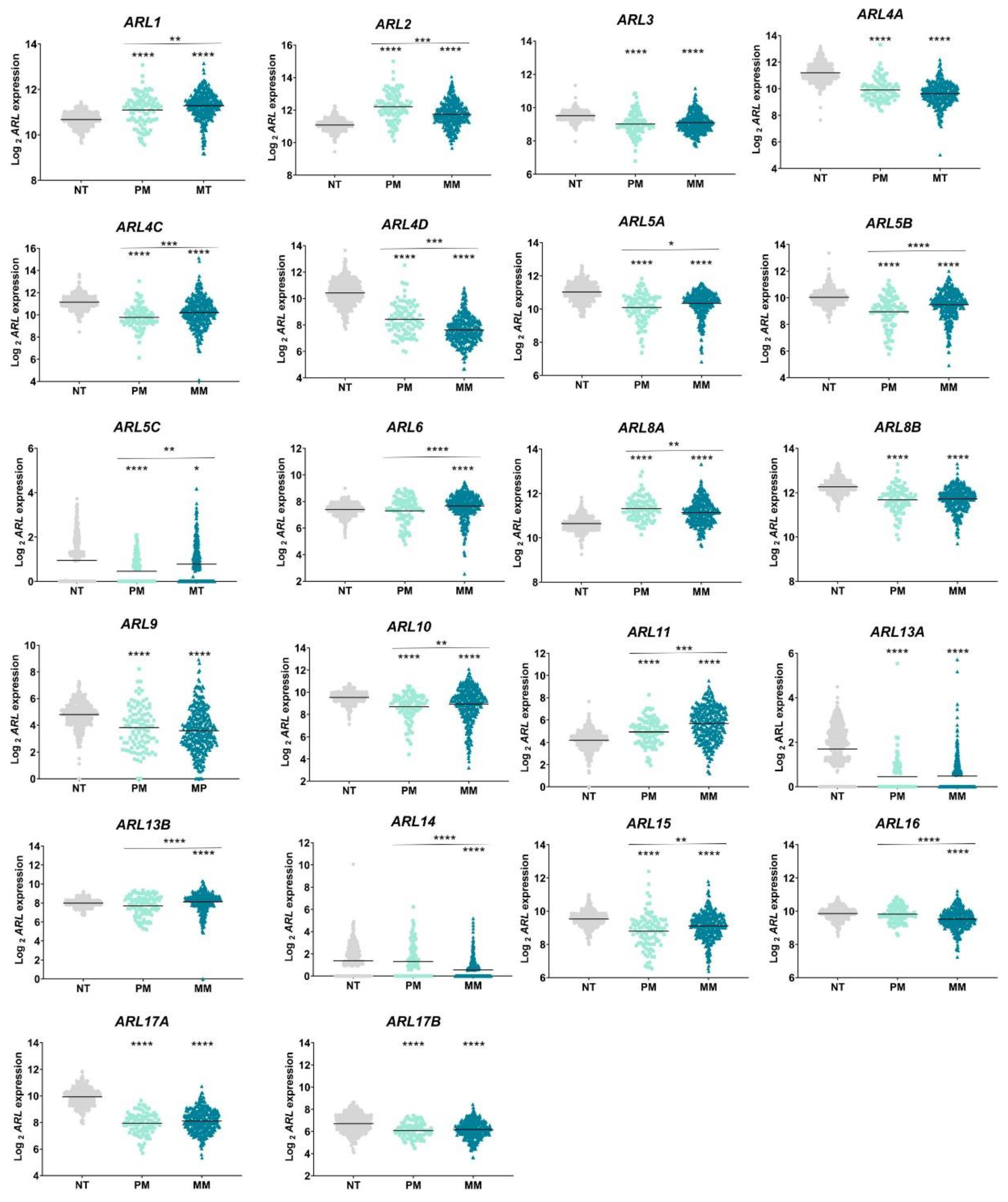
2.1. ARL Genes Are Differentially Expressed in Primary and Metastatic CM
2.2. Promoter Methylation Levels May Influence the Expression of Some ARL Genes
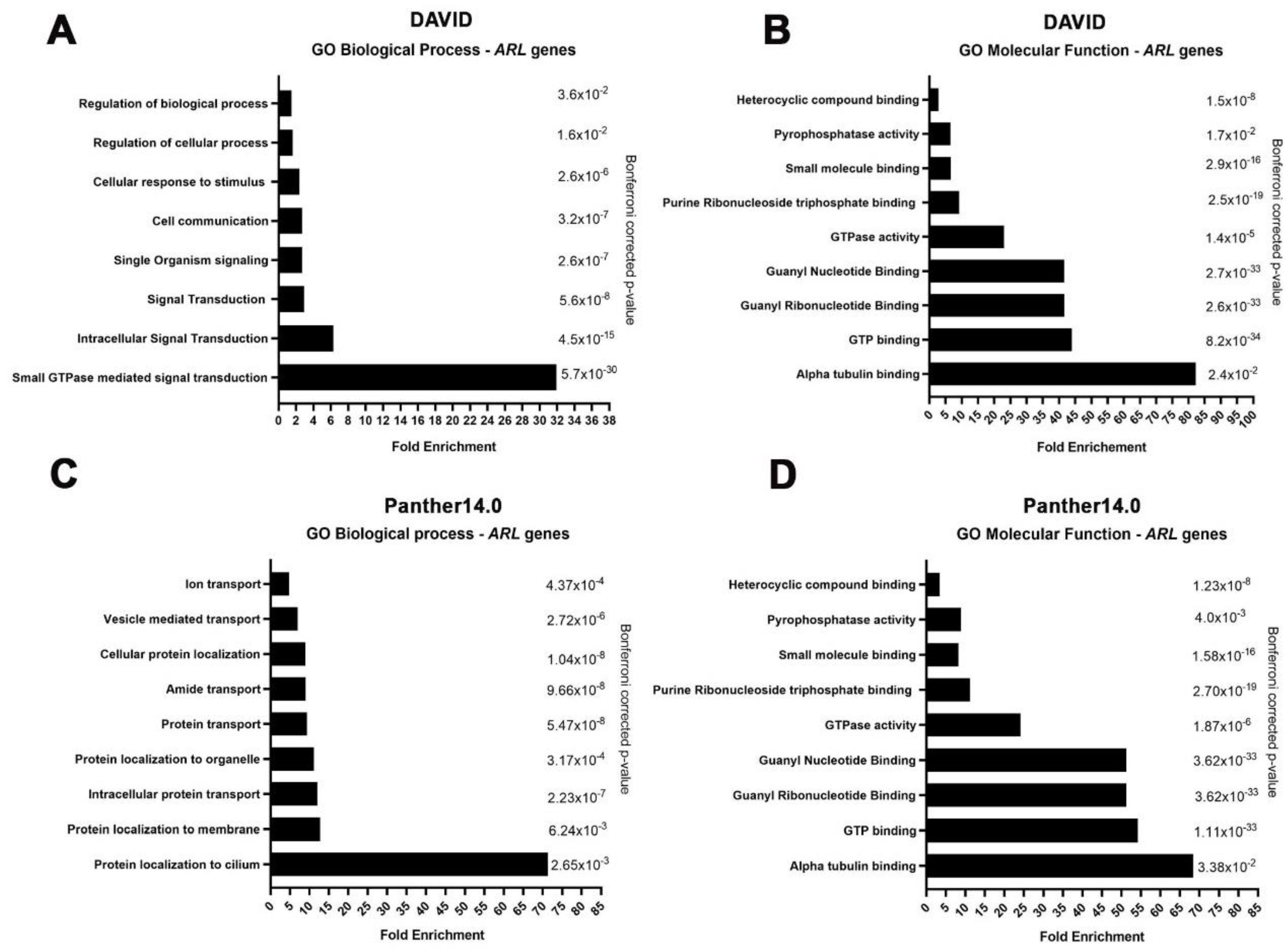
2.3. ARL GTPases Are Implicated in Cell Communication, Vesicle Transport, and Protein Recruitment in CM
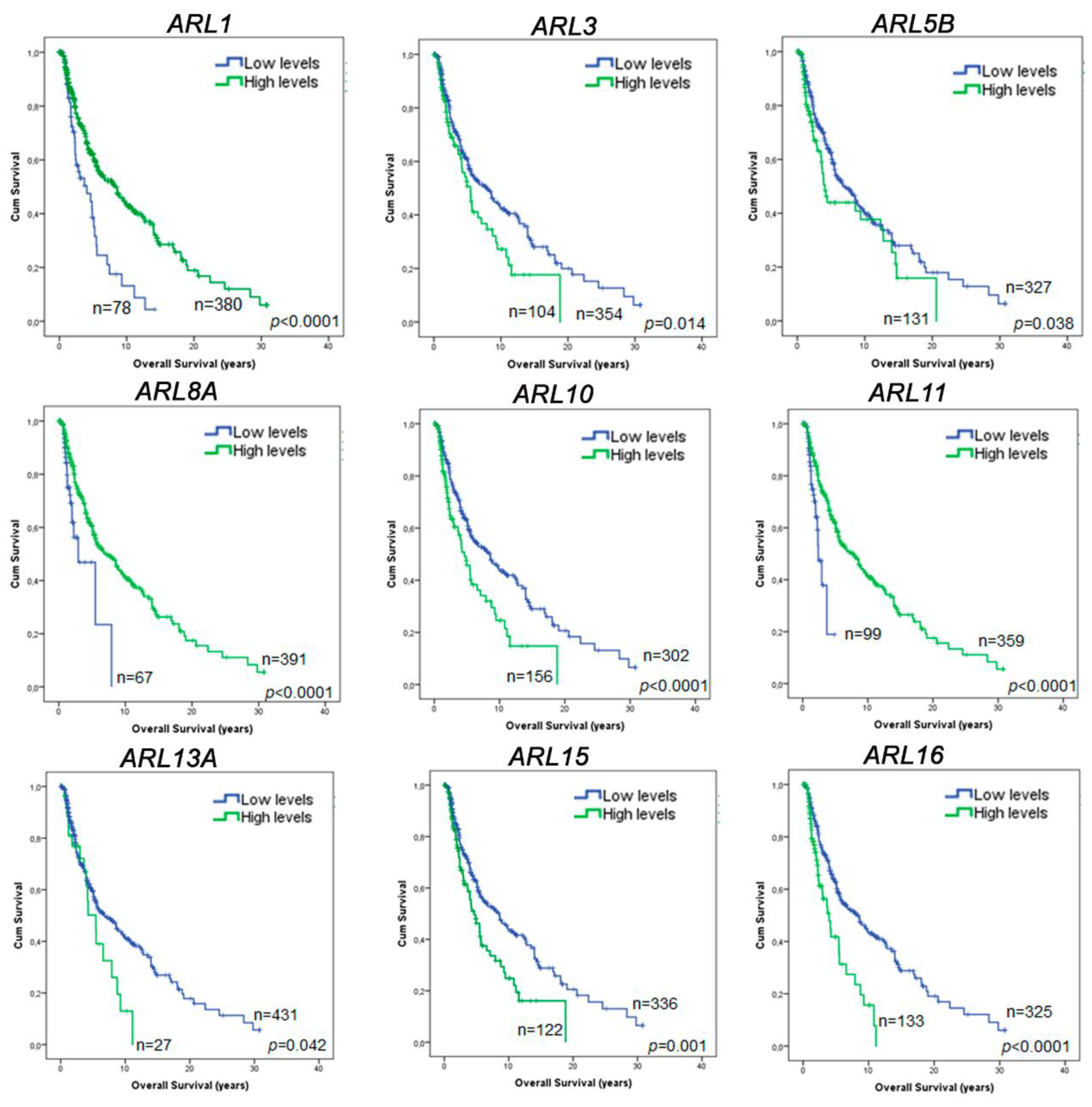
2.4. ARL1, ARL11, and ARL15 Expression Represent Prognostic Factors in CM
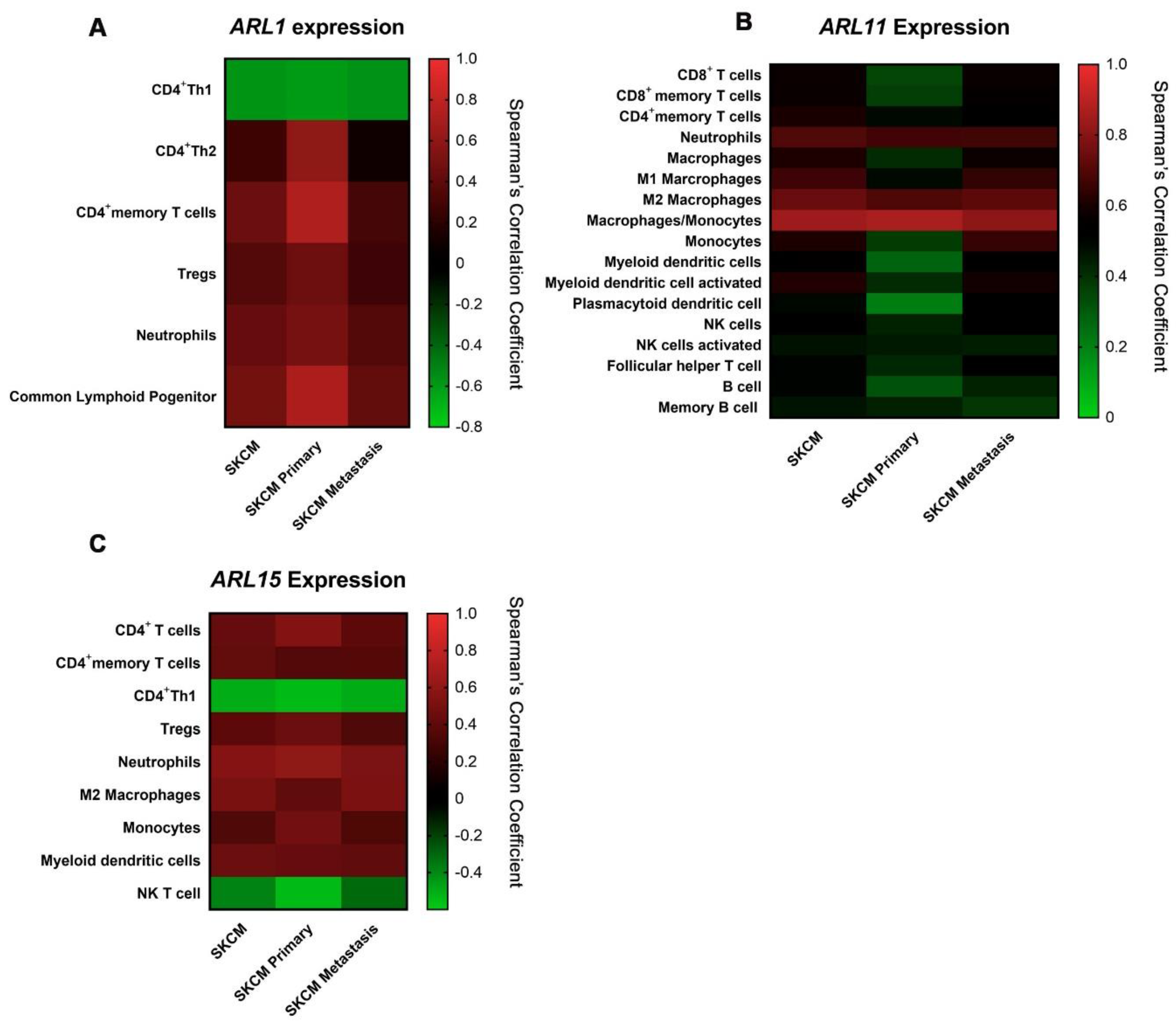
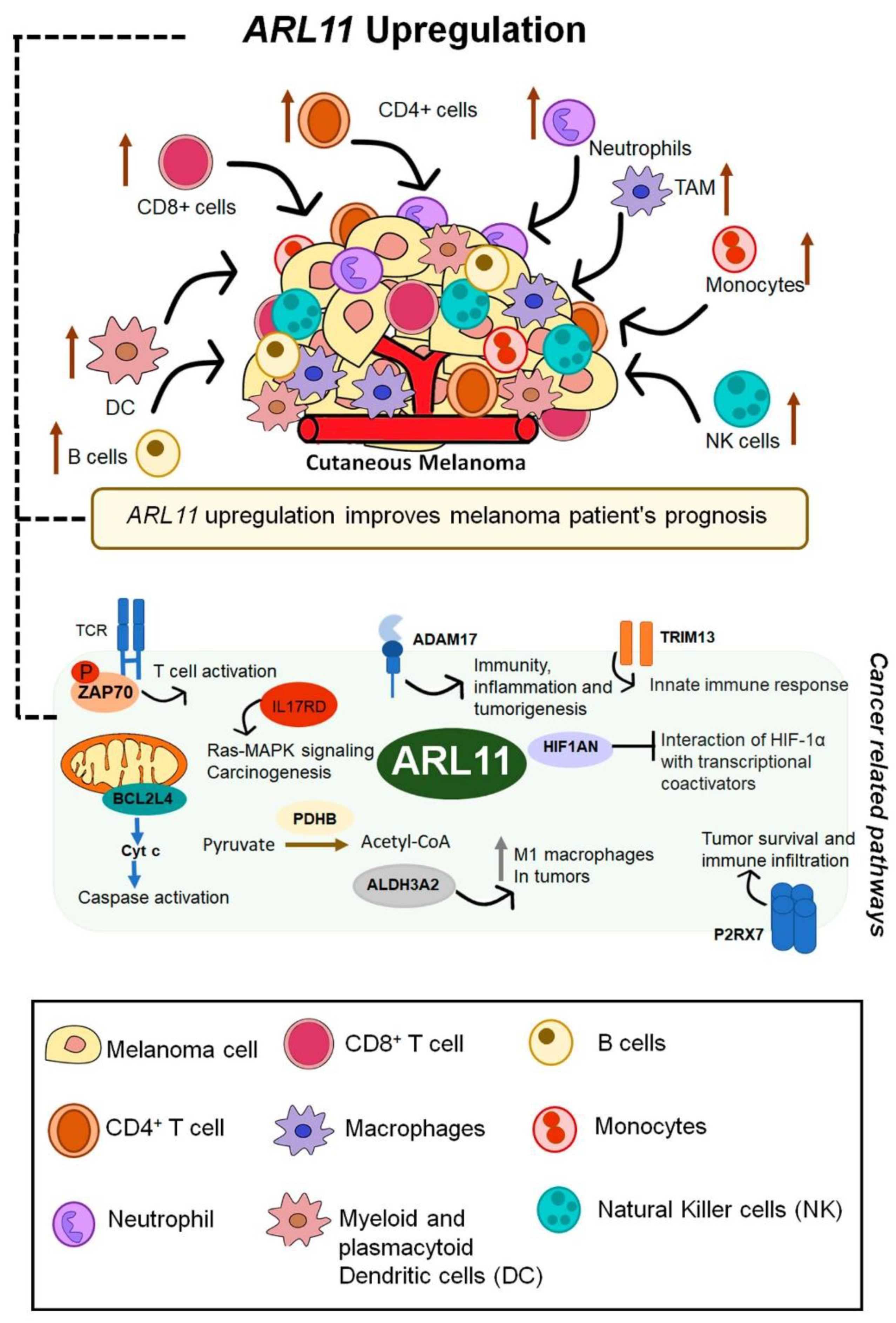
2.5. ARL11 May Have a Predominant Impact on Immune Microenvironment Remodeling
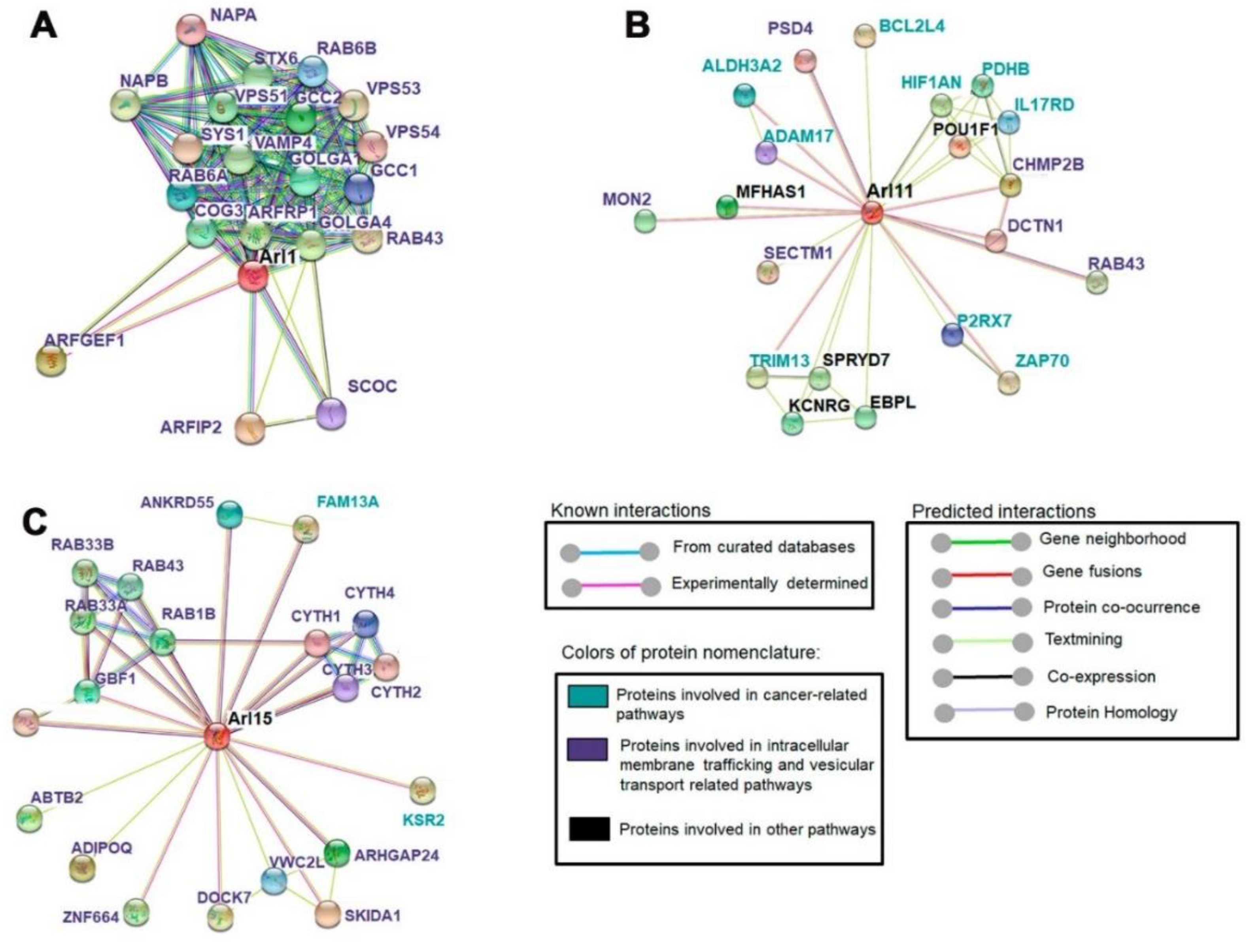
2.6. ARL11 Is Closely Interconnected with Proteins Involved in Immune Cell Activation and Recruitment
3. Discussion
4. Conclusions
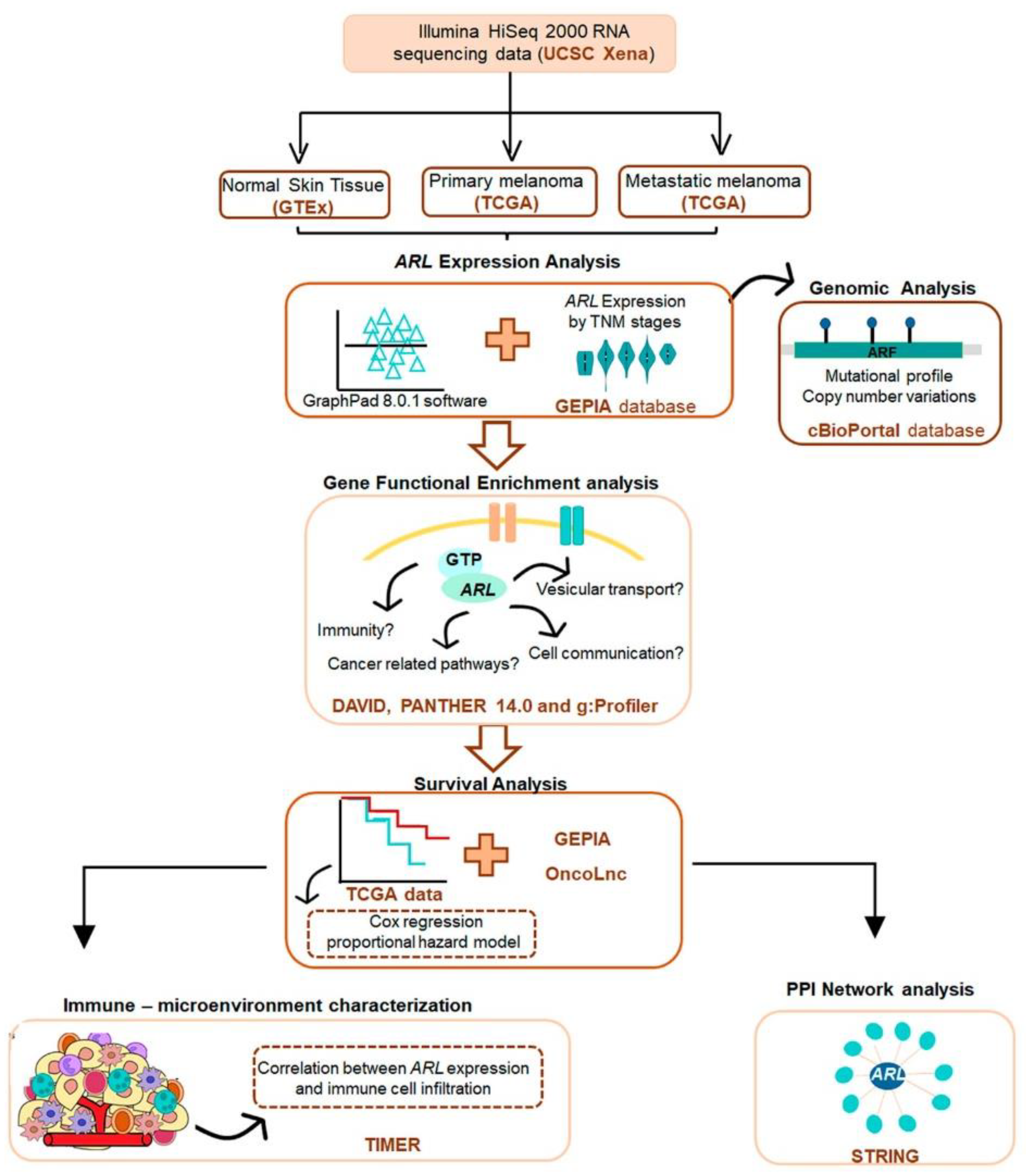
5. Materials and Methods
5.1. Study Design and Data Acquisition
5.2. Gene Expression and Promoter Methylation Analysis
5.3. Genomic Analysis
5.4. Gene Ontology Enrichment Analysis
5.5. Survival Analysis
5.6. Tumor Microenvironment Characterization
5.7. Protein–Protein Interaction (PPI) Network Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM17 | Disintegrin and metalloproteinase domain-containing protein 17 |
| ARF | Adenosine diphosphate–ribosylation factor |
| ARL | ADP-ribosylation factor-like |
| BCL2L4 | Bcl-2-like protein 14 |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 |
| CM | Cutaneous melanoma |
| DAVID | Database for Annotation, Visualization and Integrated Discovery |
| GO | Gene ontology |
| GTEx | Genotype tissue expression |
| HIF1AN | Hypoxia-inducible factor 1-alpha inhibitor |
| IL17RD | Interleukin-17 receptor D |
| NF1 | Neurofibromatosis type 1 |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| P2RX7 | P2X purinoceptor 7 |
| NK | Natural killer cells |
| PPI | Protein–protein interaction |
| STRING | Search Tool for the Retrieval of Interacting Genes |
| SKCM | Skin cutaneous melanoma |
| TCGA | The cancer genome atlas |
| TIMER | Tumor Immune Estimation Resource |
| TNM | Tumor, nodes, metastases |
| TRIM13 | E3 ubiquitin–protein ligase |
| UCSC | University of California, Santa Cruz |
| GEPIA | Gene Expression Profiling Interactive Analysis |
References
- Potrony, M.; Badenas, C.; Aguilera, P.; Puig-Butille, J.A.; Carrera, C.; Malvehy, J.; Puig, S. Update in Genetic Susceptibility in Melanoma. J. Clin. Oncol. 2015, 3, 210. [Google Scholar] [CrossRef]
- Bandarchi, B.; Ma, L.; Navab, R.; Seth, A.; Rasty, G. From Melanocyte to Metastatic Malignant Melanoma. Dermatol. Res. Pract. 2010, 2010, 583748. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Carlson, J.A.; Granese, J.; Ross, J.; Mihm, M.; Slominski, A. Current Concepts of Metastasis in Melanoma. Expert Rev. Dermatol. 2008, 3, 569–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimotty, P.A.; Guerry, D.P.; Ming, M.E.; Elenitsas, R.; Xu, X.; Czerniecki, B.; Spitz, F.; Schuchter, L.; Elder, D. Thin Primary Cutaneous Malignant Melanoma: A Prognostic Tree for 10-Year Metastasis Is More Accurate than American Joint Committee on Cancer Staging. J. Clin. Oncol. 2004, 22, 3668–3676. [Google Scholar] [CrossRef] [PubMed]
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report tto the Nation on the Status of Cancer, Part I: National Cancer Statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damsky, W.E.; Theodosakis, N.; Bosenberg, M. Melanoma Metastasis: New Concepts and Evolving Paradigms. Oncogene 2014, 33, 2413–2422. [Google Scholar] [CrossRef] [Green Version]
- Allemani, C.; Matsuda, T.; di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global Surveillance of Trends in Cancer Survival 2000–14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed with One of 18 Cancers from 322 Population-Based Registries in 71 Countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Domingues, B.; Lopes, J.; Soares, P.; Populo, H. Melanoma Treatment in Review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Yu, C.; Liu, X.; Yang, J.; Zhang, M.; Jin, H.; Ma, X.; Shi, H. Combination of Immunotherapy with Targeted Therapy: Theory and Practice in Metastatic Melanoma. Front. Immunol. 2019, 10, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eroglu, Z.; Ribas, A. Combination Therapy with BRAF and MEK Inhibitors for Melanoma: Latest Evidence and Place in Therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.J.L.; Ribas, A. Targeted Therapy for Melanoma. Cancer Treat Res. 2016, 167, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Luís, R.; Brito, C.; Pojo, M. Melanoma Metabolism: Cell Survival and Resistance to Therapy. In Tumor Microenvironment. Advances in Experimental Medicine and Biology; Serpa, J., Ed.; Springer: Cham, Switzerland, 2020; Volume 1219, pp. 203–223. [Google Scholar] [CrossRef]
- Brito, C.; Tomás, A.; Silva, S.; Bronze, M.R.; Serra, A.T.; Pojo, M. The Impact of Olive Oil Compounds on the Metabolic Reprogramming of Cutaneous Melanoma Cell Models. Molecules 2021, 26, 289. [Google Scholar] [CrossRef]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The Ras Protein Superfamily: Evolutionary Tree and Role of Conserved Amino Acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [Green Version]
- D’Souza-Schorey, C.; Chavrier, P. ARF Proteins: Roles in Membrane Traffic and Beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- Sztul, E.; Chen, P.W.; Casanova, J.E.; Cherfils, J.; Dacks, J.B.; Lambright, D.G.; Lee, F.J.S.; Randazzo, P.A.; Santy, L.C.; Schürmann, A.; et al. Arf GTPases and Their GEFs and GAPS: Concepts and Challenges. Mol. Biol. Cell 2019, 30, 1249–1271. [Google Scholar] [CrossRef]
- Huang, D.; Pei, Y.; Dai, C.; Huang, Y.; Chen, H.; Chen, X.; Zhang, X.; Lin, C.; Wang, H.; Zhang, R.; et al. Up-Regulated ADP-Ribosylation Factor 3 Promotes Breast Cancer Cell Proliferation through the Participation of FOXO1. Exp. Cell Res. 2019, 384, 111624. [Google Scholar] [CrossRef] [PubMed]
- Howley, B.V.; Link, L.A.; Grelet, S.; El-Sabban, M.; Howe, P.H. A CREB3-Regulated ER-Golgi Trafficking Signature Promotes Metastatic Progression in Breast Cancer. Oncogene 2018, 37, 1308–1325. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, C.; Aguilar, M.; Burgos, P.V.; Ehrenfeld, P.; Mardones, G.A. Functional Disruption of the Golgi Apparatus Protein ARF1 Sensitizes MDA-MB-231 Breast Cancer Cells to the Antitumor Drugs Actinomycin D and Vinblastine through ERK and AKT Signaling. PLoS ONE 2018, 13, e0195401. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, S.; Ramirez, R.A.M.; Claing, A. ARF1 Regulates Adhesion of MDA-MB-231 Invasive Breast Cancer Cells through Formation of Focal Adhesions. Cell Signal. 2015, 27, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.H.; Brady, S.W.; Acosta-Alvarez, L.; Rogers, A.; Peng, J.; Sorensen, L.K.; Wolff, R.K.; Mleynek, T.; Shin, D.; Rich, C.P.; et al. The Small GTPase ARf6 Activates PI3K in Melanoma to Induce a Prometastatic State. Cancer Res. 2019, 79, 2892–2908. [Google Scholar] [CrossRef] [PubMed]
- Tague, S.E.; Muralidharan, V.; D’Souza-Schorey, C. ADP-Ribosylation Factor 6 Regulates Tumor Cell Invasion through the Activation of the MEK/ERK Signaling Pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 9671–9676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, C.; Barral, D.C.; Pojo, M. Subversion of Ras Small GTPases in Cutaneous Melanoma Aggressiveness. Front. Cell Dev. Biol. 2020, 8, 575223. [Google Scholar] [CrossRef]
- Yu, C.J.; Lee, F.J.S. Multiple Activities of Arl1 GTPase in the Trans-Golgi Network. J. Cell Sci. 2017, 130, 1691–1699. [Google Scholar] [CrossRef] [Green Version]
- Larkins, C.E.; Gonzalez Aviles, G.D.; East, M.P.; Kahn, R.A.; Caspary, T. Arl13b Regulates Ciliogenesis and the Dynamic Localization of Shh Signaling Proteins. Mol. Biol. Cell 2011, 22, 4694–4703. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.B.; Kumar, G.; Kaur, H.; Kaur, A.; Tuli, A. ARL11 Regulates Lipopolysaccharide-Stimulated Macrophage Activation by Promoting Mitogen-Activated Protein Kinase (MAPK) Signaling. J. Biol. Chem. 2018, 293, 9892–9909. [Google Scholar] [CrossRef] [Green Version]
- Rocha, N.; Payne, F.; Huang-Doran, I.; Sleigh, A.; Fawcett, K.; Adams, C.; Stears, A.; Saudek, V.; O’Rahilly, S.; Barroso, I.; et al. The Metabolic Syndrome- Associated Small G Protein ARL15 Plays a Role in Adipocyte Differentiation and Adiponectin Secretion. Sci. Rep. 2017, 7, 17593. [Google Scholar] [CrossRef]
- Peng, R.; Men, J.; Ma, R.; Wang, Q.; Wang, Y.; Sun, Y.; Ren, J. MiR-214 down-Regulates ARL2 and Suppresses Growth and Invasion of Cervical Cancer Cells. Biochem. Biophys. Res. Commun. 2017, 484, 623–630. [Google Scholar] [CrossRef]
- Li, H.-J.; Sun, X.-M.; Li, Z.-K.; Yin, Q.-W.; Pang, H.; Pan, J.-J.; Li, X.; Chen, W. LncRNA UCA1 Promotes Mitochondrial Function of Bladder Cancer via the MiR-195/ARL2 Signaling Pathway. Cell Physiol. Biochem. 2017, 43, 2548–2561. [Google Scholar] [CrossRef]
- Casalou, C.; Ferreira, A.; Barral, D.C. The Role of ARF Family Proteins and Their Regulators and Effectors in Cancer Progression: A Therapeutic Perspective. Front. Cell Dev. Biol. 2020, 8, 217. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Su, Z.; Wang, S.; Xu, H. Clinical and Prognostic Significance of Arl4c Expression in Colorectal Cancer. Cancer Biomark. 2016, 16, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Masuda, T.; Sato, K.; Tobo, T.; Nambara, S.; Kidogami, S.; Hayashi, N.; Kuroda, Y.; Ito, S.; Eguchi, H.; et al. Identification of ARL4C as a Peritoneal Dissemination-Associated Gene and Its Clinical Significance in Gastric Cancer. Ann. Surg. Oncol. 2018, 25, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Dykes, S.S.; Gray, A.L.; Coleman, D.T.; Saxena, M.; Stephens, C.A.; Carroll, J.L.; Pruitt, K.; Cardelli, J.A. The Arf-like GTPase Arl8b Is Essential for Three-Dimensional Invasive Growth of Prostate Cancer in Vitro and Xenograft Formation and Growth in Vivo. Oncotarget 2016, 7, 31037–31052. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.-H.; Onodera, Y.; Giaccia, A.J.; Le, Q.-T.; Shimizu, S.; Shirato, H.; Nam, J.-M. Lysosomal Trafficking Mediated by Arl8b and BORC Promotes Invasion of Cancer Cells That Survive Radiation. Commun. Biol. 2020, 3, 620. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, Z.; Guo, W.; Ni, S.; Xiao, X.; Wang, L.; Huang, D.; Tan, C.; Xu, Q.; Zha, R.; et al. MicroRNA-202-3p Inhibits Cell Proliferation by Targeting Adp-Ribosylation Factor-like 5a in Human Colorectal Carcinoma. Clin. Cancer Res. 2014, 20, 1146–1157. [Google Scholar] [CrossRef] [Green Version]
- Bay, S.N.; Long, A.B.; Caspary, T. Disruption of the Ciliary GTPase Arl13b Suppresses Sonic Hedgehog Overactivation and Inhibits Medulloblastoma Formation. Proc. Natl. Acad. Sci. USA 2018, 115, 1570–1575. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Xu, L.; Chen, L.; Lu, Q.; Xie, X.; Shi, W.; Xiong, H.; Shi, C.; Huang, X.; Mei, J.; et al. Arl13b Promotes Gastric Tumorigenesis by Regulating Smo Trafficking and Activation of the Hedgehog Signaling Pathway. Cancer Res. 2017, 77, 4000–4013. [Google Scholar] [CrossRef] [Green Version]
- Casalou, C.; Faustino, A.; Silva, F.; Ferreira, I.C.; Vaqueirinho, D.; Ferreira, A.; Castanheira, P.; Barona, T.; Ramalho, J.S.; Serpa, J.; et al. Arl13b Regulates Breast Cancer Cell Migration and Invasion by Controlling Integrin-Mediated Signaling. Cancers 2019, 11, 1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, B.; Meyer, P.; Boettger, M.B.; Hemminki, K.; Stapelmann, H.; Gast, A.; Schmitt, C.; Kumar, R.; Sergi, C.; Burwinkel, B. ARLTS1 Variants and Melanoma Risk. Int. J. Cancer 2006, 119, 1736–1737. [Google Scholar] [CrossRef]
- Calin, G.A.; Trapasso, F.; Shimizu, M.; Dumitru, C.D.; Yendamuri, S.; Godwin, A.K.; Ferracin, M.; Bernardi, G.; Chatterjee, D.; Baldassarre, G.; et al. Familial Cancer Associated with a Polymorphism in ARLTS1. N. Engl. J. Med. 2005, 352, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Hass, H.G.; Vogel, U.; Scheurlen, M.; Jobst, J. Gene-Expression Analysis Identifies Specific Patterns of Dysregulated Molecular Pathways and Genetic Subgroups of Human Hepatocellular Carcinoma. Anticancer Res. 2016, 36, 5087–5095. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, W.; Liu, X.; Guan, G.; Zhuang, M. ARL3 Is Downregulated and Acts as a Prognostic Biomarker in Glioma. J. Transl. Med. 2019, 17, 210. [Google Scholar] [CrossRef] [Green Version]
- Yendamuri, S.; Trapasso, F.; Calin, G.A. ARLTS1–a Novel Tumor Suppressor Gene. Cancer Lett. 2008, 264, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Young, R.A. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, Q.; Shyr, Y. Dysregulated Transcription across Diverse Cancer Types Reveals the Importance of RNA-Binding Protein in Carcinogenesis. BMC Genom. 2015, 16, S5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Guan, G.; Cheng, W.; Jiang, Y.; Shan, F.; Wu, A.; Cheng, P.; Guo, Z. ARL2 Overexpression Inhibits Glioma Proliferation and Tumorigenicity via Down-Regulating AXL. BMC Cancer 2018, 18, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.J.; Allen, J.L.; Caswell, P.T. Vesicle Trafficking Pathways That Direct Cell Migration in 3D Matrices and in Vivo. Traffic 2018, 19, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Tagliatela, A.C.; Hempstead, S.C.; Hibshman, P.S.; Hockenberry, M.A.; Brighton, H.E.; Pecot, C.V.; Bear, J.E. Coronin 1C Inhibits Melanoma Metastasis through Regulation of MT1-MMP-Containing Extracellular Vesicle Secretion. Sci. Rep. 2020, 10, 11958. [Google Scholar] [CrossRef]
- Sinnamon, A.J.; Sharon, C.E.; Song, Y.; Neuwirth, M.G.; Elder, D.E.; Xu, X.; Chu, E.Y.; Ming, M.E.; Fraker, D.L.; Gimotty, P.A.; et al. The Prognostic Significance of Tumor-Infiltrating Lymphocytes for Primary Melanoma Varies by Sex. J. Am. Acad. Dermatol. 2018, 79, 245–251. [Google Scholar] [CrossRef]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune System and Melanoma Biology: A Balance between Immunosurveillance and Immune Escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schedel, F.; Mayer-Hain, S.; Pappelbaum, K.I.; Metze, D.; Stock, M.; Goerge, T.; Loser, K.; Sunderkötter, C.; Luger, T.A.; Weishaupt, C. Evidence and Impact of Neutrophil Extracellular Traps in Malignant Melanoma. Pigment Cell Melanoma Res. 2020, 33, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattes, J.; Hulett, M.; Xie, W.; Hogan, S.; Rothenberg, M.E.; Foster, P.; Parish, C. Immunotherapy of Cytotoxic T Cell-Resistant Tumors by T Helper 2 Cells: An Eotaxin and STAT6-Dependent Process. J. Exp. Med. 2003, 197, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Kamran, N.; Li, Y.; Sierra, M.; Alghamri, M.S.; Kadiyala, P.; Appelman, H.D.; Edwards, M.; Lowenstein, P.R.; Castro, M.G. Melanoma Induced Immunosuppression Is Mediated by Hematopoietic Dysregulation. Oncoimmunology 2018, 7, e1408750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairfax, B.P.; Taylor, C.A.; Watson, R.A.; Nassiri, I.; Danielli, S.; Fang, H.; Mahé, E.A.; Cooper, R.; Woodcock, V.; Traill, Z.; et al. Peripheral CD8+ T Cell Characteristics Associated with Durable Responses to Immune Checkpoint Blockade in Patients with Metastatic Melanoma. Nat. Med. 2020, 26, 193–199. [Google Scholar] [CrossRef]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the Crosstalk between Melanoma and Immune Cells in the Tumor Microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy—new Insights into Old Paradigms. Cancer Gene. Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Chiaruttini, G.; Mele, S.; Opzoomer, J.; Crescioli, S.; Ilieva, K.M.; Lacy, K.E.; Karagiannis, S.N. B Cells and the Humoral Response in Melanoma: The Overlooked Players of the Tumor Microenvironment. Oncoimmunology 2017, 6, e1294296. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.S.; Hsu, L.Y.; Freedman, T.S.; Weiss, A. ZAP-70: An Essential Kinase in T-Cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F. P2RX7: A Receptor with a Split Personality in Inflammation and Cancer. Mol. Cell Oncol. 2015, 3, e1010937. [Google Scholar] [CrossRef] [Green Version]
- Girondel, C.; Meloche, S. Interleukin-17 Receptor D in Physiology, Inflammation and Cancer. Front. Oncol. 2021, 11, 656004. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A Novel Biological Module-Centric Algorithm to Functionally Analyze Large Gene Lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, C.; Fragoso, S.; Luís, R.; Pinto, F.; Brito, C.; Esteves, S.; Pataco, M.; Santos, S.; Machado, P.; Vicente, J.B.; et al. High-Throughput Sequencing Identifies 3 Novel Susceptibility Genes for Hereditary Melanoma. Genes 2020, 11, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. G:Profiler—A Web-Based Toolset for Functional Profiling of Gene Lists from Large-Scale Experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J. OncoLnc: Linking TCGA Survival Data to MRNAs, MiRNAs, and LncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein–protein Interaction Networks, Integrated over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | p-Value | Exp(B) | 95.0% CI for Exp(B) | |
|---|---|---|---|---|
| Lower | Upper | |||
| ARL1 | 0.022 | 1.642 | 1.074 | 2.510 |
| ARL2 | 0.436 | 1.805 | 0.408 | 7.980 |
| ARL3 | 0.094 | 2.873 | 0.835 | 9.882 |
| ARL4A | 0.120 | 3.565 | 0.718 | 17.705 |
| ARL4C | 0.211 | 0.675 | 0.364 | 1.250 |
| ARL4D | 0.952 | 0.956 | 0.219 | 4.163 |
| ARL5A | 0.561 | 0.814 | 0.407 | 1.628 |
| ARL5B | 0.513 | 1.645 | 0.371 | 7.306 |
| ARL5C | 0.720 | 0.850 | 0.351 | 2.063 |
| ARL6 | 0.149 | 3.268 | 0.655 | 16.312 |
| ARL8A | 0.912 | 0.955 | 0.424 | 2.151 |
| ARL8B | 0.136 | 2.144 | 0.787 | 5.837 |
| ARL9 | 0.194 | 4.025 | 0.492 | 32.914 |
| ARL10 | 0.413 | 0.632 | 0.211 | 1.895 |
| ARL11 | 0.953 | 1.080 | 0.083 | 14.113 |
| ARL13A | 0.343 | 0.561 | 0.169 | 1.855 |
| ARL13B | 0.537 | 0.801 | 0.395 | 1.621 |
| ARL14 | 0.818 | 0.823 | 0.157 | 4.312 |
| ARL15 | 0.009 | 0.288 | 0.114 | 0.730 |
| ARL16 | 0.719 | 1.243 | 0.380 | 4.074 |
| ARL17A | 0.873 | 0.801 | 0.053 | 12.167 |
| ARL17B | 0.455 | 0.395 | 0.035 | 4.514 |
| Age | 0.000 | 1.023 | 1.013 | 1.034 |
| Gender | 0.691 | 1.070 | 0.767 | 1.491 |
| Tumor Grade | 0.000 | 0.200 | 0.096 | 0.417 |
| Type of Tumor | 0.840 | 1.239 | 0.154 | 9.941 |
| BRAF mutations | 0.792 | 1.050 | 0.732 | 1.504 |
| NRAS mutations | 0.402 | 0.851 | 0.583 | 1.242 |
| NF1 mutations | 0.488 | 1.194 | 0.723 | 1.972 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brito, C.; Costa-Silva, B.; Barral, D.C.; Pojo, M. Unraveling the Relevance of ARL GTPases in Cutaneous Melanoma Prognosis through Integrated Bioinformatics Analysis. Int. J. Mol. Sci. 2021, 22, 9260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179260
Brito C, Costa-Silva B, Barral DC, Pojo M. Unraveling the Relevance of ARL GTPases in Cutaneous Melanoma Prognosis through Integrated Bioinformatics Analysis. International Journal of Molecular Sciences. 2021; 22(17):9260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179260
Chicago/Turabian StyleBrito, Cheila, Bruno Costa-Silva, Duarte C. Barral, and Marta Pojo. 2021. "Unraveling the Relevance of ARL GTPases in Cutaneous Melanoma Prognosis through Integrated Bioinformatics Analysis" International Journal of Molecular Sciences 22, no. 17: 9260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179260