
Atypical Hemolytic Uremic Syndrome (aHUS) and Adenosine Deaminase (ADA)-Deficient Severe Combined Immunodeficiency (SCID)—Two Diseases That Exacerbate Each Other: Case Report

 , , , , and
, , , , and
Abstract
:1. Introduction
Aim
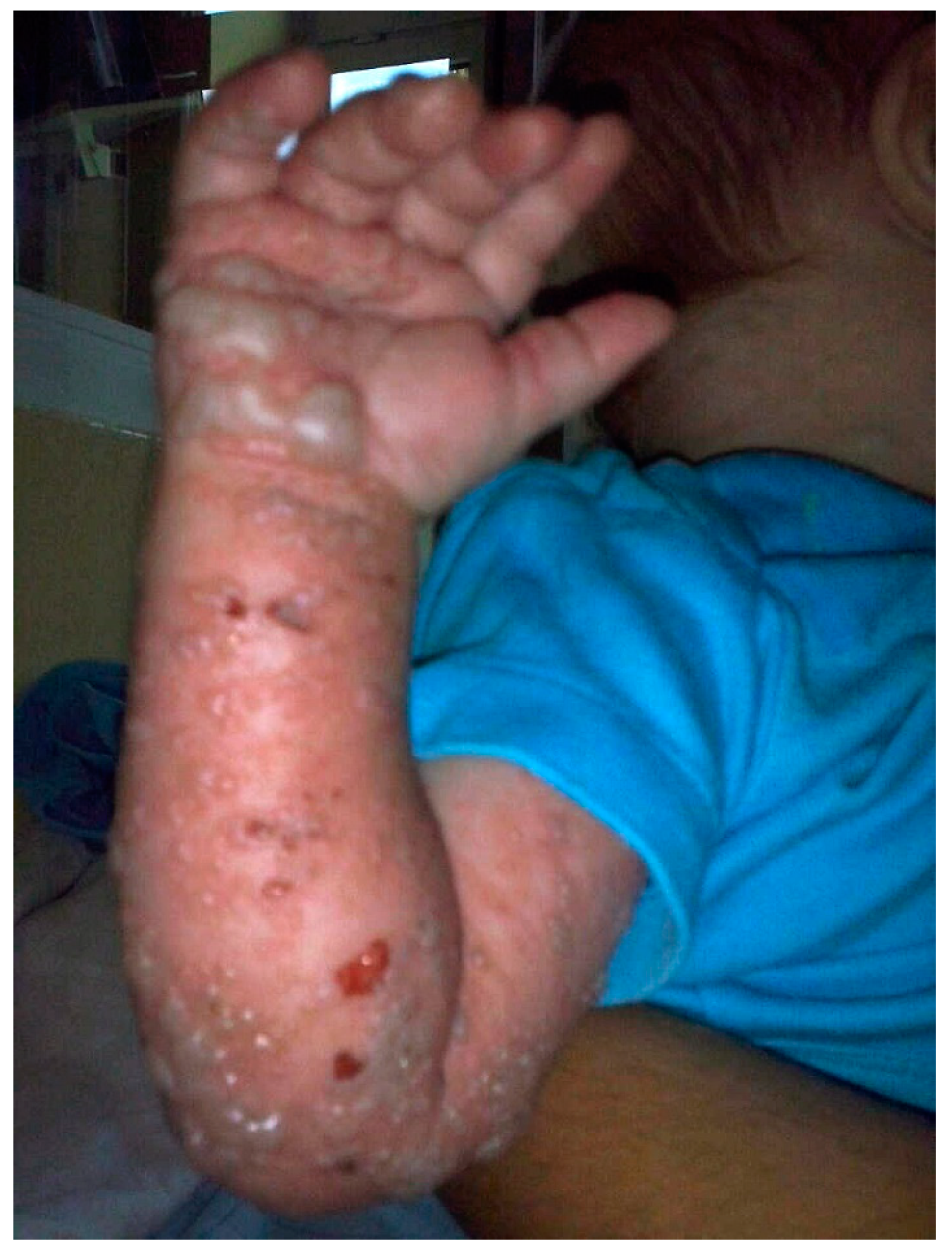
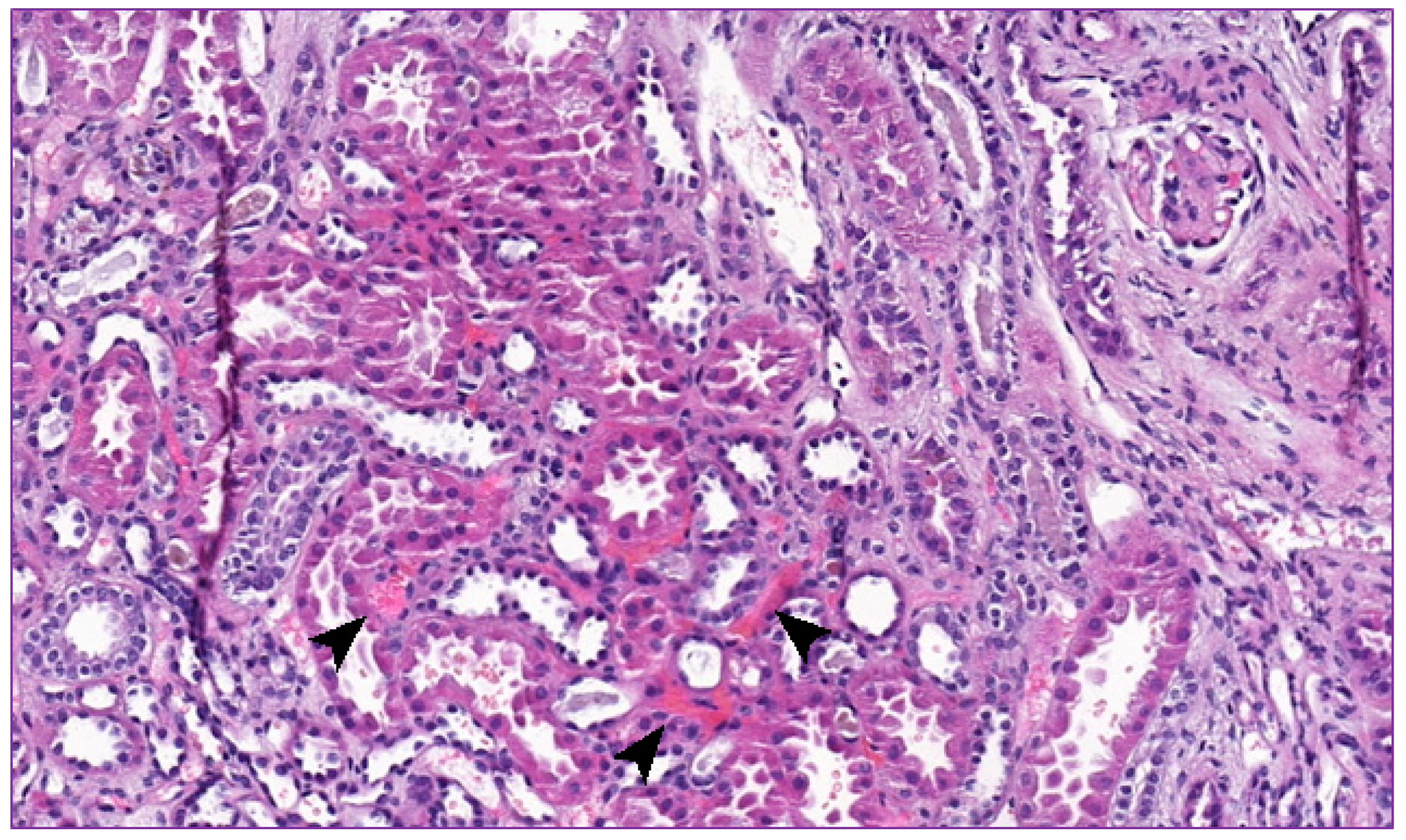
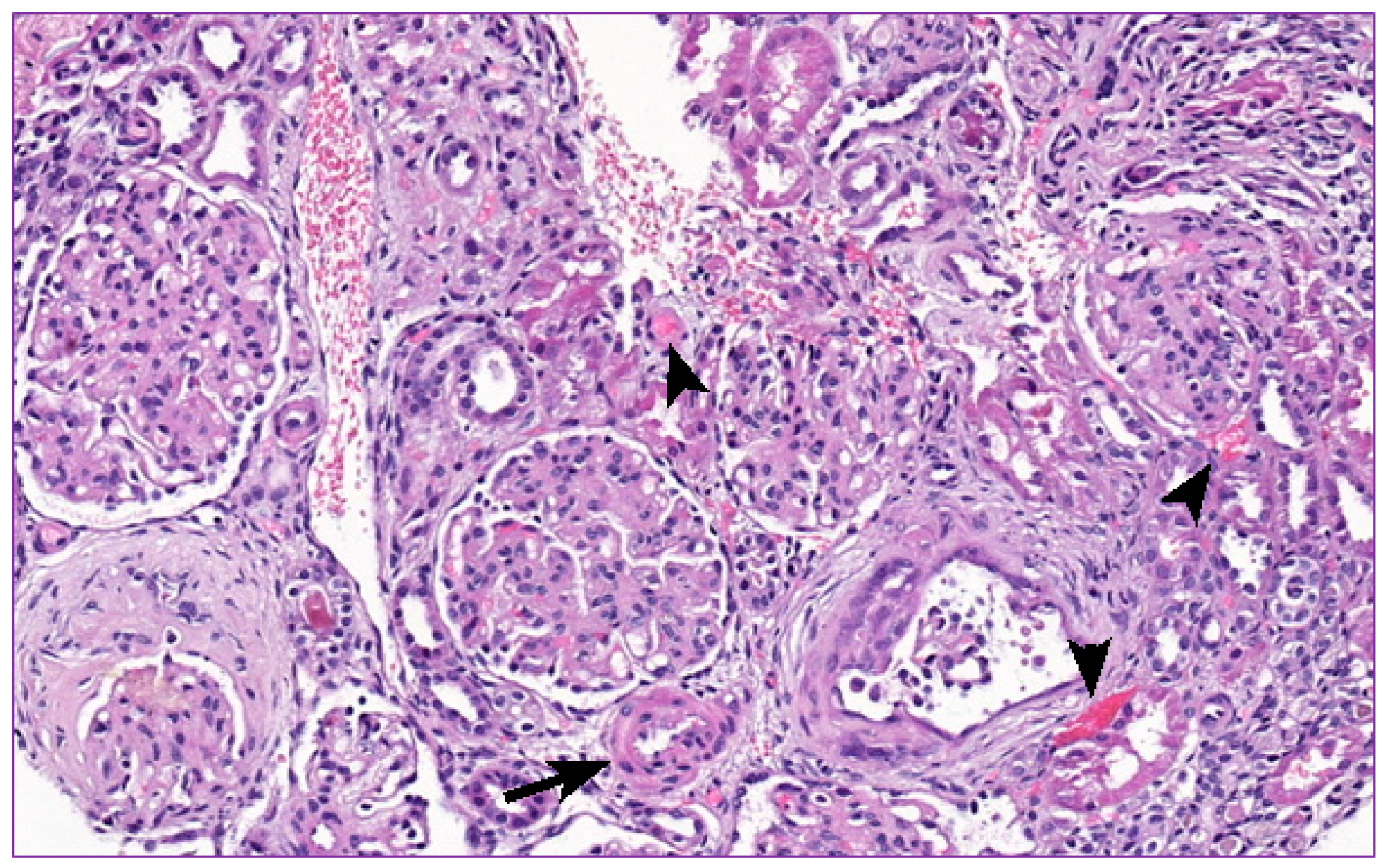
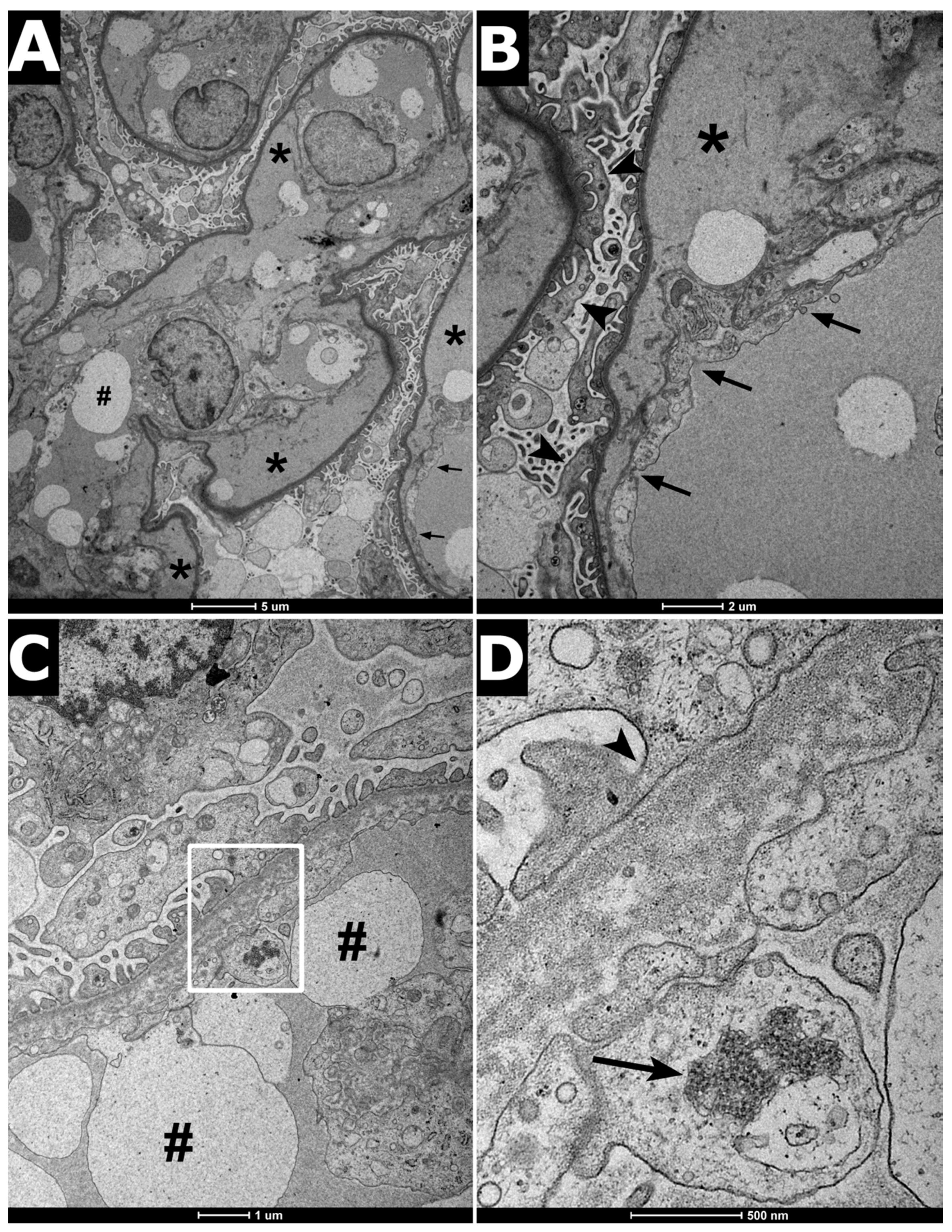
2. Clinical Report
3. Discussion
3.1. Atypical Hemolytic Uremic Syndrome
3.2. Adenosine Deaminase (ADA)-Deficient Severe Combined Immune Deficiency (SCID)
3.3. ADA-SCID and aHUS Interaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- McFarlane, P.A.; Bitzan, M.; Broome, C.; Baran, D.; Garland, J.; Girard, L.P.; Grewal, K.; Lapeyraque, A.L.; Patriquin, C.J.; Pavenski, K.; et al. Making the Correct Diagnosis in Thrombotic Microangiopathy: A Narrative Review. Can. J. Kidney Health Dis. 2021, 8. [Google Scholar] [CrossRef]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic uraemic syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [CrossRef]
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: A review. Orphanet J. Rare Dis. 2018, 13, 65. [Google Scholar] [CrossRef]
- Nikolajeva, O.; Worth, A.; Hague, R.; Martinez-Alier, N.; Smart, J.; Adams, S.; Davies, E.G.; Gaspar, H.B. Adenosine Deaminase Deficient Severe Combined Immunodeficiency Presenting as Atypical Haemolytic Uraemic Syndrome. J. Clin. Immunol. 2015, 35, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Al-Herz, W.; Bousfiha, A.; Casanova, J.; Chapel, H.; Conley, M.E.; Cunningham-Rundles, C.; Etzioni, A.; Fischer, A.; Franco, J.L.; Geha, R.S.; et al. Primary immunodeficiency diseases: An update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front. Immunol. 2011, 2, 1–26. [Google Scholar] [CrossRef] [Green Version]
- De Córdoba, S.R.; de Jorge, E.G. Translational mini-review series on complement factor H: Genetics and disease of human complement factor H. Clin. Exp. Immunol. 2008, 151, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fremeaux-Bacchi, V.; Kemp, E.J.; Goodship, J.A.; Dragon-Durey, M.A.; Strain, L.; Loirat, C.; Deng, H.W.; Goodship, T.H. The development of atypical haemolytic-uraemic syndrome is influenced by susceptibility factors in factor H and membrane cofactor protein: Evidence from two independent cohorts. J. Med. Genet. 2005, 42, 852–856. [Google Scholar] [CrossRef] [Green Version]
- Caprioli, J.; Bettinaglio, P.; Zipfel, P.F.; Amadei, B.; Daina, E.; Gamba, S.; Skerka, C.; Marziliano, N.; Remuzzi, G.; Noris, M. The molecular basis of familial hemolytic uremic syndrome: Mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J. Am. Soc. Nephrol. 2001, 12, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, M.; Xin, W.; Liu, S.; Zheng, L.; Li, Y.; Li, M.; Zhan, M.; Yang, X. Intracranial aneurysm’s association with genetic variants, transcription abnormality, and methylation changes in ADAMTS genes. Peer J. 2020, 8, e8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; van der Velden, V.H.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.; Dębska-Ślizień, A.; Durlik, M.; Hołub, T.; Karkoszka, H.; Stompór, T.; Szczepańska, M.; Żurowska, A. The management of atypical hemolytic-uremic syndrome–the statement of the Working Group of the Polish Society of Nephrology. Nefrol. Dial. Pol. 2019, 23, 55–68. [Google Scholar]
- Lemaire, M.; Frémeaux-Bacchi, V.; Schaefer, F.; Choi, M.; Tang, W.H.; Le Quintrec, M.; Fakhouri, F.; Taque, S.; Nobili, F.; Martinez, F.; et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat. Genet. 2013, 45, 531–536. [Google Scholar] [CrossRef] [Green Version]
- Ozaltin, F.; Li, B.; Rauhauser, A.; An, S.W.; Soylemezoglu, O.; Gonul, I.I.; Taskiran, E.Z.; Ibsirlioglu, T.; Korkmaz, E.; Bilginer, Y.; et al. DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J. Am. Soc. Nephrol. 2013, 24, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Kind, T.; Levy, J.; Lee, M.; Kaicker, S.; Nicholson, J.F.; Kane, S.A. Cobalamin C disease presenting as hemolytic-uremic syndrome in the neonatal period. J. Pediatr. Hematol. Oncol. 2002, 24, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Bitzan, M. Glomerular Diseases. In Manual of Pediatric Nephrology; Phadke, K.D., Goodyer, P., Bitzan, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 215–225. [Google Scholar]
- Kokame, K.; Matsumoto, M.; Soejima, K.; Yagi, H.; Ishizashi, H.; Funato, M.; Tamai, H.; Konno, M.; Kamide, K.; Kawano, Y.; et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc. Natl. Acad. Sci. USA 2002, 99, 11902–119077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Żurowska, A.; Szczepańska, M.; Dębska-Ślizień, A.; Durlik, M.; Grenda, R.; Klinger, M.; Nowicki, M.; Pańczyk-Tomaszewska, M.; Zwolińska, D.; Gellert, R. Management of patients with suspected thrombotic microangiopathy. Position of the Coordinating Group for the Treatment of Atypical Hemolytic-Uremic Syndrome. Forum. Nefrol. 2019, 12, 187–201. [Google Scholar]
- Bollinger, M.E.; Arredondo-Vega, F.X.; Santisteban, I.; Schwarz, K.; Hershfield, M.S.; Lederman, H.M. Brief report: Hepatic dysfunction as a complication of adenosine deaminase deficiency. N. Engl. J. Med. 1996, 334, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Kühl, J.S.; Schwarz, K.; Münch, A.; Schmugge, M.; Pekrun, A.; Meisel, C.; Wahn, V.; Ebell, W.; von Bernuth, H. Hyperbilirubinemia and rapid fatal hepatic failure in severe combined immunodeficiency caused by adenosine deaminase deficiency (ADA-SCID). Klin. Padiatr. 2011, 223, 85–89. [Google Scholar] [CrossRef]
- Nyhan, W.L. Disorders of purine and pyrimidine metabolism. Mol. Genet. Metab. 2005, 86, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.J.; Cooke, R. The deaminases of adenosine and adenylic acid in blood and tissues. Biochem. J. 1939, 33, 479–492. [Google Scholar] [CrossRef]
- Cristalli, G.; Costanzi, S.; Lambertucci, C.; Lupidi, G.; Vittori, S.; Volpini, R.; Camaioni, E. Adenosine deaminase: Functional implications and different classes of inhibitors. Med. Res. Rev. 2001, 21, 105–128. [Google Scholar] [CrossRef]
- Blackburn, M.R.; Thompson, L.F. Adenosine Deaminase Deficiency: Unanticipated Benefits from the Study of a Rare Immunodeficiency. J. Immunol. 2013, 188, 933–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, M.R.; Kellems, R.E. Adenosine deaminase deficiency: Metabolic basis of immune deficiency and pulmonary inflammation. Adv. Immunol. 2005, 86, 1–41. [Google Scholar] [CrossRef]
- Pham-Huy, A.; Kim, V.; Nizalik, E.; Weiler, G.; Vethamuthu, J.; Grunebaum, E. Atypical hemolytic-uremic syndrome in a patient with adenosine deaminase deficiency. LymphoSign J. 2015, 2, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Churchill, P.C.; Bidani, A.K. Hypothesis: Adenosine mediates hemodynamic changes in renal failure. Med. Hypotheses 1982, 8, 275–285. [Google Scholar] [CrossRef]
- Fredholm, B.B. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007, 14, 1315–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratech, H.; Greco, M.A.; Gallo, G.; Rimoin, D.L.; Kamino, H.; Hirschhorn, R. Pathologic findings in adenosine-deaminase-deficient severe combined immunodeficiency. I. kidney, adrenal, and chondro-osseous tissue alterations. Am. J. Pathol. 1985, 120, 157–169. [Google Scholar] [PubMed]
- Hirt-Minkowski, P.; Dickenmann, M.; Schifferli, J.A. Atypical hemolytic uremic syndrome: Update on the complement system and what is new. Nephron. Clin. Pract. 2010, 114, 219–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, F.; Borsa, N.; Gianluigi, A.; Smith, R.J. Familial atypical hemolytic uremic syndrome: A review of its genetic and clinical aspects. Clin. Dev. Immunol. 2012, 2012, 370426. [Google Scholar] [CrossRef]
- Ansari, R.; Rosen, L.B.; Lisco, A.; Gilden, D.; Holland, S.M.; Zerbe, C.S.; Bonomo, R.A.; Cohen, J.I. Primary and Acquired Immunodeficiencies Associated with Severe Varicella-Zoster Infections. Clin. Infect. Dis. 2020, ciaa1274. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laboratory Tests | A | B | Reference Ranges |
|---|---|---|---|
| Complete Blood Count On admission On discharge | |||
| Hematocrit [%] | 32.5, 30.7 | 32.25 | 32–42 |
| Red blood cell count [106/uL] | 4.98, 3.52 | 4.05 | 3.8–5.4 |
| Hemoglobin [g/dL] | 11.3, 10.2 | 11.98 | 10.5–14 |
| White cell count [103/uL] | 3.79, 5.26 | 4.53 | 6–14 |
| Granulocytes [103/uL] | 1.35, 1.76 | 2.7 | 1.6–6 |
| Lymphocytes [103/uL] | 0.46, 0.93 | 0.4 | 1.0–3.3 |
| Monocytes [103/uL] | 1.56, 0.42 | 1.2 | 0.15–0.6 |
| Mean corpuscular volume [fL] | 65.3, 87.22 | 87.13 | 72–88 |
| Mean corpuscular hemoglobin [pg] | 22.7, 28.92 | 29.62 | 24–30 |
| Mean corpuscular hemoglobin concentration [g/dL] | 34.8, 33.16 | 34 | 32–36 |
| Platelet count [103/uL] | 50, 361 | 379 | 150–450 |
| Reticulocyte count [109/L] | 87.6 | 70.2 | 26–85 |
| Serum laboratory tests | |||
| Albumins [g/L] | 29.84, 38.44 | 29.65 | 30–50 |
| Aspartate aminotransferase [U/L] | 660.6, 104.3 | 51.2 | 0.40 |
| Alanin aminotransferase [U/L] | 178.1, 49.2 | 33.2 | 0–40 |
| Bilirubin (total) [umol/L] | 22.8, 3.9 | 0.5 | 3.4–22 |
| Complement component 3 [g/L] | 0.84, 0.86 | - | 0.9–1.8 |
| Complement component 4 [g/L] | 0.1, 0.28 | - | 0.1–0.4 |
| C-reactive protein [mg/L] | 23.56, 2.63 | 37.95 | 0–5 |
| Gamma glutamyltranspeptidase [U/L] | 198, 127 | 360 | 5–65 |
| Total protein [g/L] | 41.1, 62.8 | 62.2 | 60–80 |
| Creatinine [umol/L] | 149, 61 | 55 | 21–53 |
| Uric acid [umol/L] | -, 226 | 243 | 202.3–416.5 |
| Urea [mmol/L] | 23.1, 4.9 | 20.7 | 3.2–7.1 |
| Lactate dehydrogenase [U/L] | 3757, 591 | - | 180–435 |
| Sodium [mmol/L] | 121.1, 136 | 135 | 135–148 |
| Potassium [mmol/L] | 5.43, 5.84 | 3.74 | 3.5–5.0 |
| Magnesium [mmol/L] | 0.78, 0.87 | 1.05 | 0.7–1.1 |
| Phosphate [mmol/L] | 1.15, 1.74 | 1.02 | 0.81–1.45 |
| Ionised calcium [mmol/L] | 0.86, 1.19 | 1.11 | 1.1–1.35 |
| Proteinuria [g/L] | 26.0, 1.35 | 0.75 | 0–0.15 |
| Hematuria [RBC/HPF] | Massive, 5–10 | 1–3 | 0–3 |
| Coagulation profile | |||
| Activated partial thromboplastin time [s] | 40.2, 38.1 | 27.8 | 28–40 |
| Prothrombin time [s] | 16.3, 13.1 | 12.5 | 11–16 |
| International normalized ratio [INR] | 1.31, 0.99 | 0.95 | 0.9–1.3 |
| Fibrinogen [mg/dL] | 178, 347 | 252 | 200–400 |
| D-dimer [mg/mL] | 3.1, 1.76 | 1.05 | 0–0.5 |
| Antithrombin III [%] | 90, 110 | 81 | 80–120 |
| Gene | Description | Inheritance | Additional Information |
|---|---|---|---|
| ADAMTS13 | allele G rs2301612 | mutations in both copies of gene | maternal and paternal |
| CD46 | allele A rs2796267 | mutations in both copies of gene | maternal and paternal |
| CD46 | allele A rs2796268 | mutations in both copies of gene | maternal and paternal |
| CD46 | allele G rs859705 | mutations in both copies of gene | maternal and paternal |
| CFB | allele T rs13194698 | mutations in single copy of gene | maternal |
| CFH | allele T rs3753394 | mutations in single copy of gene | paternal |
| CFH | allele G rs800292 | mutations in both copies of gene | paternal |
| CFH | allele T rs1061170 | mutations in single copy of gene | paternal |
| CFH | allele G rs3753396 | mutations in single copy of gene | paternal |
| CFH | allele T rs1065489 | mutations in single copy of gene | paternal |
| CFH | allele A rs2274700 | mutations in single copy of gene | maternal and paternal |
| Lymphocyte Immunophenotyping | Immunoglobulins | [g/L] | ||
|---|---|---|---|---|
| Parameter | Absolute Number [103/uL] | Percentage of Lymphocytes | ||
| T-cells | 0.11 [N: 1.6–6.7] | 79% | IgG | 3.09 [N: 4.53–9.16] |
| B-cells | 0.0056 [N: 0.6–2.7] | 0.04% | IgA | 0.11 [N: 0.19–1.46] |
| NK-cells | 0.0287 [N; 0.2–1.2] | 20.5% | IgM | 0.1 [N: 0.2–1] |
| CD4 T-cells | 0.00002 [N: 1.4–5.1] | 1.2% of T-cells | ||
| CD8 T-cells | 0.10 [N: 0.6–2.2] | 95.5% of T-cells | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdał, A.; Badeński, A.; Pac, M.; Wójcicka, A.; Badeńska, M.; Didyk, A.; Trembecka-Dubel, E.; Dąbrowska-Leonik, N.; Walaszczyk, M.; Matysiak, N.; et al. Atypical Hemolytic Uremic Syndrome (aHUS) and Adenosine Deaminase (ADA)-Deficient Severe Combined Immunodeficiency (SCID)—Two Diseases That Exacerbate Each Other: Case Report. Int. J. Mol. Sci. 2021, 22, 9479. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179479
Bogdał A, Badeński A, Pac M, Wójcicka A, Badeńska M, Didyk A, Trembecka-Dubel E, Dąbrowska-Leonik N, Walaszczyk M, Matysiak N, et al. Atypical Hemolytic Uremic Syndrome (aHUS) and Adenosine Deaminase (ADA)-Deficient Severe Combined Immunodeficiency (SCID)—Two Diseases That Exacerbate Each Other: Case Report. International Journal of Molecular Sciences. 2021; 22(17):9479. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179479
Chicago/Turabian StyleBogdał, Anna, Andrzej Badeński, Małgorzata Pac, Anna Wójcicka, Marta Badeńska, Agnieszka Didyk, Elżbieta Trembecka-Dubel, Nel Dąbrowska-Leonik, Małgorzata Walaszczyk, Natalia Matysiak, and et al. 2021. "Atypical Hemolytic Uremic Syndrome (aHUS) and Adenosine Deaminase (ADA)-Deficient Severe Combined Immunodeficiency (SCID)—Two Diseases That Exacerbate Each Other: Case Report" International Journal of Molecular Sciences 22, no. 17: 9479. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179479