
P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Cardiac Fibrosis Induces PCAF Activity in Cardiac Fibroblasts In Vivo
2.2. TGF-β1 Reduces the PCAF Amount but Increases Activity in Human Cardiac Fibroblasts
2.3. PCAF Is Required for TGF-β1-Mediated α-SMA and COL1A1 Induction
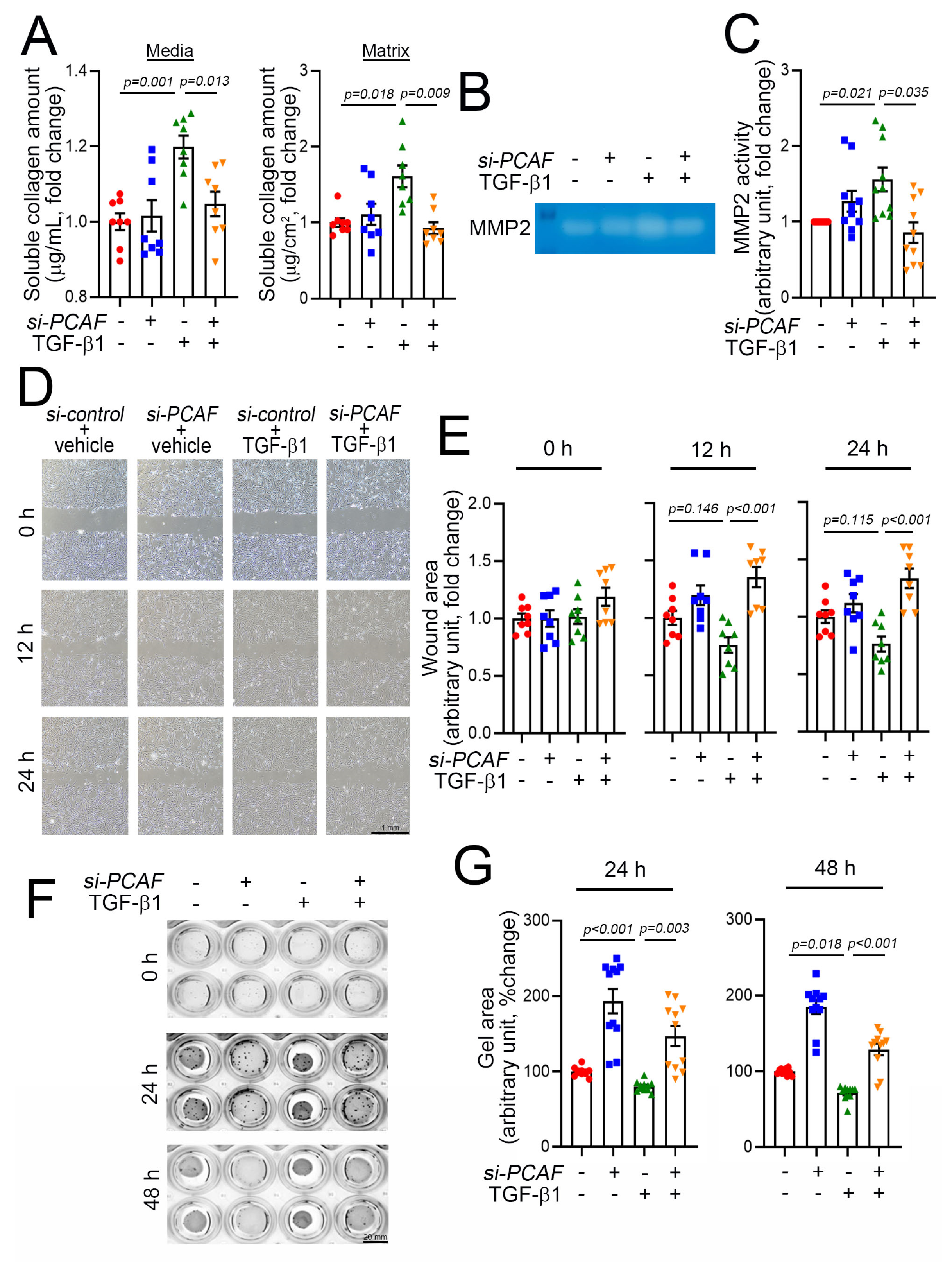
2.4. PCAF Regulates Cardiac Fibroblast Contraction, Migration, and Matrix Metalloproteinase (MMP) Activity
2.5. PCAF Acetylates SMAD2 to Induce Its Phosphorylation and Nuclear Translocation
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Isoproterenol Administration
4.3. Isolation of Mouse Cardiac Fibroblasts and Myocytes
4.4. Histologic Analysis
4.5. Cell Culture and Differentiation
4.6. Small Interfering RNA and Transfection
4.7. RNA Extraction and Quantitative Real-Time PCR
4.8. Western Blot Analysis and Immunoprecipitation
4.9. PCAF Activity Assay
4.10. Sircol Collagen Assay
4.11. Gelatin Zymography Assay
4.12. Wound Healing Assay
4.13. Collagen Gel Contraction Assay
4.14. Immunocytochemistry
4.15. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| α-SMA | alpha-smooth muscle actin |
| COL1A1 | collagen type I alpha 1 |
| CTGF | connective tissue growth factor |
| ECM | extracellular matrix |
| EndMT | endothelial-to-mesenchymal transition |
| EMT | epithelial-to-mesenchymal transition |
| FN1 | fibronectin 1 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HDAC2 | histone deacetylase 2 |
| HDAC5 | histone deacetylase 5 |
| HRP | horseradish peroxidase |
| ISP | isoproterenol |
| MMP | matrix metalloproteinase |
| PCAF | p300/CBP-associated factor |
| SMAD | small mothers against decapentaplegic |
| TGF-β | transforming growth factor-β |
References
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis–A short review of causes and therapeutic strategies. Adv. Drug. Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Mandawat, A.; Chattranukulchai, P.; Mandawat, A.; Blood, A.J.; Ambati, S.; Hayes, B.; Rehwald, W.; Kim, H.W.; Heitner, J.F.; Shah, D.J.; et al. Progression of myocardial fibrosis in nonischemic DCM and association with mortality and heart failure outcomes. JACC Cardiovasc. Imaging 2021. [Google Scholar] [CrossRef]
- Weng, Z.; Yao, J.; Chan, R.H.; He, J.; Yang, X.; Zhou, Y.; He, Y. Prognostic value of LGE-CMR in HCM: A meta-analysis. JACC Cardiovasc. Imaging 2016, 9, 1392–1402. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, A.; Schelbert, E.B.; Diez, J.; Butler, J. Myocardial interstitial fibrosis in heart failure: Biological and translational perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The fibroblast awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The role of vascular smooth muscle cells in arterial remodeling: Focus on calcification-related processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Zeng, H.; Liu, B.; Chen, J.X. Sirtuin 3 is essential for hypertension-induced cardiac fibrosis via mediating pericyte transition. J. Cell. Mol. Med. 2020, 24, 8057–8068. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Quijada, P.; Trembley, M.A.; Small, E.M. The role of the epicardium during heart development and repair. Circ. Res. 2020, 126, 377–394. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug. Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [Green Version]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell. Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [Green Version]
- Papait, R.; Serio, S.; Condorelli, G. Role of the epigenome in heart failure. Physiol. Rev. 2020, 100, 1753–1777. [Google Scholar] [CrossRef]
- Ameer, S.S.; Hossain, M.B.; Knoll, R. Epigenetics and heart failure. Int. J. Mol. Sci. 2020, 21, 9010. [Google Scholar] [CrossRef] [PubMed]
- Kee, H.J.; Eom, G.H.; Joung, H.; Shin, S.; Kim, J.-R.; Cho, Y.; Choe, N.; Sim, B.-W.; Jo, D.; Jeong, M.H.; et al. Activation of histone deacetylase 2 by inducible heat shock protein 70 in cardiac hypertrophy. Circ. Res. 2008, 103, 1259–1269. [Google Scholar] [CrossRef]
- Eom, G.H.; Cho, Y.K.; Ko, J.-H.; Shin, S.; Choe, N.; Kim, Y.; Joung, H.; Kim, H.-S.; Nam, K.-I.; Kee, H.J.; et al. Casein kinase-2alpha1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation 2011, 123, 2392–2403. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.-H.; Eom, G.H.; Ko, J.H.; Shin, S.; Joung, H.; Choe, N.; Nam, Y.S.; Min, H.-K.; Kook, T.; Yoon, S.; et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat. Commun. 2016, 7, 10492. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Kim, M.; Lee, H.; Kang, G.; Bedi, K.; Margulies, K.B.; Jain, R.; Nam, K.I.; Kook, H.; Eom, G.H. S-nitrosylation of histone deacetylase 2 by neuronal nitric oxide synthase as a mechanism of diastolic dysfunction. Circulation 2021, 143, 1912–1925. [Google Scholar] [CrossRef]
- Eom, G.H.; Nam, Y.S.; Oh, J.G.; Choe, N.; Min, H.K.; Yoo, E.K.; Kang, G.; Nguyen, V.H.; Min, J.J.; Kim, J.K.; et al. Regulation of acetylation of histone deacetylase 2 by p300/CBP-associated factor/histone deacetylase 5 in the development of cardiac hypertrophy. Circ. Res. 2014, 114, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, D.; Trompet, S.; de Craen, A.J.; Thijssen, P.E.; Quax, P.H.; de Vries, M.R.; Wierda, R.J.; van den Elsen, P.J.; Monraats, P.S.; Ewing, M.M.; et al. Genetic variation in PCAF, a key mediator in epigenetics, is associated with reduced vascular morbidity and mortality: Evidence for a new concept from three independent prospective studies. Heart 2011, 97, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Kim, S.; Son, M.; Kim, M.; Koh, E.S.; Shin, S.J.; Park, C.W.; Kim, H.S. Inhibition of p300/CBP-associated factor attenuates renal tubulointerstitial fibrosis through modulation of NF-kB and Nrf2. Int. J. Mol. Sci. 2019, 20, 1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.; Ericsson, J.; Nishikawa, J.; Heldin, C.H.; ten Dijke, P. The transcriptional co-activator P/CAF potentiates TGF-beta/Smad signaling. Nucleic Acids Res. 2000, 28, 4291–4298. [Google Scholar] [CrossRef] [Green Version]
- Simonsson, M.; Kanduri, M.; Gronroos, E.; Heldin, C.H.; Ericsson, J. The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. J. Biol. Chem. 2006, 281, 39870–39880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Panahi, M.; Baxan, N.; Ng, F.S.; Boyle, J.J.; Branca, J.; Bedard, O.; Hasham, M.G.; Benson, L.; Harding, S.E.; et al. Type 2 MI induced by a single high dose of isoproterenol in C57BL/6J mice triggers a persistent adaptive immune response against the heart. J. Cell. Mol. Med. 2020, 25, 229–243. [Google Scholar] [CrossRef]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Scolnick, D.M.; Trievel, R.C.; Zhang, H.B.; Marmorstein, R.; Halazonetis, T.D.; Berger, S.L. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999, 19, 1202–1209. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.A.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasek, J.J.; Hay, E.D.; Fujiwara, K. Collagen modulates cell shape and cytoskeleton of embryonic corneal and fibroma fibroblasts: Distribution of actin, alpha-actinin, and myosin. Dev. Biol. 1982, 92, 107–122. [Google Scholar] [CrossRef]
- Petrov, V.V.; Fagard, R.H.; Lijnen, P.J. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension 2002, 39, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell-Garcia, T.; Rodriguez-Pascual, F. Enhancement of collagen deposition and cross-linking by coupling lysyl oxidase with bone morphogenetic protein-1 and its application in tissue engineering. Sci. Rep. 2018, 8, 10780. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracaglia, L.G.; Fisher, J.P. Extracellular matrix-based biohybrid materials for engineering compliant, matrix-dense tissues. Adv. Health Mater. 2015, 4, 2475–2487. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Ngo, P.; Ramalingam, P.; Phillips, J.A.; Furuta, G.T. Collagen gel contraction assay. Methods Mol. Biol. 2006, 341, 103–109. [Google Scholar]
- McVicker, B.L.; Bennett, R.G. Novel Anti-fibrotic Therapies. Front. Pharmacol. 2017, 8, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Wang, M.; Liu, X.; Luo, L.; Li, K.; Zhang, S.; Wang, Y.; Yang, Y.; Ding, F.; Gu, X. PCAF improves glucose homeostasis by suppressing the gluconeogenic activity of PGC-1alpha. Cell Rep. 2014, 9, 2250–2262. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug. Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attisano, L.; Wrana, J.L. Signal transduction by the TGF-beta superfamily. Science 2002, 296, 1646–1647. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, A.W.; Luo, K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J. Biol. Chem. 2007, 282, 21187–21196. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Itoh, Y.; Abe, K.; Okamoto, T.; Daitoku, H.; Fukamizu, A.; Onozaki, K.; Hayashi, H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 2006, 26, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-M.; Hur, W.-H.; Kang, B.-Y.; Lee, S.-W.; Roh, P.-R.; Park, D.-J.; Sung, P.-S.; Yoon, S.-K. Death-associated protein 6 (Daxx) alleviates liver fibrosis by modulating Smad2 acetylation. Cells 2021, 10, 1742. [Google Scholar] [CrossRef]
- Bugyei-Twum, A.; Advani, A.; Advani, S.L.; Zhang, Y.; Thai, K.; Kelly, D.J.; Connelly, K.A. High glucose induces Smad activation via the transcriptional coregulator p300 and contributes to cardiac fibrosis and hypertrophy. Cardiovasc. Diabetol. 2014, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Qu, X.; Ricardo, S.D.; Bertram, J.F.; Nikolic-Paterson, D.J. Resveratrol inhibits renal fibrosis in the obstructed kidney: Potential role in deacetylation of Smad3. Am. J. Pathol. 2010, 177, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Zhang, Y.; Wang, X.; Fan, X.F.; Zhang, Y.; Li, X.; Gong, Y.S.; Han, L.P. SIRT1 activation attenuates cardiac fibrosis by endothelial-to-mesenchymal transition. Biomed. Pharmacother. 2019, 118, 109227. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Liu, Q.; Huang, Y.; Li, R.; Wu, T.; Zhang, Z.; Zhou, J.; Huang, H.; Tang, Q.; et al. Sirt6 alleviated liver fibrosis by deacetylating conserved lysine 54 on Smad2 in hepatic stellate cells. Hepatology 2020, 73, 1140–1157. [Google Scholar] [CrossRef] [PubMed]
- YYoon, S.; Kim, M.; Min, H.-K.; Lee, Y.-U.; Kwon, D.-H.; Lee, M.; Lee, S.; Kook, T.; Joung, H.; Nam, K.-I.; et al. Inhibition of heat shock protein 70 blocks the development of cardiac hypertrophy by modulating the phosphorylation of histone deacetylase 2. Cardiovasc. Res. 2018, 115, 1850–1860. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | ||
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
| PCAF | GAAGAGAACAGAAGCTCCAGG | GCAATTGGTAAAGACTCGCTG |
| ACTA2 | CCATCATGCGTCTGGATCTG | ACGCTCAGCAGTAGTAACGA |
| COL1A1 | TCTGCAACATGGAGACTGGT | TCGAACTGGAATCCATCGGT |
| CTGF | ATTAGAGCCAACTGCCTGGT | AGGAGGCGTTGTCATTGGTA |
| FN1 | GGTACAGGGTGACCTACTCG | GGAATAGCTGTGGACTGGGT |
| GAPDH | GTCGGAGTCAACGGATTTGG | TGACGGTGCCATGGAATTTG |
| Mouse | ||
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
| Pcaf | GCCGTGTCATTGGTGGTATC | GGGTTCCATAGCCCTTGACT |
| Acta2 | CTCCCTGGAGAAGAGCTACG | CGCTGACTCCATCCCAATGA |
| Col1a1 | TCCCTGGAATGAAGGGACAC | CTCTCCCTTAGGACCAGCAG |
| Ctgf | AGTGTGCACTGCCAAAGATG | CCAGGCAAGTGCATTGGTAT |
| Fn1 | ACCCTTGGCCTCCAAGTATC | CAGAGGCTGCAGGGTAGTAA |
| Tgfb1 | TTGCTTCAGCTCCACAGAGA | CAGAAGTTGGCATGGTAGCC |
| Gapdh | GCATGGCCTTCCGTGTTCCT | CCCTGTTGCTGTAGCCGTATTCAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Y.; Jeong, A.; Kwon, D.-H.; Lee, Y.-U.; Kim, Y.-K.; Ahn, Y.; Kook, T.; Park, W.-J.; Kook, H. P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. Int. J. Mol. Sci. 2021, 22, 9944. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189944
Lim Y, Jeong A, Kwon D-H, Lee Y-U, Kim Y-K, Ahn Y, Kook T, Park W-J, Kook H. P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation. International Journal of Molecular Sciences. 2021; 22(18):9944. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189944
Chicago/Turabian StyleLim, Yongwoon, Anna Jeong, Duk-Hwa Kwon, Yeong-Un Lee, Young-Kook Kim, Youngkeun Ahn, Taewon Kook, Woo-Jin Park, and Hyun Kook. 2021. "P300/CBP-Associated Factor Activates Cardiac Fibroblasts by SMAD2 Acetylation" International Journal of Molecular Sciences 22, no. 18: 9944. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189944