
C9ORF72 Repeat Expansion Affects the Proteome of Primary Skin Fibroblasts in ALS

 ,
,  , , , , , ,
, , , , , , 
Abstract
:1. Introduction
2. Results
2.1. Experimental Design: Exploring the Impact of C9ORF72 Repeat Expansion on the Proteome of Primary Skin Fibroblasts of ALS Patients
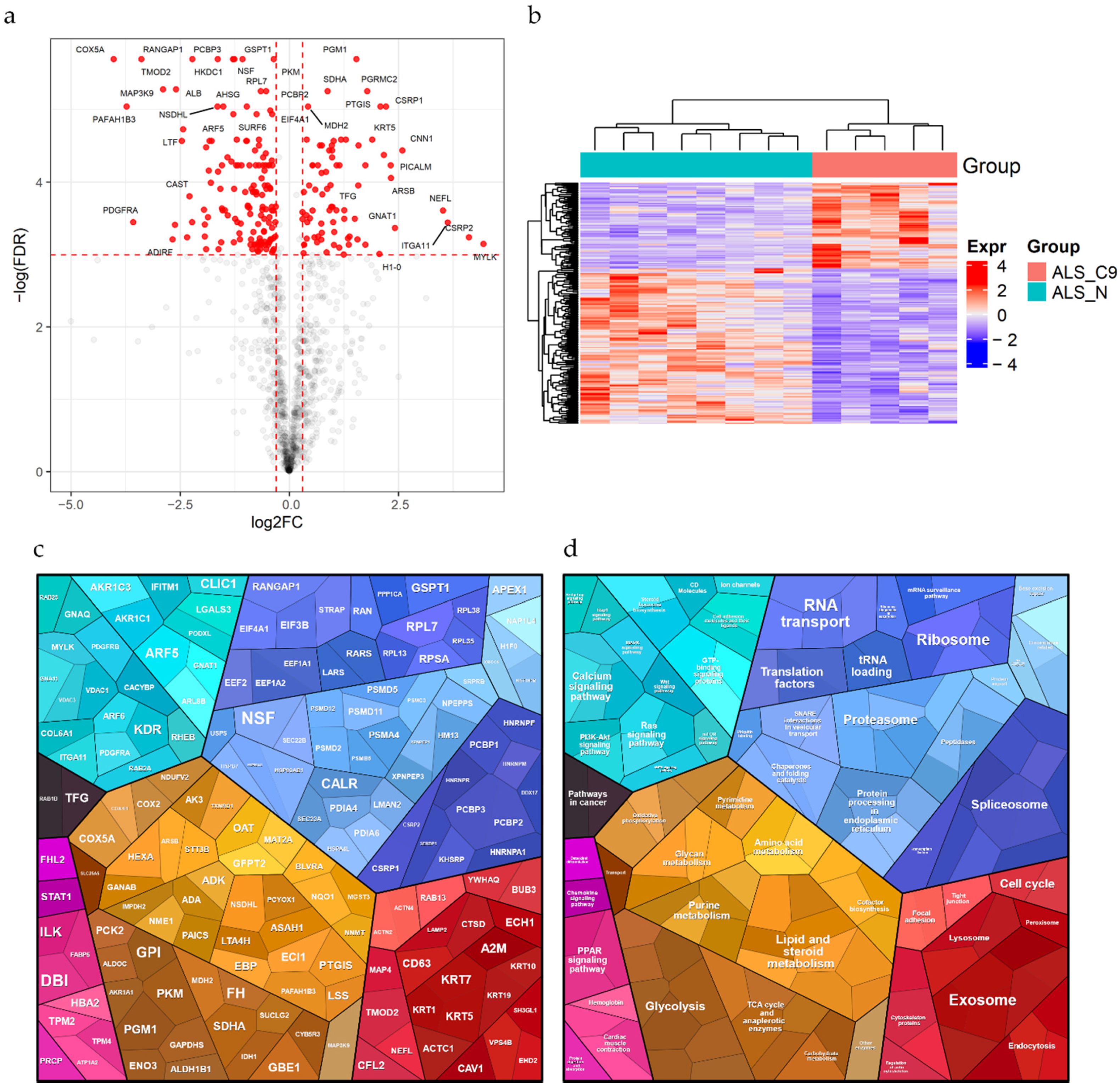
2.2. Differential Expression Analysis
2.3. Functional Enrichment Analyses
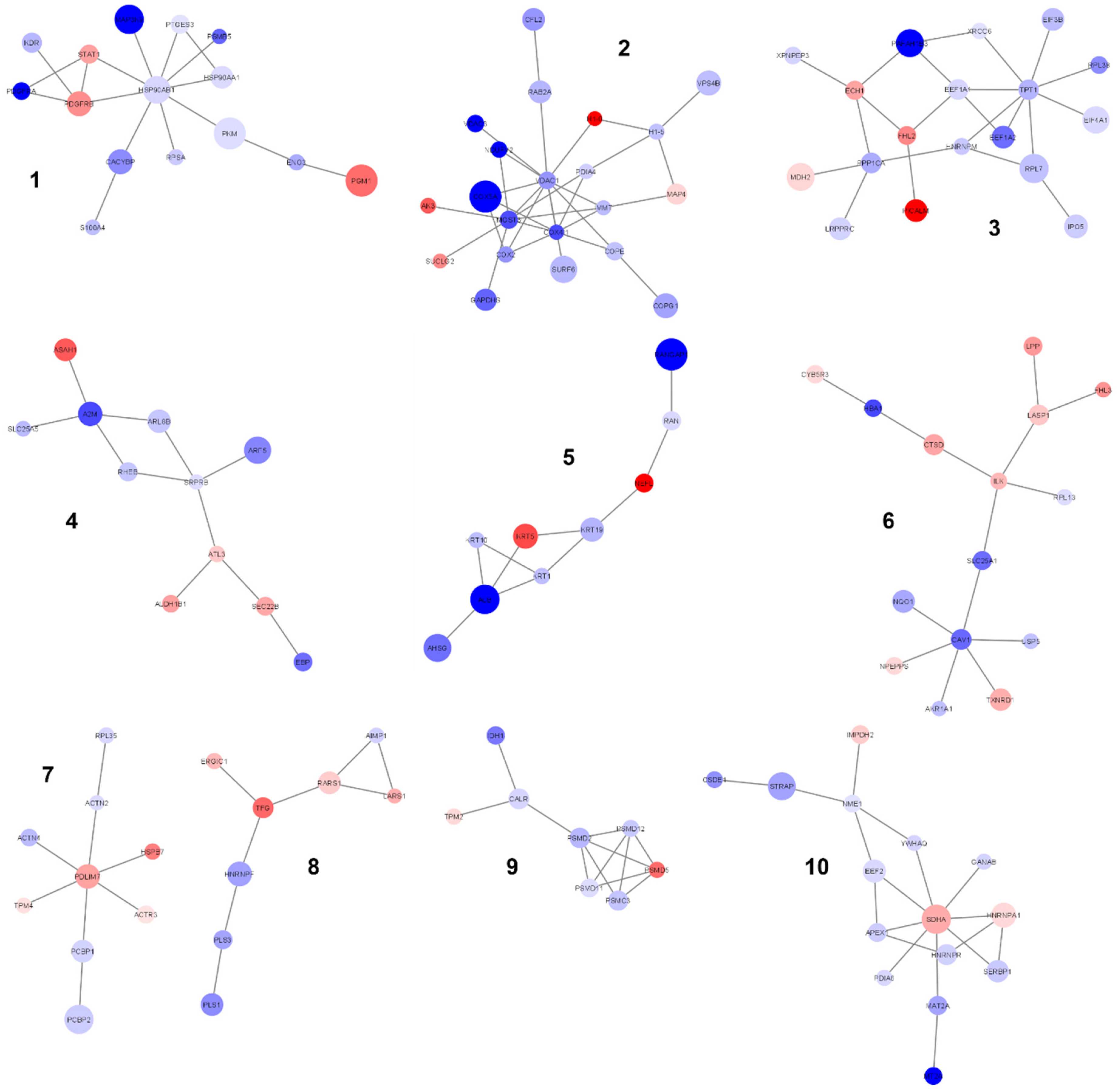
2.4. PPI Network Generation and Analysis
2.5. Comparison between ALS and FTD as Both Are C9ORF72-Associated Diseases
3. Discussion
4. Materials and Methods
4.1. Patients Recruitment, Skin Biopsy and Primary Fibroblast Cell Lines
4.2. Cell Culture
4.3. Quantitative Proteomics by LC-MS/MS
4.4. Network Analysis
4.5. Supervised Multivariate Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic Lateral Sclerosis |
| FTD | Frontotemporal Dementia |
| C9ORF72 | Chromosome 9 Open Reading Frame 72 |
| PPI | Protein-Protein Interaction |
| LC-MS/MS | Liquid Chromatography Tandem Mass Spectrometry |
| sPLS-DA | Sparse Partial Least Squares Discriminant Analysis |
| ORA | Over-Representation Analysis |
| BH | Benjamini–Hochberg |
References
- Van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How Do C9ORF72 Repeat Expansions Cause ALS and FTD: Can We Learn from Other Non-Coding Repeat Expansion Disorders? Curr. Opin. Neurol. 2012, 25, 689–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, L.A.; Salajegheh, M.K. Motor Neuron Disease: Pathophysiology, Diagnosis, and Management. Am. J. Med. 2019, 132, 32–37. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Gilmer, H.F.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Non-Coding Region of C9ORF72 Causes Chromosome 9p-Linked Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Kaivola, K.; Kiviharju, A.; Jansson, L.; Rantalainen, V.; Eriksson, J.G.; Strandberg, T.E.; Laaksovirta, H.; Renton, A.E.; Traynor, B.J.; Myllykangas, L.; et al. C9ORF72 Hexanucleotide Repeat Length in Older Population: Normal Variation and Effects on Cognition. Neurobiol. Aging 2019, 84, 242.e7–242.e12. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-B.; Chen, H.-J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide Repeats in ALS/FTD Form Length-Dependent RNA Foci, Sequester RNA Binding Proteins, and Are Neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, J.D.; Isaacs, A.M.; Mizielinska, S.; Mead, S.; Lashley, T.; Wray, S.; Sidle, K.; Fratta, P.; Orrell, R.W.; Hardy, J.; et al. C9ORF72 Expansions in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Lancet Neurol. 2015, 14, 291–301. [Google Scholar] [CrossRef]
- Corbier, C.; Sellier, C. C9ORF72 Is a GDP/GTP Exchange Factor for Rab8 and Rab39 and Regulates Autophagy. Small GTPases 2017, 8, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.; Subramanian, V.; Acharya, K.R. C9ORF72, a Protein Associated with Amyotrophic Lateral Sclerosis (ALS) Is a Guanine Nucleotide Exchange Factor. PeerJ 2018, 6, e5815. [Google Scholar] [CrossRef] [Green Version]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, Implicated in Amytrophic Lateral Sclerosis and Frontotemporal Dementia, Regulates Endosomal Trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef]
- Sivadasan, R.; Hornburg, D.; Drepper, C.; Frank, N.; Jablonka, S.; Hansel, A.; Lojewski, X.; Sterneckert, J.; Hermann, A.; Shaw, P.J.; et al. C9ORF72 Interaction with Cofilin Modulates Actin Dynamics in Motor Neurons. Nat. Neurosci. 2016, 19, 1610–1618. [Google Scholar] [CrossRef]
- Maharjan, N.; Künzli, C.; Buthey, K.; Saxena, S. C9ORF72 Regulates Stress Granule Formation and Its Deficiency Impairs Stress Granule Assembly, Hypersensitizing Cells to Stress. Mol. Neurobiol. 2017, 54, 3062–3077. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.D.; Ichida, J.K. C9ORF72 Protein Function and Immune Dysregulation in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 713, 134523. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balendra, R.; Isaacs, A.M. C9ORF72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, C.; Marcone, A.; Crespi, C.; Iannaccone, S.; Marangoni, C.; Dodich, A.; Giusti, M.C.; Zamboni, M.; Golzi, V.; Cappa, S.F. Motor Neuron Dysfunctions in the Frontotemporal Lobar Degeneration Spectrum: A Clinical and Neurophysiological Study. J. Neurol. Sci. 2015, 351, 72–77. [Google Scholar] [CrossRef]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The Overlap of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Neurology 2002, 59, 1077–1079. [Google Scholar] [CrossRef]
- Souza, P.V.S.; de Pinto, W.B.V.; de Oliveira, A.S.B. C9ORF72-Related Disorders: Expanding the Clinical and Genetic Spectrum of Neurodegenerative Diseases. Arq. Neuropsiquiatr. 2015, 73, 246–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourinaris, T.; Houlden, H. C9ORF72 and Its Relevance in Parkinsonism and Movement Disorders: A Comprehensive Review of the Literature. Mov. Disord. Clin. Pract. 2018, 5, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary Skin Fibroblasts as a Model of Parkinson’s Disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, K.Y.; Kariawasam, R.; Bax, M.; Fifita, J.A.; Ooi, L.; Yerbury, J.J.; Nicholson, G.A.; Blair, I.P. Evaluation of Skin Fibroblasts from Amyotrophic Lateral Sclerosis Patients for the Rapid Study of Pathological Features. Neurotox. Res. 2015, 28, 138–146. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Magalhaes, J.; Beavan, M.S.; McNeill, A.; Gegg, M.E.; Cleeter, M.W.J.; Bloor-Young, D.; Churchill, G.C.; Duchen, M.R.; Schapira, A.H.; et al. Endoplasmic Reticulum and Lysosomal Ca2+ Stores Are Remodelled in GBA1-Linked Parkinson Disease Patient Fibroblasts. Cell Calcium 2016, 59, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskin, I.; Forsgren, E.; Lange, D.J.; Weber, M.; Birve, A.; Synofzik, M.; Gilthorpe, J.D.; Andersen, P.M.; Marklund, S.L. Effects of Cellular Pathway Disturbances on Misfolded Superoxide Dismutase-1 in Fibroblasts Derived from ALS Patients. PLoS ONE 2016, 11, e0150133. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 Pathology Disrupts Nuclear Pore Complexes and Nucleocytoplasmic Transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, J.; Dębska-Vielhaber, G.; Vielhaber, S.; Szymański, J.; Charzyńska, A.; Duszyński, J.; Szczepanowska, J. Distinction of Sporadic and Familial Forms of ALS Based on Mitochondrial Characteristics. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 4388–4403. [Google Scholar] [CrossRef]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic Stress Induces Formation of Stress Granules and Pathological TDP-43 Aggregates in Human ALS Fibroblasts and IPSC-Motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef]
- Zilocchi, M.; Colugnat, I.; Lualdi, M.; Meduri, M.; Marini, F.; Corasolla Carregari, V.; Moutaoufik, M.T.; Phanse, S.; Pieroni, L.; Babu, M.; et al. Exploring the Impact of PARK2 Mutations on the Total and Mitochondrial Proteome of Human Skin Fibroblasts. Front. Cell Dev. Biol. 2020, 8, 423. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Conlon, E.; Manley, J.; Rio, D. Widespread Intron Retention Impairs Protein Homeostasis in C9ORF72 ALS Brains. Genome Res. 2020, 26, 5298. [Google Scholar] [CrossRef]
- Dickson, D.W.; Baker, M.C.; Jackson, J.L.; DeJesus-Hernandez, M.; Finch, N.A.; Tian, S.; Heckman, M.G.; Pottier, C.; Gendron, T.F.; Murray, M.E.; et al. Extensive Transcriptomic Study Emphasizes Importance of Vesicular Transport in C9ORF72 Expansion Carriers. Acta Neuropathol. Commun. 2019, 7, 150. [Google Scholar] [CrossRef]
- Hedl, T.J.; San Gil, R.; Cheng, F.; Rayner, S.L.; Davidson, J.M.; De Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, M.; Seeley, K.W.; Jinwal, U.K. Identification of Apo B48 and Other Novel Biomarkers in Amyotrophic Lateral Sclerosis Patient Fibroblasts. Biomark. Med. 2016, 10, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA Stability in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, C.; Kawamata, H.; Bredvik, K.G.; Arreguin, A.J.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.M.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast Bioenergetics to Classify Amyotrophic Lateral Sclerosis Patients. Mol. Neurodegener. 2017, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial Bioenergetic Deficits in C9ORF72 Amyotrophic Lateral Sclerosis Motor Neurons Cause Dysfunctional Axonal Homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Talbot, K. The Role of Mitochondrial Dysfunction and ER Stress in TDP-43 and C9ORF72 ALS. Front. Cell. Neurosci. 2021, 15, 97. [Google Scholar] [CrossRef]
- Tong, M.; Jun, T.; Nie, Y.; Hao, J.; Fan, D. The Role of the Slit/Robo Signaling Pathway. J. Cancer 2019, 10, 2694–2705. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Fontelonga, T.M.; Lee, C.H.; Barnum, S.J.; Mastick, G.S. Motor Axons Are Guided to Exit Points in the Spinal Cord by Slit and Netrin Signals. Dev. Biol. 2017, 432, 178–191. [Google Scholar] [CrossRef]
- Kaneko, N.; Herranz-Pérez, V.; Otsuka, T.; Sano, H.; Ohno, N.; Omata, T.; Nguyen, H.B.; Thai, T.Q.; Nambu, A.; Kawaguchi, Y.; et al. New Neurons Use Slit-Robo Signaling to Migrate through the Glial Meshwork and Approach a Lesion for Functional Regeneration. Sci. Adv. 2018, 4, 618. [Google Scholar] [CrossRef] [Green Version]
- Dominici, C.; Rappeneau, Q.; Zelina, P.; Fouquet, S.; Chédotal, A. Non-Cell Autonomous Control of Precerebellar Neuron Migration by Slit and Robo Proteins. Dev. Camb. Engl. 2018, 145, 150375. [Google Scholar] [CrossRef] [Green Version]
- Marini, F.; Carregari, V.C.; Greco, V.; Ronci, M.; Iavarone, F.; Persichilli, S.; Castagnola, M.; Urbani, A.; Pieroni, L. Exploring the HeLa Dark Mitochondrial Proteome. Front. Cell Dev. Biol. 2020, 8, 137. [Google Scholar] [CrossRef]
- Distler, U.; Kuharev, J.; Navarro, P.; Tenzer, S. Label-Free Quantification in Ion Mobility-Enhanced Data-Independent Acquisition Proteomics. Nat. Protoc. 2016, 11, 795–812. [Google Scholar] [CrossRef]
- Silva, J.C.; Gorenstein, M.V.; Li, G.-Z.; Vissers, J.P.C.; Geromanos, S.J. Absolute Quantification of Proteins by LCMSE: A Virtue of Parallel MS Acquisition. Mol. Cell. Proteom. MCP 2006, 5, 144–156. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Bolstad, B. Preprocesscore: A Collection of Pre-Processing Functions. R Package Version 1.54.0. 2021. Available online: https://github.com/bmbolstad/preprocessCore (accessed on 21 September 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Kuchinsky, A.; Morris, J.H.; States, D.J.; Meng, F. GLay: Community Structure Analysis of Biological Networks. Bioinforma. Oxf. Engl. 2010, 26, 3135–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual Account of Protein Investment in Cellular Functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [Green Version]
- Rohart, F.; Gautier, B.; Singh, A.; Cao, K.-A.L. MixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Lualdi, M.; Alberio, T.; Fasano, M. Proteostasis and Proteotoxicity in the Network Medicine Era. Int. J. Mol. Sci. 2020, 21, 6405. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Group | Age at Biopsy | Sex | Cell Passage | Mutation | Note |
|---|---|---|---|---|---|---|
| A610 | ALS_C9 | 69 | M | 6 | C9ORF72 expansion | / |
| A670 | ALS_C9 | 64 | M | 4 | C9ORF72 expansion | / |
| A863 | ALS_C9 | 45 | M | 4 | C9ORF72 expansion | / |
| A899 | ALS_C9 | 52 | F | 5 | C9ORF72 expansion | / |
| 9848 | ALS_C9 | 57 | F | 3 | C9ORF72 expansion | Dementia |
| 10008 | ALS_C9 | 59 | M | 3 | C9ORF72 expansion | / |
| 10879 | ALS_C9 | 80 | M | 4 | C9ORF72 expansion | Familial history of FTD |
| 10881 | ALS_C9 | 52 | F | 4 | C9ORF72 expansion | Familial history of FTD |
| 2284 | ALS_N | 42 | F | 3 | Neg. four ALS genes | / |
| 2293 | ALS_N | 50 | F | 3 | Neg. four ALS genes | / |
| 5786 | ALS_N | 53 | M | 3 | Neg. four ALS genes | / |
| 8729 | ALS_N | 65 | M | 2 | Fus 3′-UTR (c.87 C > G) | / |
| 8873 | ALS_N | 53 | M | 3 | SOD1 ex 5 p.I149T | / |
| 9718 | ALS_N | 49 | F | 3 | SOD1 ex 5 p.L145F | / |
| 9799 | ALS_N | 70 | M | 4 | SOD1, intron 1 | Schizophrenia |
| 9847 | ALS_N | 57 | F | 3 | Neg. four ALS genes | / |
| 10234 | FTD_C9 | 57 | M | 3 | C9ORF72 expansion | Dementia |
| 10946 | FTD_C9 | 64 | M | 8 | C9ORF72 expansion | Dementia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lualdi, M.; Shafique, A.; Pedrini, E.; Pieroni, L.; Greco, V.; Castagnola, M.; Cucina, G.; Corrado, L.; Di Pierro, A.; De Marchi, F.; et al. C9ORF72 Repeat Expansion Affects the Proteome of Primary Skin Fibroblasts in ALS. Int. J. Mol. Sci. 2021, 22, 10385. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910385
Lualdi M, Shafique A, Pedrini E, Pieroni L, Greco V, Castagnola M, Cucina G, Corrado L, Di Pierro A, De Marchi F, et al. C9ORF72 Repeat Expansion Affects the Proteome of Primary Skin Fibroblasts in ALS. International Journal of Molecular Sciences. 2021; 22(19):10385. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910385
Chicago/Turabian StyleLualdi, Marta, Adeena Shafique, Edoardo Pedrini, Luisa Pieroni, Viviana Greco, Massimo Castagnola, Giorgia Cucina, Lucia Corrado, Alice Di Pierro, Fabiola De Marchi, and et al. 2021. "C9ORF72 Repeat Expansion Affects the Proteome of Primary Skin Fibroblasts in ALS" International Journal of Molecular Sciences 22, no. 19: 10385. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910385