
Bacterial Luciferases from Vibrio harveyi and Photobacterium leiognathi Demonstrate Different Conformational Stability as Detected by Time-Resolved Fluorescence Spectroscopy

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
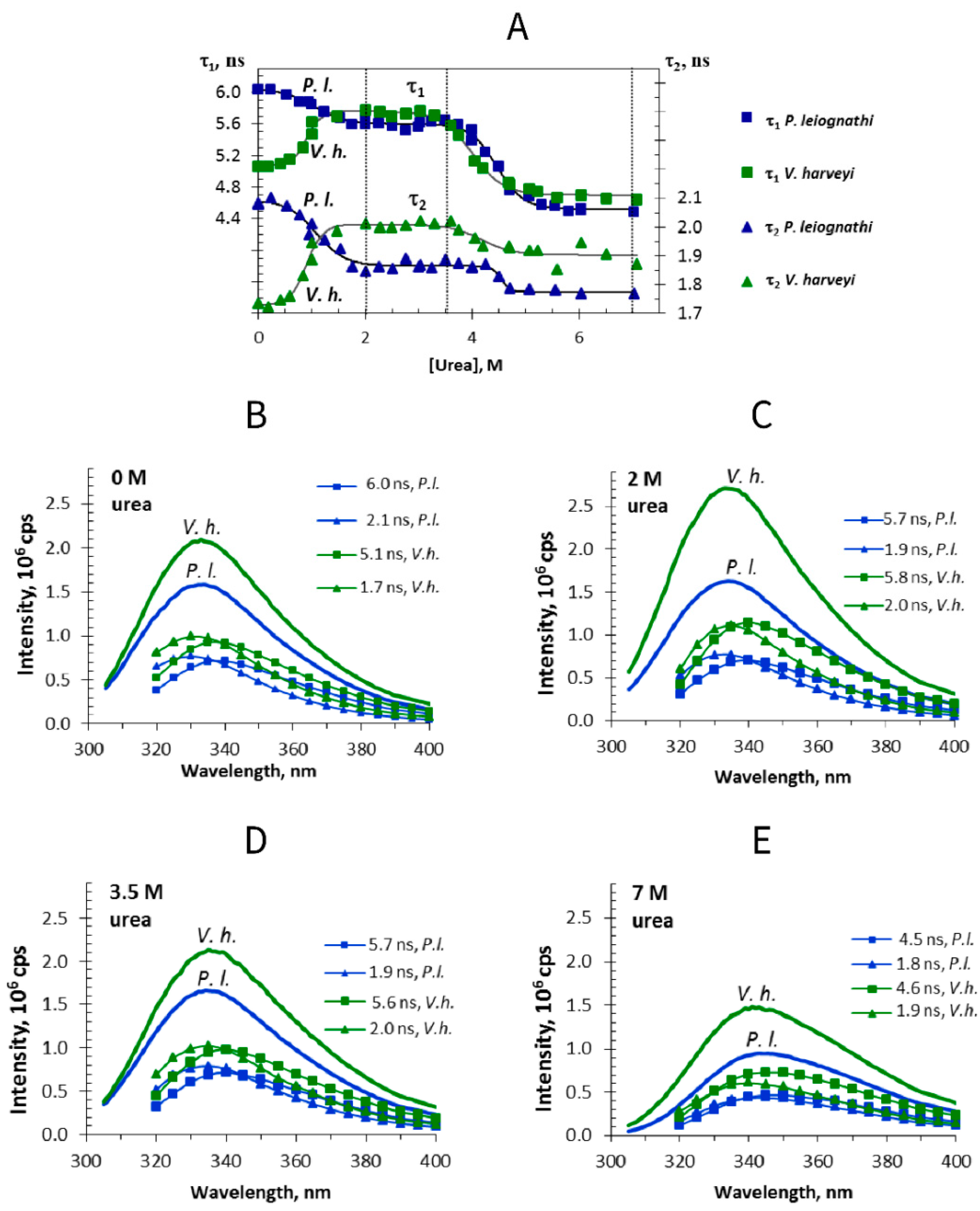
2.1. Fluorescence Lifetime Change during Urea-Induced Unfolding of Luciferases
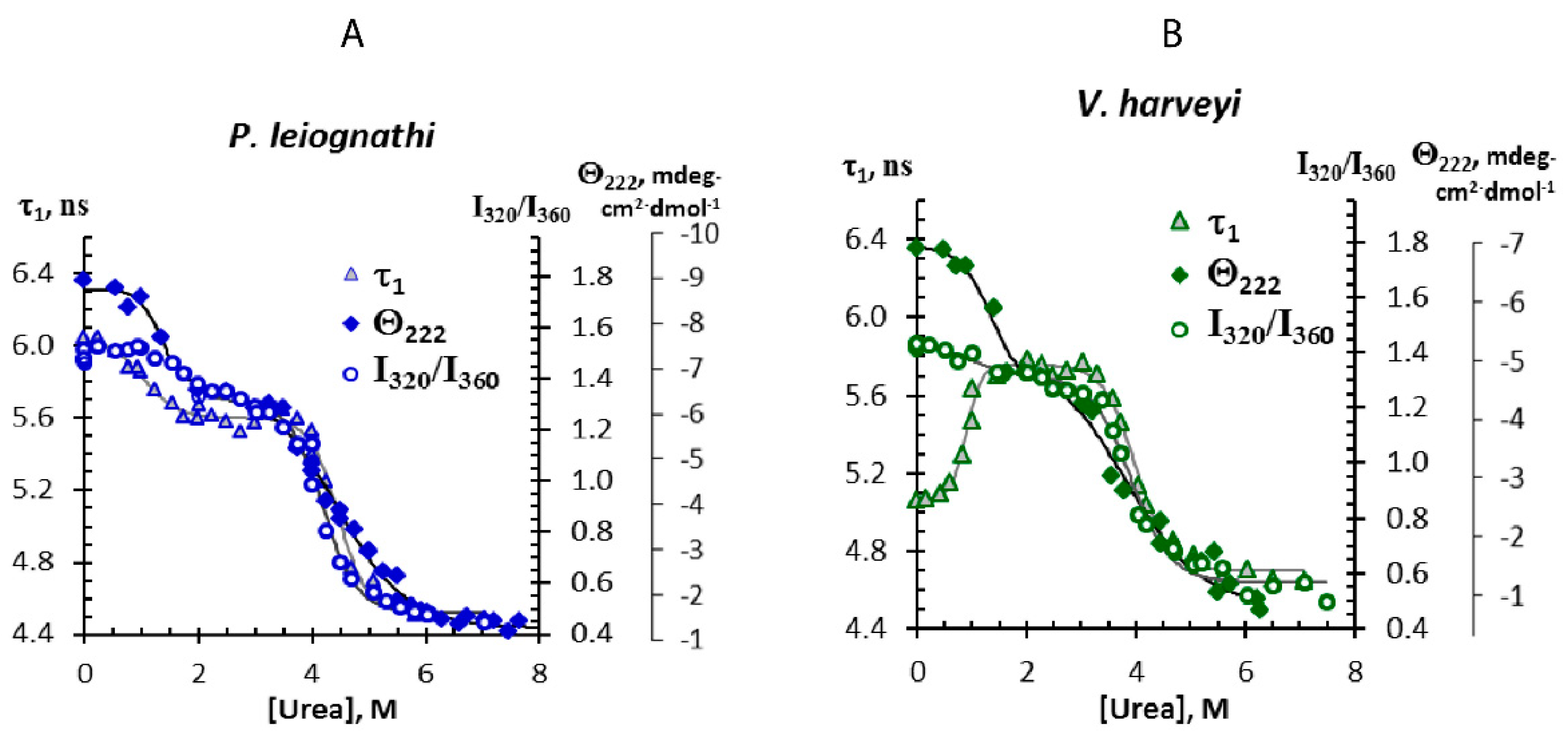
2.2. Tryptophans of the Bacterial Luciferases as Fluorescent Reporters
3. Discussion
3.1. The Origin of the Difference in Tryptophan Fluorescence of the Two Luciferases
3.2. Comparison of the Stability of the Luciferases Structures
3.3. The Origin of the Difference in Stability of Two Luciferases
3.4. Informativeness of Different Experimental Techniques on Luciferase Unfolding
4. Materials and Methods
4.1. Materials
4.2. Expression and Purification of V. harveyi luciferase
4.3. Steady-State Fluorescence
4.4. Time-Resolved Fluorescence
4.5. Circular Dichroism Spectroscopy
4.6. Data Treatment
4.7. Förster Resonance Energy Transfer (FRET)
4.8. Molecular Dynamics of the Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blancas-Mejía, L.M.; Ramirez-Alvarado, M. Systemic amyloidoses. Annu. Rev. Biochem. 2013, 82, 745–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. (Ed.) Topics in Fluorescence Spectroscopy: Protein Fluorescence; Kluwer Academic/Plenum: New York, NY, USA, 2000; Volume 6. [Google Scholar]
- Krishnamoorthy, G. Fluorescence spectroscopy for revealing mechanisms in biology: Strengths and pitfalls. J. Biosci. 2018, 43, 555–567. [Google Scholar] [CrossRef]
- Tinikul, R.; Chunthaboon, P.; Phonbuppha, J.; Paladkong, T. Bacterial luciferase: Molecular mechanisms and applications. Enzymes 2020, 47, 427–455. [Google Scholar]
- Gregor, C.; Gwosch, K.C.; Sahl, S.J.; Hell, S.W. Strongly enhanced bacterial bioluminescence with the ilux operon for single-cell imaging. Proc. Natl. Acad. Sci. USA 2018, 115, 962–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratasyuk, V.A.; Esimbekova, E.N. Applications of luminous bacteria enzymes in toxicology. Comb. Chem. High Throughput Screen. 2015, 18, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Gui, Q.; Lawson, T.; Shan, S.; Yan, L.; Liu, Y. The application of whole cell-based biosensors for use in environmental analysis and in medical diagnostics. Sensors 2017, 17, 1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeva, A.A.; Temlyakova, E.A.; Sorokin, A.A.; Nemtseva, E.V.; Kratasyuk, V.A. Structural distinctions of fast and slow bacterial luciferases revealed by phylogenetic analysis. Bioinformatics 2016, 32, 3053–3057. [Google Scholar] [CrossRef]
- Nealson, K.H.; Hastings, J.W. Bacterial bioluminescence: Its control and ecological significance. Microbiol. Rev. 1979, 43, 496–518. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, Y.J. Revisiting the origin of bacterial bioluminescence: QM/MM study on oxygenation reaction of reduced flavin in protein. Chem. Phys. Chem. 2019, 20, 405–409. [Google Scholar] [CrossRef]
- Fisher, A.J.; Thompson, T.B.; Thoden, J.B.; Baldwin, T.O.; Rayment, I. The 1.5-Å resolution crystal structure of bacterial luciferase in low salt conditions. J. Biol. Chem. 1996, 271, 21956–21968. [Google Scholar] [CrossRef] [Green Version]
- Campbell, Z.T.; Weichsel, A.; Montfort, W.R.; Baldwin, T.O. Crystal structure of the bacterial luciferase/flavin complex provides insight into the function of the β subunit. Biochemistry 2009, 48, 6085–6094. [Google Scholar] [CrossRef]
- Apuy, J.L.; Park, Z.Y.; Swartz, P.D.; Dangott, L.J.; Russell, D.H.; Baldwin, T.O. Pulsed-alkylation mass spectrometry for the study of protein folding and dynamics: Development and application to the study of a folding/unfolding intermediate of bacterial luciferase. Biochemistry 2001, 40, 15153–15163. [Google Scholar] [CrossRef]
- Campbell, Z.T.; Baldwin, T.O. Two lysine residues in the bacterial luciferase mobile loop stabilize reaction intermediates. J. Biol. Chem. 2009, 284, 32827–32834. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, T.O.; Ziegler, M.M.; Powers, D.A. Covalent structure of subunits of bacterial luciferase: NH2-terminal sequence demonstrates subunit homology. Proc. Natl. Acad. Sci. USA 1979, 76, 4887–4889. [Google Scholar] [CrossRef] [Green Version]
- Sparks, J.M.; Baldwin, T.O. Functional implications of the unstructured loop in the (β/α)8 barrel structure of the bacterial luciferase a subunit. Biochemistry 2001, 40, 15436–15443. [Google Scholar] [CrossRef] [PubMed]
- Thoden, J.B.; Holden, H.M.; Fisher, A.J.; Sinclair, J.F.; Wesenberg, G.; Baldwin, T.O.; Rayment, I. Structure of the β2 homodimer of bacterial luciferase from Vibrio harveyi: X-ray analysis of a kinetic protein folding trap. Protein Sci. 1997, 6, 13–23. [Google Scholar] [CrossRef]
- Xin, X.; Xi, L.; Tu, S.C. Functional consequences of site-directed mutation of conserved histidyl residues of the bacterial luciferase. alpha. subunit. Biochemistry 1991, 30, 11255–11262. [Google Scholar] [CrossRef] [PubMed]
- Inlow, J.K.; Baldwin, T.O. Mutational analysis of the subunit interface of Vibrio harveyi bacterial luciferase. Biochemistry 2002, 41, 3906–3915. [Google Scholar] [CrossRef]
- Nemtseva, E.V.; Gerasimova, M.A.; Melnik, T.N.; Melnik, B.S. Experimental approach to study the effect of mutations on the protein folding pathway. PLoS ONE 2019, 14, e0210361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, I.; Yakusheva, T.; Turoverov, K. Contribution of separate tryptophan residues to intrinsic fluorescence of actin. Analysis of 3D structure. FEBS Lett. 1999, 452, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Noland, B.W.; Dangott, L.J.; Baldwin, T.O. Folding, stability, and physical properties of the α subunit of bacterial luciferase. Biochemistry 1999, 38, 16136–16145. [Google Scholar] [CrossRef] [PubMed]
- Ghisaidoobe, A.B.; Chung, S.J. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on Förster resonance energy transfer techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Valkova, N.; Meighen, E. Spectral properties of Trp182, Trp194, and Trp250 on the α subunit of bacterial luciferase. Biochem. Biophys. Res. Commun. 1999, 263, 820–824. [Google Scholar] [CrossRef]
- Eisinger, J.; Feuer, B.; Lamola, A.A. Intramolecular singlet excitation transfer. Applications to polypeptides. Biochemistry 1969, 8, 3908–3915. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Barkley, M.D. Toward understanding tryptophan fluorescence in proteins. Biochemistry 1998, 37, 9976–9982. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer Science & Business Media: New York, NY, USA, 2006. [Google Scholar]
- Holzman, T.F.; Baldwin, T.O. The effects of phosphate on the structure and stability of the luciferases from Beneckea harveyi, Photobacterium, fischeri, and, Photobacterium phosphoreum. Biochem. Biophys. Res. Commun. 1980, 94, 1199–1206. [Google Scholar] [CrossRef]
- Clark, A.; Sinclair, J.; Baldwin, T. Folding of bacterial luciferase involves a non-native heterodimeric intermediate in equilibrium with the native enzyme and the unfolded subunits. J. Biol. Chem. 1993, 268, 10773–10779. [Google Scholar] [CrossRef]
- Ahmad, A.; Mishra, R. Different unfolding pathways of homologous alpha amylases from Bacillus licheniformis (BLA) and Bacillus amyloliquefaciens (BAA) in GdmCl and urea. Int. J. Biol. Macromol. 2020, 159, 667–674. [Google Scholar] [CrossRef]
- Nagibina, G.S.; Glukhova, K.A.; Uversky, V.N.; Melnik, T.N.; Melnik, B.S. Intrinsic disorder-based design of stable globular proteins. Biomolecules 2020, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins Struct. Funct. Bioinform. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Callis, P.R.; Vivian, J.T. Understanding the variable fluorescence quantum yield of tryptophan in proteins using QM-MM simulations. Quenching by charge transfer to the peptide backbone. Chem. Phys. Lett. 2003, 369, 409–414. [Google Scholar] [CrossRef]
- Gill, S.C.; Von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Baldwin, T.O.; Ziegler, M.M.; Green, V.A.; Thomas, M.D. Overexpression of bacterial luciferase and purification from recombinant sources. Methods Enzymol. Acad. Press 2000, 305, 135–152. [Google Scholar]
- O’Connor, D. Time-Correlated Single Photon Counting; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Förster, T. Transfer mechanisms of electronic excitation energy. Radiat. Res. Suppl. 1960, 2, 326–339. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone f, y and side-chain c1 and c2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]


 ). Solid lines represent fitting to a double Boltzmann function.
). Solid lines represent fitting to a double Boltzmann function.
). Solid lines represent fitting to a double Boltzmann function.
). Solid lines represent fitting to a double Boltzmann function.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Method | Fraction of Transition # | ΔGH2O, kcal/mol * | mU, kcal/M/mol * | [Urea]50%, M * | ||||
|---|---|---|---|---|---|---|---|---|---|
| P. l. | V. h. | P. l. | V. h. | P. l. | V. h. | P. l. | V. h. | ||
| 1 | I320/I360 | 0.18 0.82 | 0.15 0.85 | 5.2 ± 2.3 8.4 ± 0.7 | 1.6 ± 2.0 6.6 ± 0.6 | 2.8 ± 1.3 2.0 ± 0.2 | 1.5 ± 0.3 1.7 ± 0.2 | 1.8 ± 0.1 4.2 ± 0.1 | 1.0 ± 0.4 3.9 ± 0.1 |
| 2 | Θ222 | 0.27 0.73 | 0.34 0.66 | 4.2 ± 0.9 4.3 ± 0.6 | 4.1 ± 0.8 3.8 ± 0.6 | 3.2 ± 0.9 1.0 ± 0.2 | 3.0 ± 1.2 1.0 ± 0.3 | 1.3 ± 0.1 4.4 ± 0.1 | 1.4 ± 0.2 3.7 ± 0.2 |
| 3 | τ1 | 0.30 0.70 | 0.39 0.61 | 2.1 ± 0.3 10.8 ± 0.9 | 4.0 ± 0.6 8.2 ± 0.7 | 2.0 ± 0.3 2.4 ± 0.2 | 4.4 ± 0.7 2.0 ± 0.2 | 1.0 ± 0.1 4.5 ± 0.1 | 0.9 ± 0.1 4.0 ± 0.1 |
| 4 | τ2 | 0.70 0.30 | 0.73 0.27 | 2.5 ± 0.4 11.0 ± 4.3 | 3.5 ± 0.8 8.5 ± 4.2 | 2.3 ± 0.3 2.4 ± 0.9 | 3.9 ± 0.9 2.0 ± 1.0 | 1.1 ± 0.1 4.6 ± 0.2 | 0.9 ± 0.1 4.2 ± 0.2 |
| Tryptophan ↓ | Characteristics of Microenvironment in 7 Å * & | SASA, Å2 (%) # | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NΣ, pcs | Npol, pcs, (%) & | Nsec (Nα + Nβ), pcs, (%) & | NC-term, pcs, (%) & | Nβ-sub, pcs, (%) & | |||||||||
| Subunit | Position | P.l. | V.h. | P.l. | V.h. | P.l. | V.h. | P.l. | V.h. | P.l. | V.h. | P.l. | V.h. |
| α | 40 | 159 | 177 | 64 (40) | 63 (36) | 159 (5 + 154), (100) | 170 (0 + 170), (96) | 14 (9) | 18 (10) | 0 | 0 | 14.8 ± 7.9 (5.6 ± 3.0) | 8.6 ± 6.2 (3.2 ± 2.4) |
| α | 131 | - | 162 | - | 59 (36) | - | 99 (84 + 15), (61) | - | 12 (7) | - | 0 | - | 7.2 ± 5.9 (2.7 ± 2.2) |
| α | 182 | 135 | 148 | 54 (40) | 56 (38) | 121 (107 + 14), (90) | 130 (111 + 19), (88) | 0 | 0 | 0 | 0 | 25.4 ± 13.1 (9.6 ± 5.0) | 33.0 ± 8.3 (12.5 ± 3.2) |
| α | 194 | 127 | 141 | 39 (31) | 44 (31) | 78 (34 + 44), (61) | 84 (36 + 48), (60) | 50 (39) | 60 (43) | 0 | 0 | 19.3 ± 7.9 (7.3 ± 3.0) | 20.8 ± 7.9 (7.9 ± 3.0) |
| α | 250 | 121 | 151 | 33 (27) | 48 (32) | 79 (79 + 0), (65) | 83 (82 + 1), (55) | 93 (77) | 106 (70) | 0 | 0 | 39.8 ± 11.7 (15.1 ± 4.4) | 34.3 ± 13.6 (13.0 ± 5.1) |
| α | 277 | 93 | 121 | 39 (42) | 52 (43) | 67 (0 + 67), (72) | 20 (20 + 0), (17) | 63 (68) | 64 (53) | 28 (30) | 0 | 114.2 ± 22.8 (43.3 ± 8.6) | 99.4 ± 26.8 (37.7 ± 10.2) |
| β | 182 | 146 | 148 | 48 (32) | 47 (32) | 118 (101 + 17), (81) | 132 (111 + 21), (89) | 0 | 0 | N/A | N/A | 25.7 ± 10.3 (9.7 ± 3.9) | 22.6 ± 9.1 (8.6 ± 3.5) |
| β | 194 | 145 | 152 | 41 (28) | 44 (29) | 87 (42 + 45), (60) | 92 (38 + 54), (61) | 0 | 0 | N/A | N/A | 17.8 ± 6.5 (6.7 ± 2.5) | 6.1 ± 5.1 (2.3 ± 1.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemtseva, E.V.; Gulnov, D.V.; Gerasimova, M.A.; Sukovatyi, L.A.; Burakova, L.P.; Karuzina, N.E.; Melnik, B.S.; Kratasyuk, V.A. Bacterial Luciferases from Vibrio harveyi and Photobacterium leiognathi Demonstrate Different Conformational Stability as Detected by Time-Resolved Fluorescence Spectroscopy. Int. J. Mol. Sci. 2021, 22, 10449. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910449
Nemtseva EV, Gulnov DV, Gerasimova MA, Sukovatyi LA, Burakova LP, Karuzina NE, Melnik BS, Kratasyuk VA. Bacterial Luciferases from Vibrio harveyi and Photobacterium leiognathi Demonstrate Different Conformational Stability as Detected by Time-Resolved Fluorescence Spectroscopy. International Journal of Molecular Sciences. 2021; 22(19):10449. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910449
Chicago/Turabian StyleNemtseva, Elena V., Dmitry V. Gulnov, Marina A. Gerasimova, Lev A. Sukovatyi, Ludmila P. Burakova, Natalya E. Karuzina, Bogdan S. Melnik, and Valentina A. Kratasyuk. 2021. "Bacterial Luciferases from Vibrio harveyi and Photobacterium leiognathi Demonstrate Different Conformational Stability as Detected by Time-Resolved Fluorescence Spectroscopy" International Journal of Molecular Sciences 22, no. 19: 10449. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910449