
Mechanisms Involved in the Promoting Activity of Fibroblasts in HTLV-1-Mediated Lymphomagenesis: Insights into the Plasticity of Lymphomatous Cells

 ,
,  , ,
, , 
Abstract
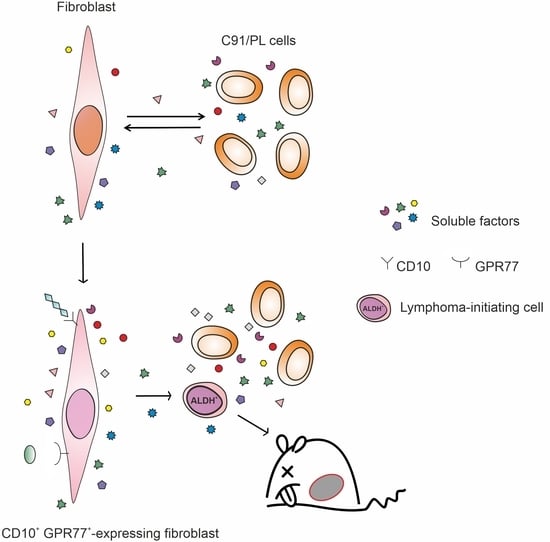
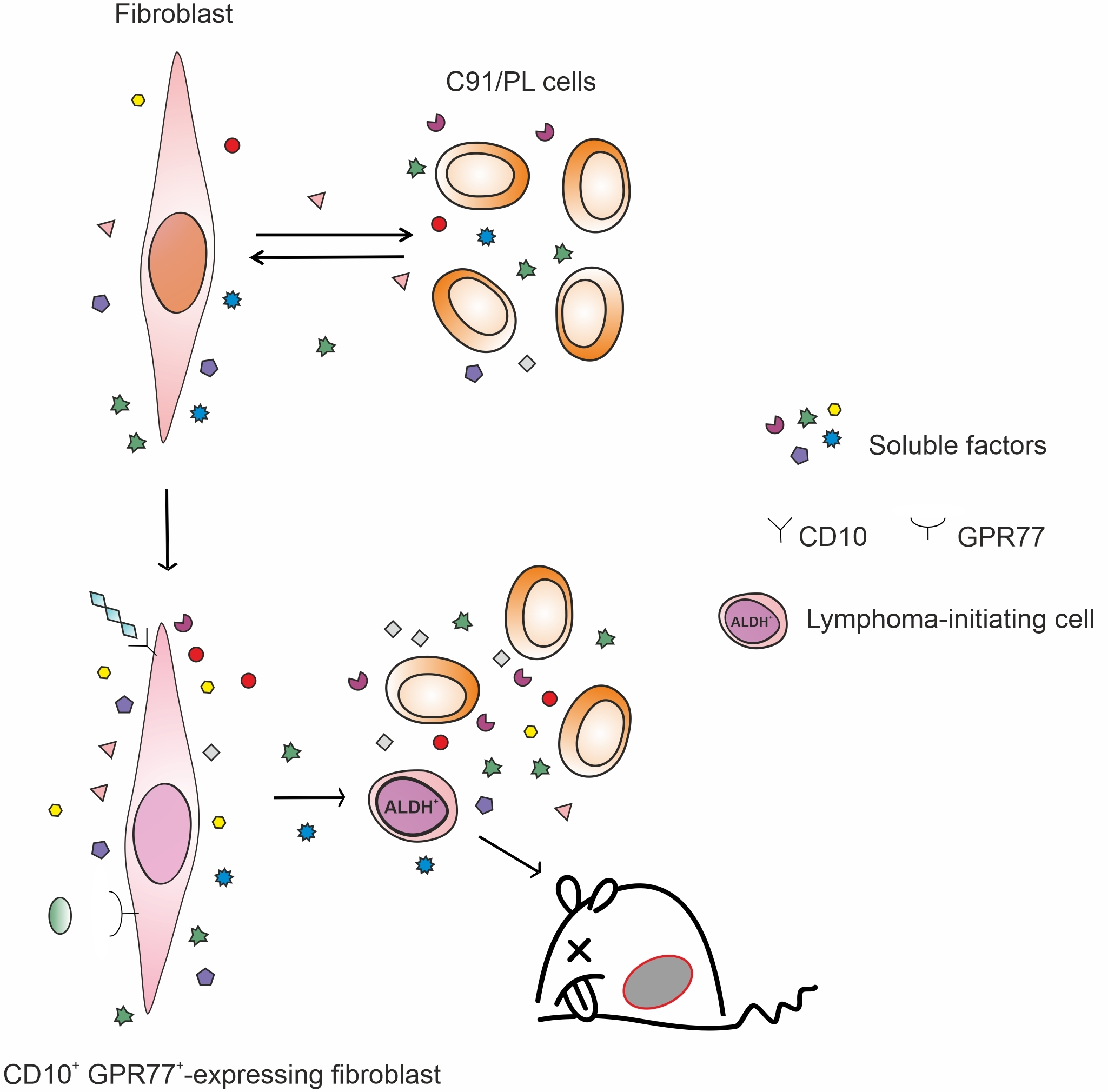
:
1. Introduction
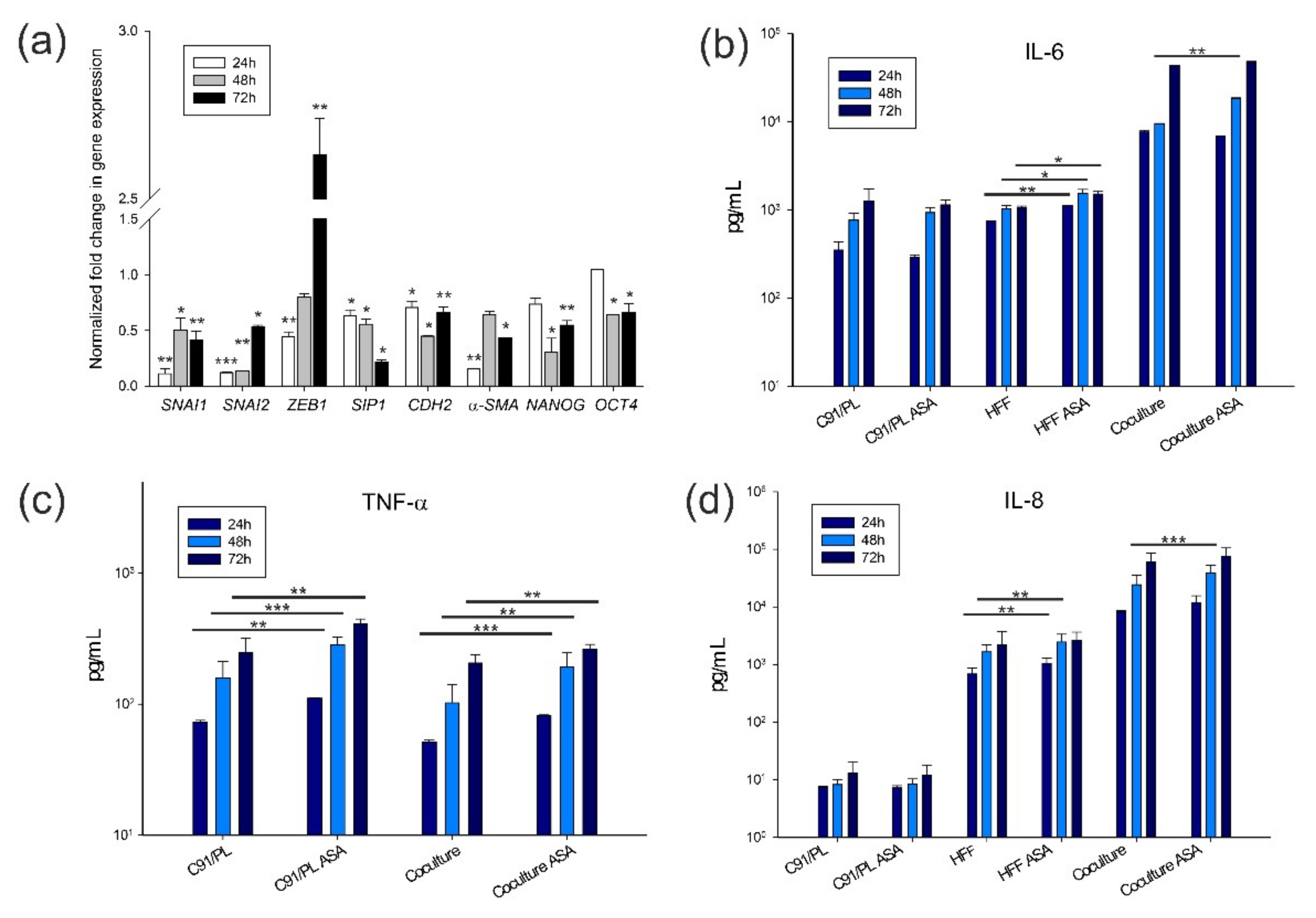
2. Results
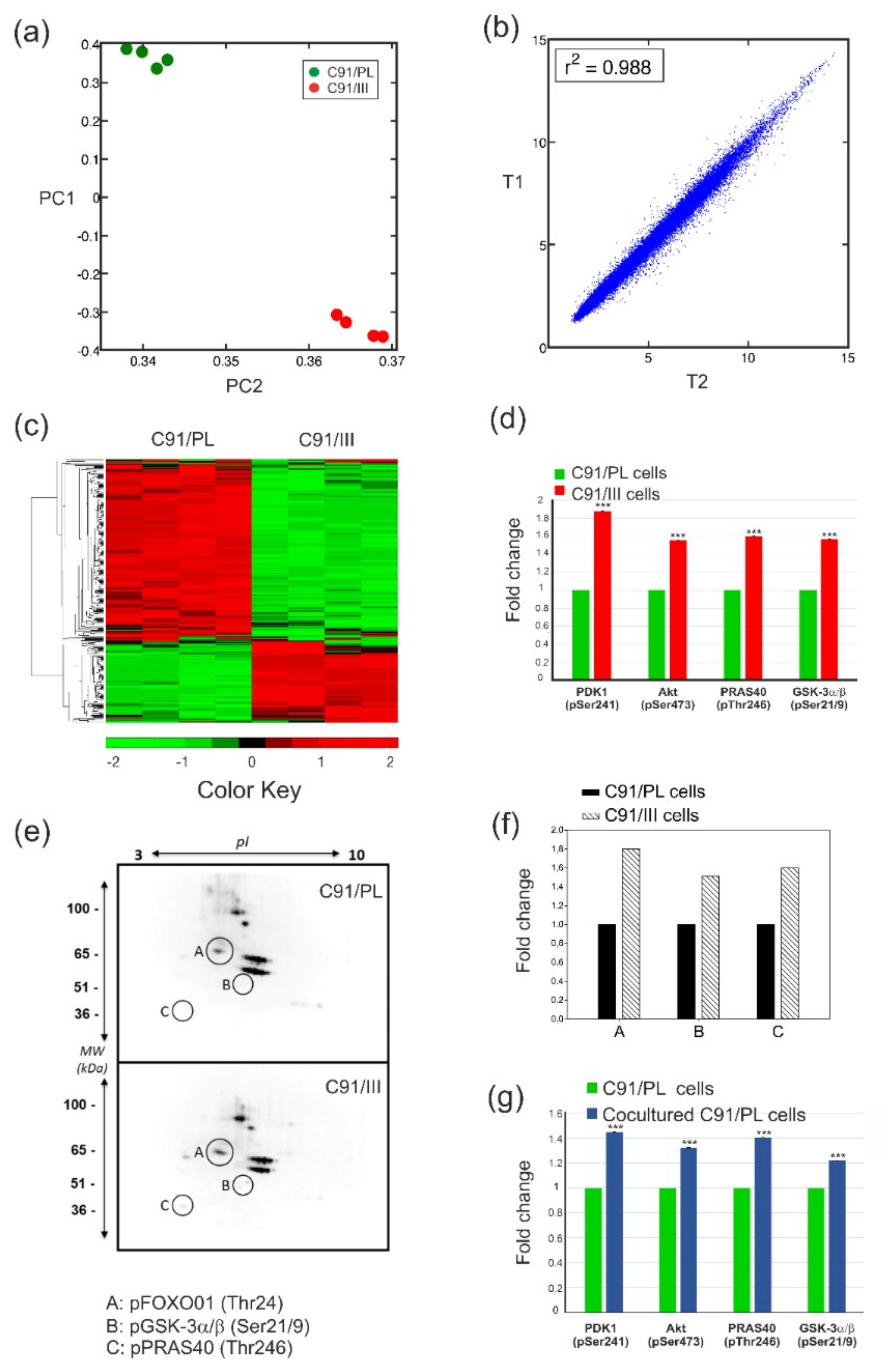
2.1. C91/III Cells Display Transcriptional Reprogramming and Hyperactivation of the PI3K/Akt Signaling Pathway
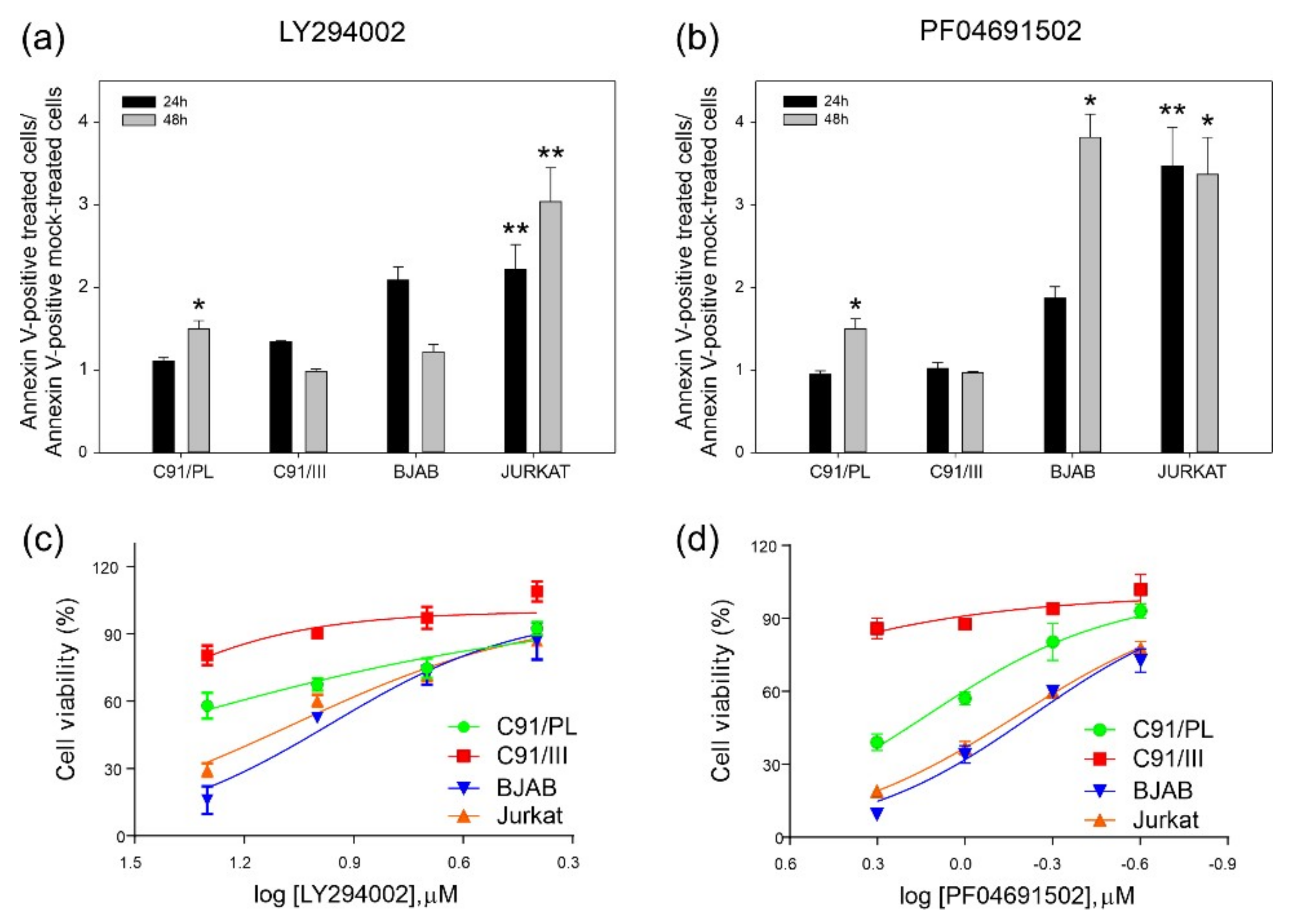
2.2. Blockade of PI3K/Akt Signaling Pathway
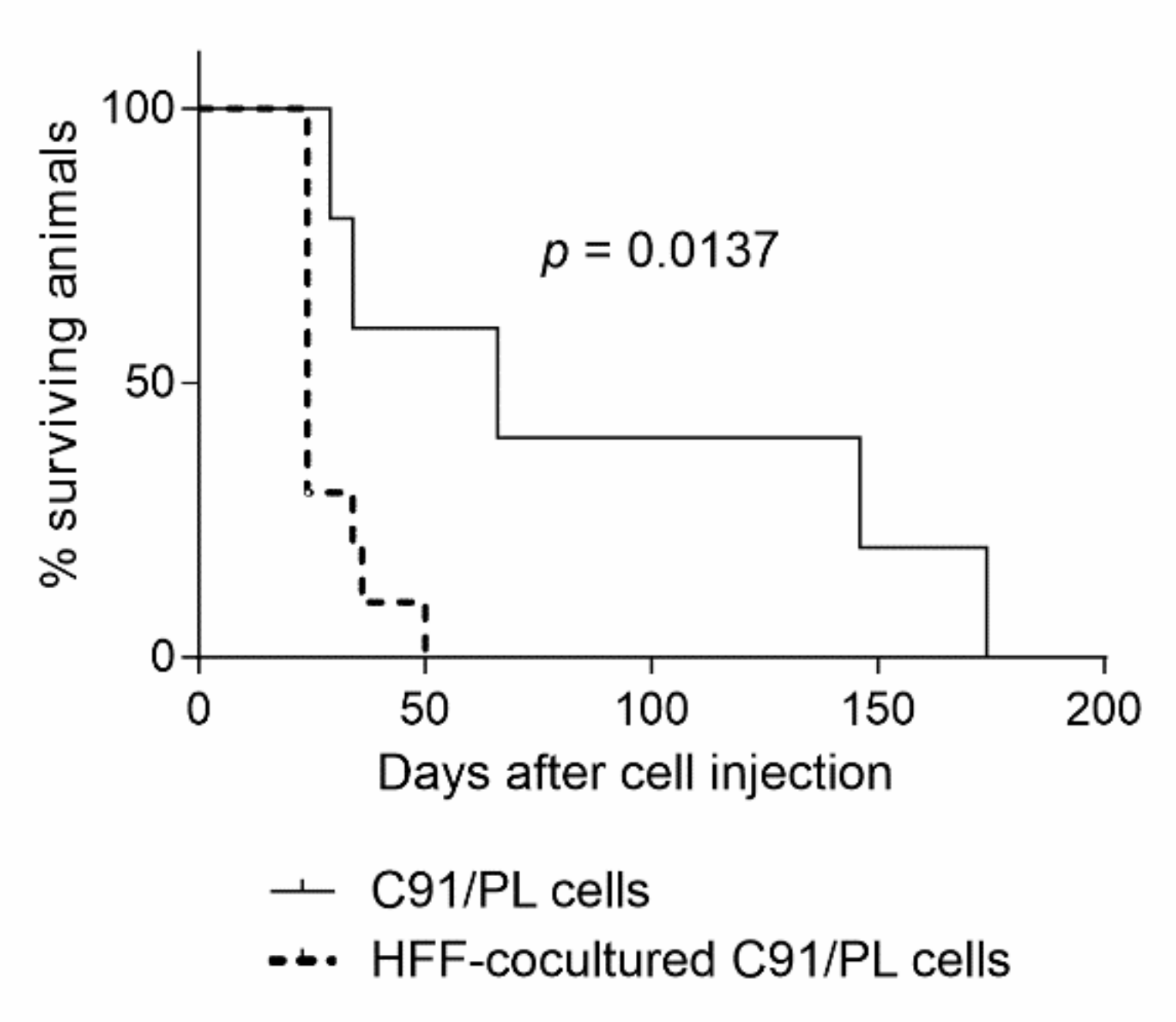
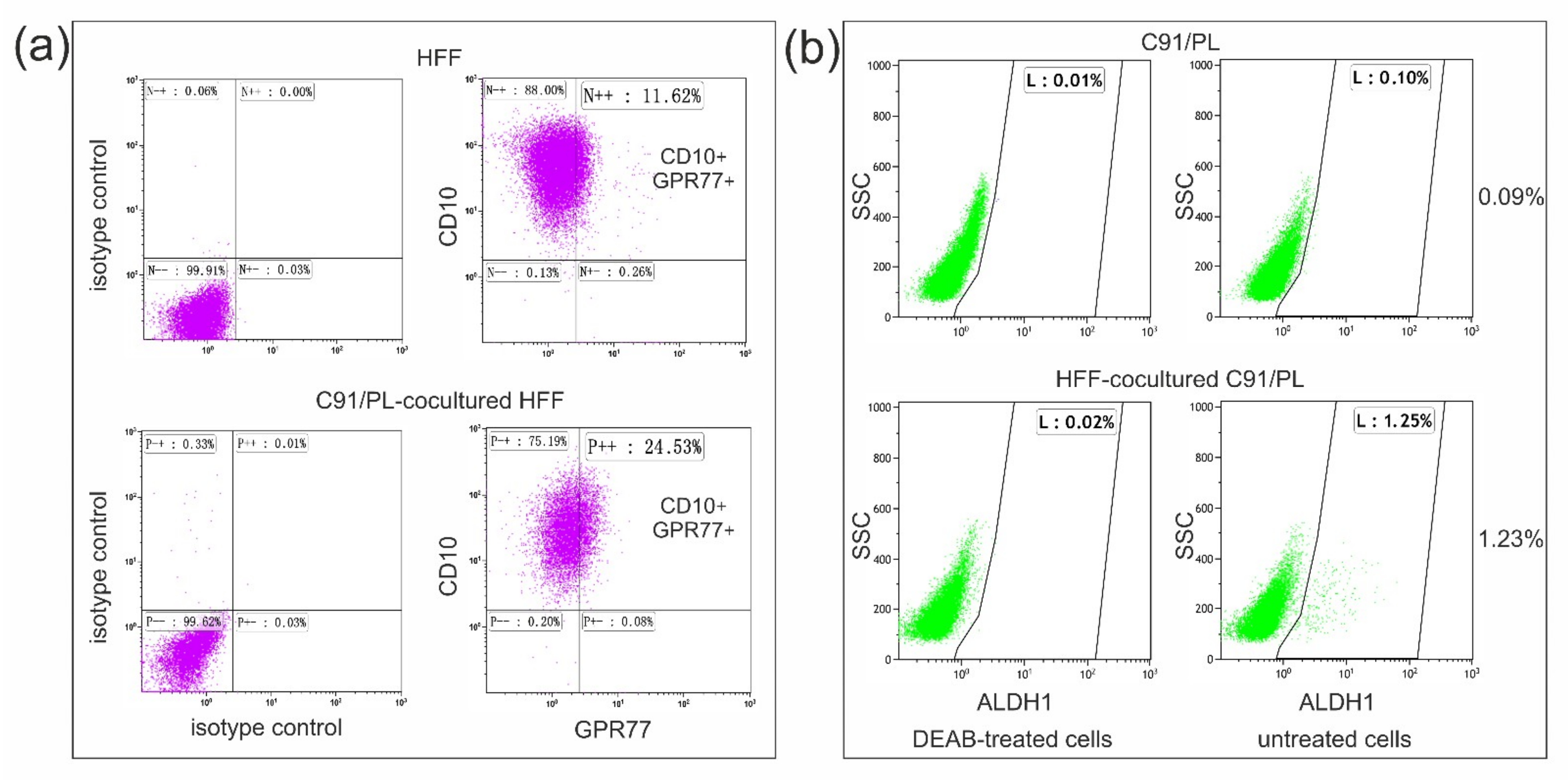
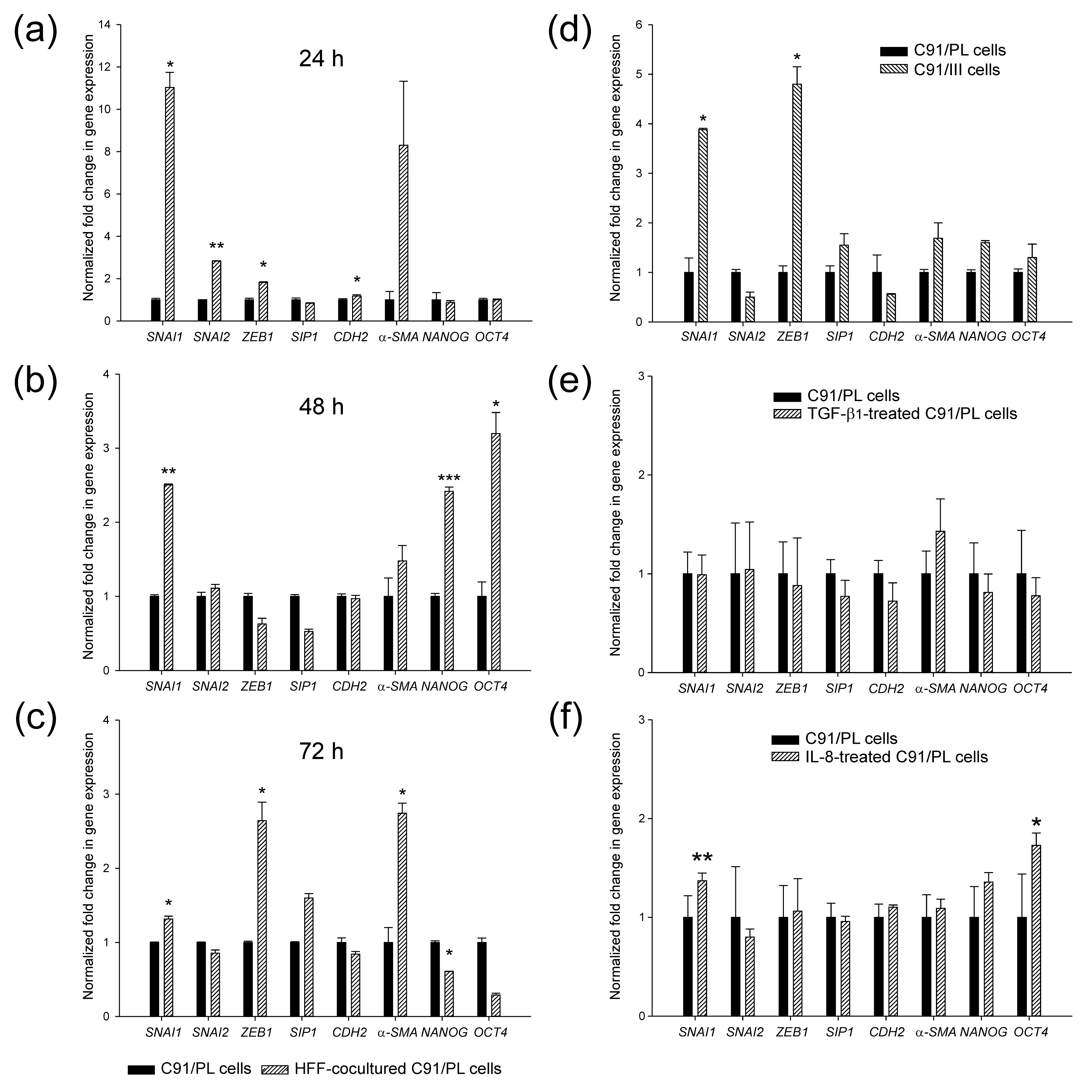
2.3. In Vivo and In Vitro Effects of Short-Term Coculture of C91/PL Cells with HFF
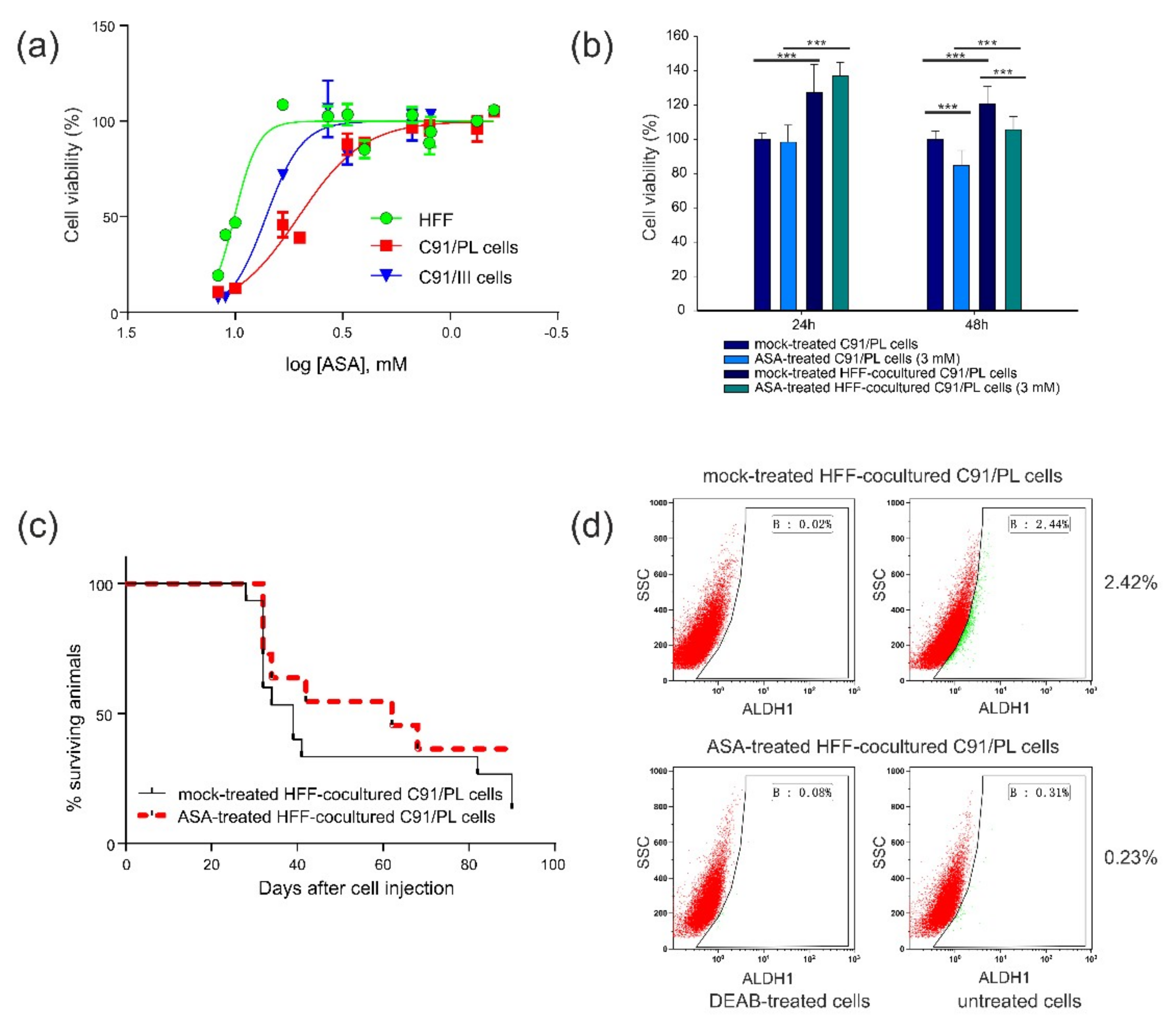
2.4. In Vitro and In Vivo Activity of Acetylsalicylic Acid (ASA, Aspirin) Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cocultures and Mice
4.2. Transcriptomic and Bioinformatics Analyses
4.3. Analysis of the PI3K/Akt Signaling Pathway
4.4. In vivo Experiments
4.5. Cytofluorimetric Analyses
4.6. Quantitative mRNA Analyses
4.7. Quantitative Analyses of Soluble Factors
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALDH1 | aldehyde dehydrogenase 1 |
| APC | allophycocyanin |
| ASA | acetylsalicylic acid, aspirin |
| ATLL | Adult T-cell Leukaemia/Lymphoma |
| CAF | cancer-associated fibroblasts |
| COX-1 | cyclooxygenase 1 |
| COX-2 | cyclooxygenase 2 |
| CRC | colorectal carcinoma |
| CSCs | cancer stem cells |
| DEAB | N,N-diethylaminobenzaldehyde |
| DMSO | dimethyl sulfoxide |
| ECM | extracellular matrix |
| EMT | epithelial-mesenchymal transition |
| GSK-3α/β | glycogen synthase kinase 3 |
| HFF | human foreskin fibroblasts |
| HTLV-1 | Human T-cell Leukemia Virus type 1 |
| IL | interleukin |
| i.p. | intraperitoneally |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| NSAID | nonsteroidal anti-inflammatory drug |
| NSG | NOD/SCID/IL2Rγc KO |
| PCA | principal component analysis |
| PDK1 | Akt upstream regulator phosphoinositide-dependent kinase 1 |
| PE | phycoerythrin |
| PRAS40 | proline-rich Akt substrate of 40 kDa |
| qRT-PCR | quantitative real-time PCR |
| SD | standard deviation |
| SSC | side scatter signal |
| TFs | transcription factors |
| TGF-β1 | transforming growth factor beta1 |
| TNF-α | tumor necrosis factor alpha |
| 2-DE | two-dimensional electrophoresis |
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017; Volume 2. [Google Scholar]
- Watanabe, T. Adult T-cell leukemia: Molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 2017, 129, 1071–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagaya, Y.; Matsuoka, M.; Gallo, R. 40 years of the human T-cell leukemia virus: Past, present, and future. F1000 Research 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Schierhout, G.; McGregor, S.; Gessain, A.; Einsiedel, L.; Martinello, M.; Kaldor, J. Association between HTLV-1 infection and adverse health outcomes: A systematic review and meta-analysis of epidemiological studies. Lancet Infect. Dis. 2020, 20, 133–143. [Google Scholar] [CrossRef]
- Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–1987). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Hermine, O.; Bazarbachi, A.; Ratner, L.; Ramos, J.C.; Harrington, W., Jr.; O’Mahony, D.; Janik, J.E.; Bittencourt, A.L.; Taylor, G.P.; et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: A proposal from an international consensus meeting. J. Clin. Oncol. 2009, 27, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.B.; Fuji, S.; Hermine, O.; Bazarbachi, A.; Ramos, J.C.; Ratner, L.; Horwitz, S.; Fields, P.; Tanase, A.; Bumbea, H.; et al. Revised Adult T-Cell Leukemia-Lymphoma International Consensus Meeting Report. J. Clin. Oncol. 2019, 37, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Hirons, A.; Khoury, G.; Purcell, D.F.J. Human T-cell lymphotropic virus type-1: A lifelong persistent infection, yet never truly silent. Lancet Infect Dis. 2021, 21, e2–e10. [Google Scholar] [CrossRef]
- Verdiere, L.; Mourcin, F.; Tarte, K. Microenvironment signaling driving lymphomagenesis. Curr. Opin. Hematol. 2018, 25, 335–345. [Google Scholar] [CrossRef]
- Menter, T.; Tzankov, A. Lymphomas and Their Microenvironment: A Multifaceted Relationship. Pathobiology 2019, 86, 225–236. [Google Scholar] [CrossRef]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Bu, L.; Baba, H.; Yoshida, N.; Miyake, K.; Yasuda, T.; Uchihara, T.; Tan, P.; Ishimoto, T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 2019, 38, 4887–4901. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019, 178, 160–175.e127. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e816. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef]
- Vicario, M.; Mattiolo, A.; Montini, B.; Piano, M.A.; Cavallari, I.; Amadori, A.; Chieco-Bianchi, L.; Calabro, M.L. A Preclinical Model for the ATLL Lymphoma Subtype With Insights Into the Role of Microenvironment in HTLV-1-Mediated Lymphomagenesis. Front. Microbiol. 2018, 9, 1215. [Google Scholar] [CrossRef] [PubMed]
- Calabro, M.L.; Gasperini, P.; Di Gangi, I.M.; Indraccolo, S.; Barbierato, M.; Amadori, A.; Chieco-Bianchi, L. Antineoplastic activity of lentiviral vectors expressing interferon-alpha in a preclinical model of primary effusion lymphoma. Blood 2009, 113, 4525–4533. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Hayashi, A.; Utsunomiya, A.; Nukada, Y.; Fukui, R.; Itoh, K.; Tezuka, K.; Ohashi, K.; Mizuno, K.; Sakamoto, M.; et al. Alteration of phosphatidylinositol 3-kinase cascade in the multilobulated nuclear formation of adult T cell leukemia/lymphoma (ATLL). Proc. Natl. Acad. Sci. USA 2005, 102, 15213–15218. [Google Scholar] [CrossRef] [Green Version]
- Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Yang, Y.; Kuwayama, Y.; Adachi, Y.; Takeuchi, T.; Koeffler, H.P.; Taguchi, H. Longitudinal inhibition of PI3K/Akt/mTOR signaling by LY294002 and rapamycin induces growth arrest of adult T-cell leukemia cells. Leuk. Res. 2007, 31, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Katsuya, H.; Cook, L.B.M.; Rowan, A.G.; Satou, Y.; Taylor, G.P.; Bangham, C.R.M. Phosphatidylinositol 3-kinase-delta (PI3K-delta) is a potential therapeutic target in adult T-cell leukemia-lymphoma. Biomark Res. 2018, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafin, V.; Lissandron, V.; Buldini, B.; Bresolin, S.; Paganin, M.; Grillo, F.; Andriano, N.; Palmi, C.; Cazzaniga, G.; Marmiroli, S.; et al. Phosphoproteomic analysis reveals hyperactivation of mTOR/STAT3 and LCK/Calcineurin axes in pediatric early T-cell precursor ALL. Leukemia 2017, 31, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Douville, J.; Beaulieu, R.; Balicki, D. ALDH1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev. 2009, 18, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Ma, J.; Cai, Z.; Wei, H.; Liu, X.; Zhao, Q.; Zhang, T. The anti-tumor effect of aspirin: What we know and what we expect. Biomed. Pharmacother 2017, 95, 656–661. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Bertacchini, J.; Toker, A.; Marmiroli, S. Cross-talk between the CK2 and AKT signaling pathways in cancer. Adv. Biol. Regul. 2017, 64, 1–8. [Google Scholar] [CrossRef]
- Bertacchini, J.; Mediani, L.; Beretti, F.; Guida, M.; Ghalali, A.; Brugnoli, F.; Bertagnolo, V.; Petricoin, E.; Poti, F.; Arioli, J.; et al. Clusterin enhances AKT2-mediated motility of normal and cancer prostate cells through a PTEN and PHLPP1 circuit. J. Cell Physiol. 2019, 234, 11188–11199. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, M.; Schmitz, R.; Xiao, W.; Goldman, C.K.; Xu, W.; Yang, Y.; Yu, X.; Waldmann, T.A.; Staudt, L.M. Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. J. Exp. Med. 2014, 211, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Braglia, L.; Zavatti, M.; Vinceti, M.; Martelli, A.M.; Marmiroli, S. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target? Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118731. [Google Scholar] [CrossRef]
- Hillmann, P.; Fabbro, D. PI3K/mTOR Pathway Inhibition: Opportunities in Oncology and Rare Genetic Diseases. Int. J. Mol. Sci. 2019, 20, 5792. [Google Scholar] [CrossRef] [Green Version]
- Mediani, L.; Gibellini, F.; Bertacchini, J.; Frasson, C.; Bosco, R.; Accordi, B.; Basso, G.; Bonora, M.; Calabro, M.L.; Mattiolo, A.; et al. Reversal of the glycolytic phenotype of primary effusion lymphoma cells by combined targeting of cellular metabolism and PI3K/Akt/ mTOR signaling. Oncotarget 2016, 7, 5521–5537. [Google Scholar] [CrossRef]
- Bertacchini, J.; Frasson, C.; Chiarini, F.; D’Avella, D.; Accordi, B.; Anselmi, L.; Barozzi, P.; Forghieri, F.; Luppi, M.; Martelli, A.M.; et al. Dual inhibition of PI3K/mTOR signaling in chemoresistant AML primary cells. Adv. Biol. Regul. 2018, 68, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, C.; Senba, M.; Mori, N. Effects of NVP-BEZ235, a dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, on HTLV-1-infected T-cell lines. Oncol. Lett. 2018, 15, 5311–5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, M.D.; Carter, M.J.; Larrayoz, M.; Smith, L.D.; Aguilar-Hernandez, M.; Cox, K.L.; Tipton, T.; Reynolds, M.; Murphy, S.; Lemm, E.; et al. The PI3K/mTOR inhibitor PF-04691502 induces apoptosis and inhibits microenvironmental signaling in CLL and the Emicro-TCL1 mouse model. Blood 2015, 125, 4032–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Shigemori, K.; Donaldson, N.; Trevisani, C.; Cordero, N.A.; Stevenson, K.E.; Bu, X.; Arakawa, F.; Takeuchi, M.; Ohshima, K.; et al. Genomic landscape of young ATLL patients identifies frequent targetable CD28 fusions. Blood 2020, 135, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Guida, M.; Accordi, B.; Mediani, L.; Martelli, A.M.; Barozzi, P.; Petricoin, E., 3rd; Liotta, L.; Milani, G.; Giordan, M.; et al. Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia 2014, 28, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Darici, S.; Alkhaldi, H.; Horne, G.; Jorgensen, H.G.; Marmiroli, S.; Huang, X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Wright, S.C.E.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P.J.A. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538. [Google Scholar] [CrossRef]
- Mukherjee, R.; Vanaja, K.G.; Boyer, J.A.; Gadal, S.; Solomon, H.; Chandarlapaty, S.; Levchenko, A.; Rosen, N. Regulation of PTEN translation by PI3K signaling maintains pathway homeostasis. Mol. Cell 2021, 81, 708–723.e705. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, R.R. PI3K in stemness regulation: From development to cancer. Biochem. Soc. Trans. 2020, 48, 301–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, B.; Wang, J.; Li, J.; Gong, Y.; Li, S.; Wang, C.; Cui, B.; Xue, X.; Yang, M.; et al. Reduction of NANOG Mediates the Inhibitory Effect of Aspirin on Tumor Growth and Stemness in Colorectal Cancer. Cell Physiol. Biochem. 2017, 44, 1051–1063. [Google Scholar] [CrossRef]
- Olejniczak-Keder, A.; Szarynska, M.; Wronska, A.; Siedlecka-Kroplewska, K.; Kmiec, Z. Effects of 5-FU and anti-EGFR antibody in combination with ASA on the spherical culture system of HCT116 and HT29 colorectal cancer cell lines. Int. J. Oncol. 2019, 55, 223–242. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Liao, T.T.; Yang, M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Joseph, J.V.; Kruyt, F.A.E. EMT- and MET-related processes in nonepithelial tumors: Importance for disease progression, prognosis, and therapeutic opportunities. Mol. Oncol. 2017, 11, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Liao, T.T.; Yang, M.H. Emerging roles of epithelial-mesenchymal transition in hematological malignancies. J. Biomed. Sci. 2018, 25, 37. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, D.; Nakagawa, S.; Hori, M.; Kurokawa, N.; Soejima, A.; Nakano, K.; Yamochi, T.; Nakashima, M.; Kobayashi, S.; Tanaka, Y.; et al. Polycomb-dependent epigenetic landscape in adult T-cell leukemia. Blood 2016, 127, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, S.; Yamazaki, S.; Nakauchi, H.; Morishita, K. Downregulation of ZEB1 and overexpression of Smad7 contribute to resistance to TGF-beta1-mediated growth suppression in adult T-cell leukemia/lymphoma. Oncogene 2010, 29, 4157–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykie, A.; Wijesinghe, T.; Rabson, A.B.; Madugula, K.; Farinas, C.; Wilson, S.; Abraham, D.; Jain, P. Human T-cell Leukemia Virus Type 1 and Strongyloides stercoralis: Partners in Pathogenesis. Pathogens 2020, 9, 904. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Bosetti, C.; Santucci, C.; Gallus, S.; Martinetti, M.; La Vecchia, C. Aspirin and the risk of colorectal and other digestive tract cancers: An updated meta-analysis through 2019. Ann. Oncol. 2020, 31, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.D.; Chen, W.Y.; Li, L.; Hertzmark, E.; Spiegelman, D.; Hankinson, S.E. Aspirin intake and survival after breast cancer. J. Clin. Oncol. 2010, 28, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Liebow, M.; Larson, M.C.; Thompson, C.A.; Nowakowski, G.S.; Call, T.G.; Macon, W.R.; Kay, N.E.; Habermann, T.M.; Slager, S.L.; Cerhan, J.R. Aspirin and other nonsteroidal anti-inflammatory drugs, statins and risk of non-Hodgkin lymphoma. Int. J. Cancer 2021, 149, 535–545. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Pillinger, M.H.; Capodici, C.; Rosenthal, P.; Kheterpal, N.; Hanft, S.; Philips, M.R.; Weissmann, G. Modes of action of aspirin-like drugs: Salicylates inhibit erk activation and integrin-dependent neutrophil adhesion. Proc. Natl. Acad. Sci. USA 1998, 95, 14540–14545. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Mukherjee, S.; Khan, P.; Banerjee, S.; Dutta, A.; Banerjee, N.; Sengupta, D.; Basak, U.; Chakraborty, S.; Dutta, A.; et al. SMAR1 repression by pluripotency factors and consequent chemoresistance in breast cancer stem-like cells is reversed by aspirin. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Saha, S.; Mukherjee, S.; Khan, P.; Kajal, K.; Mazumdar, M.; Manna, A.; Mukherjee, S.; De, S.; Jana, D.; Sarkar, D.K.; et al. Aspirin Suppresses the Acquisition of Chemoresistance in Breast Cancer by Disrupting an NFkappaB-IL6 Signaling Axis Responsible for the Generation of Cancer Stem Cells. Cancer Res. 2016, 76, 2000–2012. [Google Scholar] [CrossRef] [Green Version]
- Khan, P.; Manna, A.; Saha, S.; Mohanty, S.; Mukherjee, S.; Mazumdar, M.; Guha, D.; Das, T. Aspirin inhibits epithelial-to-mesenchymal transition and migration of oncogenic K-ras-expressing non-small cell lung carcinoma cells by down-regulating E-cadherin repressor Slug. BMC Cancer 2016, 16, 39. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Wu, X. Aspirin inhibits colon cancer cell line migration through regulating epithelial-mesenchymal transition via Wnt signaling. Oncol. Lett. 2019, 17, 4675–4682. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Zhou, H.Y.; Liu, P.; You, Q.; Kuang, F.; Shen, Y.N.; Hu, Z.Q. Aspirin inhibited the metastasis of colon cancer cells by inhibiting the expression of toll-like receptor 4. Cell Biosci. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Wang, C.H. Aspirin Disrupts the Crosstalk of Angiogenic and Inflammatory Cytokines between 4T1 Breast Cancer Cells and Macrophages. Mediat. Inflamm. 2018, 2018, 6380643. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef] [PubMed]
- Khoo, B.L.; Grenci, G.; Lim, J.S.Y.; Lim, Y.P.; Fong, J.; Yeap, W.H.; Bin Lim, S.; Chua, S.L.; Wong, S.C.; Yap, Y.S.; et al. Low-dose anti-inflammatory combinatorial therapy reduced cancer stem cell formation in patient-derived preclinical models for tumour relapse prevention. Br. J. Cancer 2019, 120, 407–423. [Google Scholar] [CrossRef] [Green Version]
- Page, T.H.; Turner, J.J.; Brown, A.C.; Timms, E.M.; Inglis, J.J.; Brennan, F.M.; Foxwell, B.M.; Ray, K.P.; Feldmann, M. Nonsteroidal anti-inflammatory drugs increase TNF production in rheumatoid synovial membrane cultures and whole blood. J. Immunol. 2010, 185, 3694–3701. [Google Scholar] [CrossRef] [Green Version]
- Cenni, V.; Bertacchini, J.; Beretti, F.; Lattanzi, G.; Bavelloni, A.; Riccio, M.; Ruzzene, M.; Marin, O.; Arrigoni, G.; Parnaik, V.; et al. Lamin A Ser404 is a nuclear target of Akt phosphorylation in C2C12 cells. J. Proteome Res. 2008, 7, 4727–4735. [Google Scholar] [CrossRef]
- Bertacchini, J.; Beretti, F.; Cenni, V.; Guida, M.; Gibellini, F.; Mediani, L.; Marin, O.; Maraldi, N.M.; de Pol, A.; Lattanzi, G.; et al. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 2013, 27, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Piano, M.A.; Brunello, A.; Cappellesso, R.; Del Bianco, P.; Mattiolo, A.; Fritegotto, C.; Montini, B.; Zamuner, C.; Del Fiore, P.; Rastrelli, M.; et al. Periostin and Epithelial-Mesenchymal Transition Score as Novel Prognostic Markers for Leiomyosarcoma, Myxofibrosarcoma, and Undifferentiated Pleomorphic Sarcoma. Clin. Cancer Res. 2020, 26, 2921–2931. [Google Scholar] [CrossRef] [Green Version]
- Lignitto, L.; Mattiolo, A.; Negri, E.; Persano, L.; Gianesello, L.; Chieco-Bianchi, L.; Calabro, M.L. Crosstalk between the mesothelium and lymphomatous cells: Insight into the mechanisms involved in the progression of body cavity lymphomas. Cancer Med. 2014, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, D.; Senbanjo, L.; Majumdar, S.; Franklin, R.B.; Chellaiah, M.A. Androgen receptor expression reduces stemness characteristics of prostate cancer cells (PC3) by repression of CD44 and SOX2. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | Description 1 | p-Value 2 | N-Path 3 | N-Sign 4 |
|---|---|---|---|---|
| hsa04144 | Endocytosis | 1.55 × 10−10 | 255 | 92 |
| hsa04512 | ECM-receptor interaction | 1.23 × 10−8 | 103 | 45 |
| hsa04510 | Focal adhesion | 8.55 × 10−8 | 222 | 76 |
| hsa04151 | PI3K/Akt signalling pathway | 6.73 × 10−6 | 366 | 105 |
| hsa04020 | Calcium signalling pathway | 8.06 × 10−5 | 191 | 59 |
| hsa03040 | Spliceosome | 0.000838 | 169 | 50 |
| hsa05340 | Primary immunodeficiency | 0.00218 | 51 | 19 |
| hsa04612 | Antigen processing and presentation | 0.00219 | 219 | 60 |
| hsa04110 | Cell cycle | 0.00231 | 126 | 38 |
| hsa04010 | MAPK signalling pathway | 0.0153 | 284 | 70 |
| hsa04514 | Cell adhesion molecules (CAMs) | 0.0168 | 253 | 63 |
| hsa04660 | T cell receptor signalling pathway | 0.0172 | 115 | 32 |
| hsa05200 | Pathways in cancer | 0.0203 | 334 | 80 |
| hsa04115 | p53 signalling pathway | 0.0483 | 67 | 19 |
| hsa04370 | VEGF signalling pathway | 0.0483 | 67 | 19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigotto, G.; Montini, B.; Mattiolo, A.; Lazzari, N.; Piano, M.A.; Remondini, D.; Marmiroli, S.; Bertacchini, J.; Chieco-Bianchi, L.; Calabrò, M.L. Mechanisms Involved in the Promoting Activity of Fibroblasts in HTLV-1-Mediated Lymphomagenesis: Insights into the Plasticity of Lymphomatous Cells. Int. J. Mol. Sci. 2021, 22, 10562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910562
Rigotto G, Montini B, Mattiolo A, Lazzari N, Piano MA, Remondini D, Marmiroli S, Bertacchini J, Chieco-Bianchi L, Calabrò ML. Mechanisms Involved in the Promoting Activity of Fibroblasts in HTLV-1-Mediated Lymphomagenesis: Insights into the Plasticity of Lymphomatous Cells. International Journal of Molecular Sciences. 2021; 22(19):10562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910562
Chicago/Turabian StyleRigotto, Giulia, Barbara Montini, Adriana Mattiolo, Nayana Lazzari, Maria Assunta Piano, Daniel Remondini, Sandra Marmiroli, Jessika Bertacchini, Luigi Chieco-Bianchi, and Maria Luisa Calabrò. 2021. "Mechanisms Involved in the Promoting Activity of Fibroblasts in HTLV-1-Mediated Lymphomagenesis: Insights into the Plasticity of Lymphomatous Cells" International Journal of Molecular Sciences 22, no. 19: 10562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910562