
Preclinical Evaluation of Sodium Selenite in Mice: Toxicological and Tumor Regression Studies after Striatum Implantation of Human Glioblastoma Stem Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. In Vitro Study
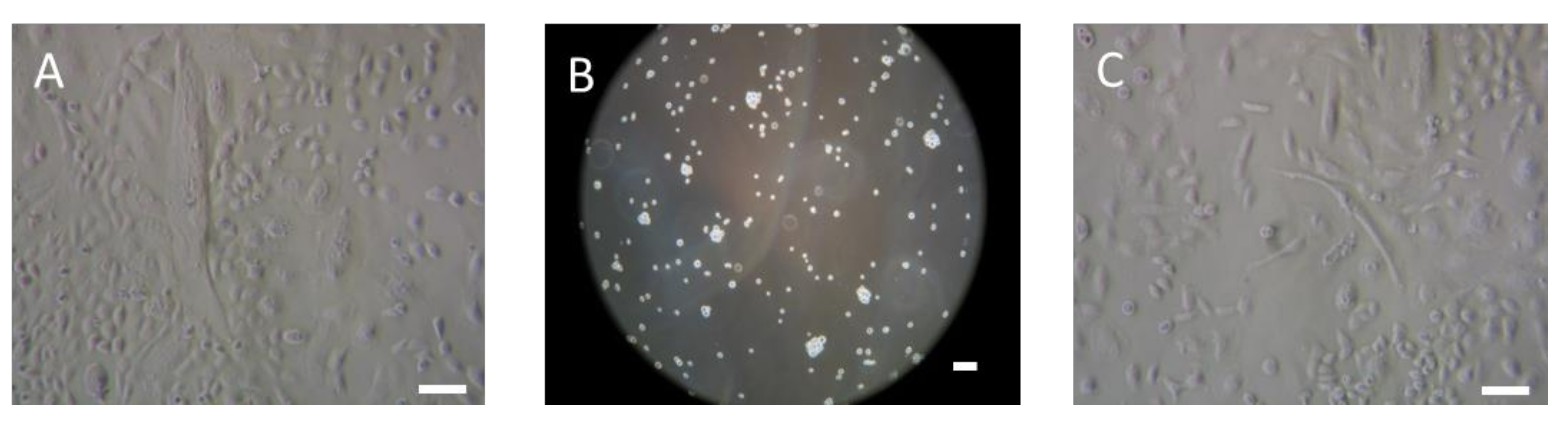
2.1.1. R2J-GS Characteristics and Plasticity in Culture
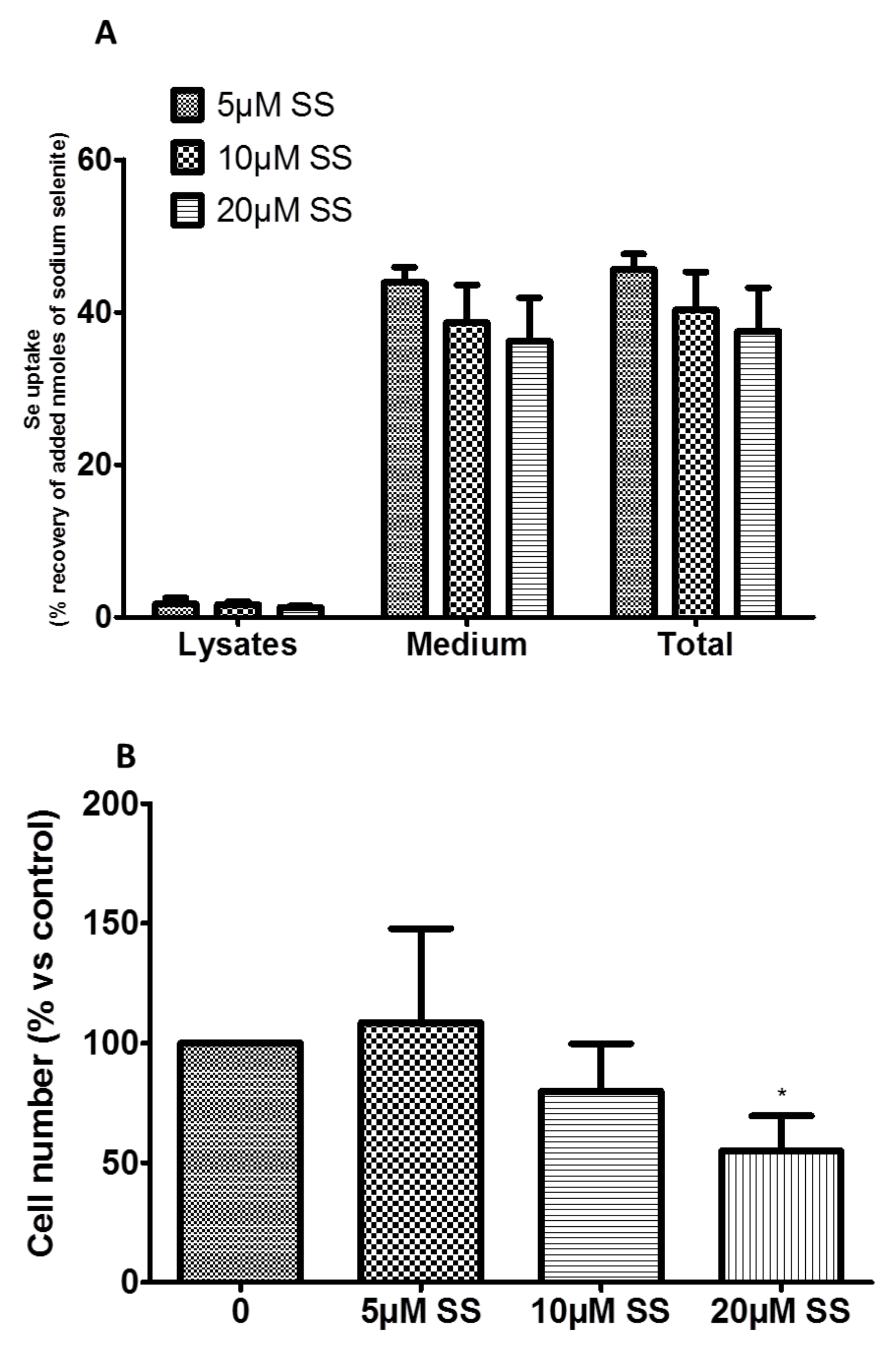
2.1.2. Se Was Uptaken and Metabolized in Se(0) by R2J-GS
2.1.3. SS Decreased Cell Invasion and Induced Both Apoptosis and Necrosis in R2J-GS Cells
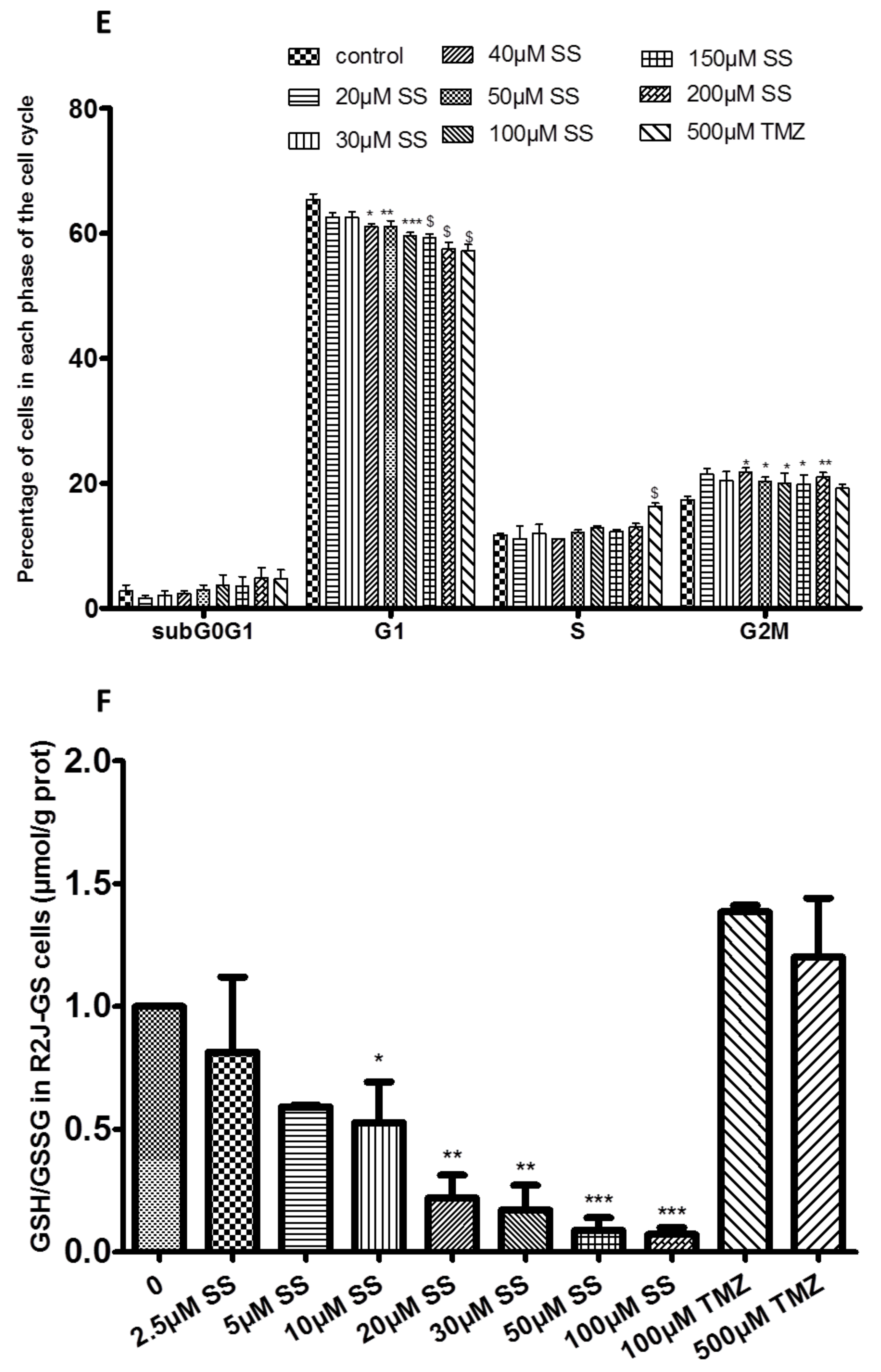
2.1.4. SS Induced Cell Cycle Arrest in R2J Spheres
2.1.5. SS Induced Oxidative Stress in R2J Spheres
2.2. In Vivo Study: Evaluation of SS Toxicity
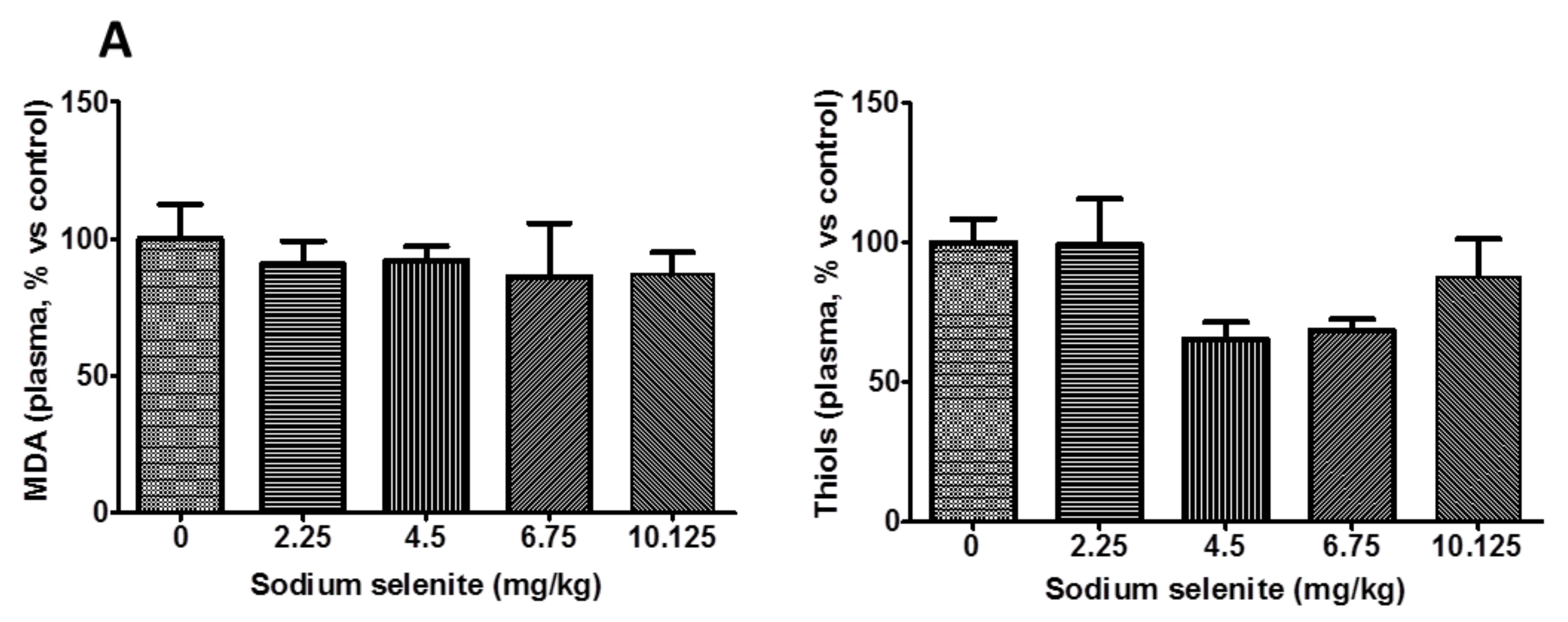
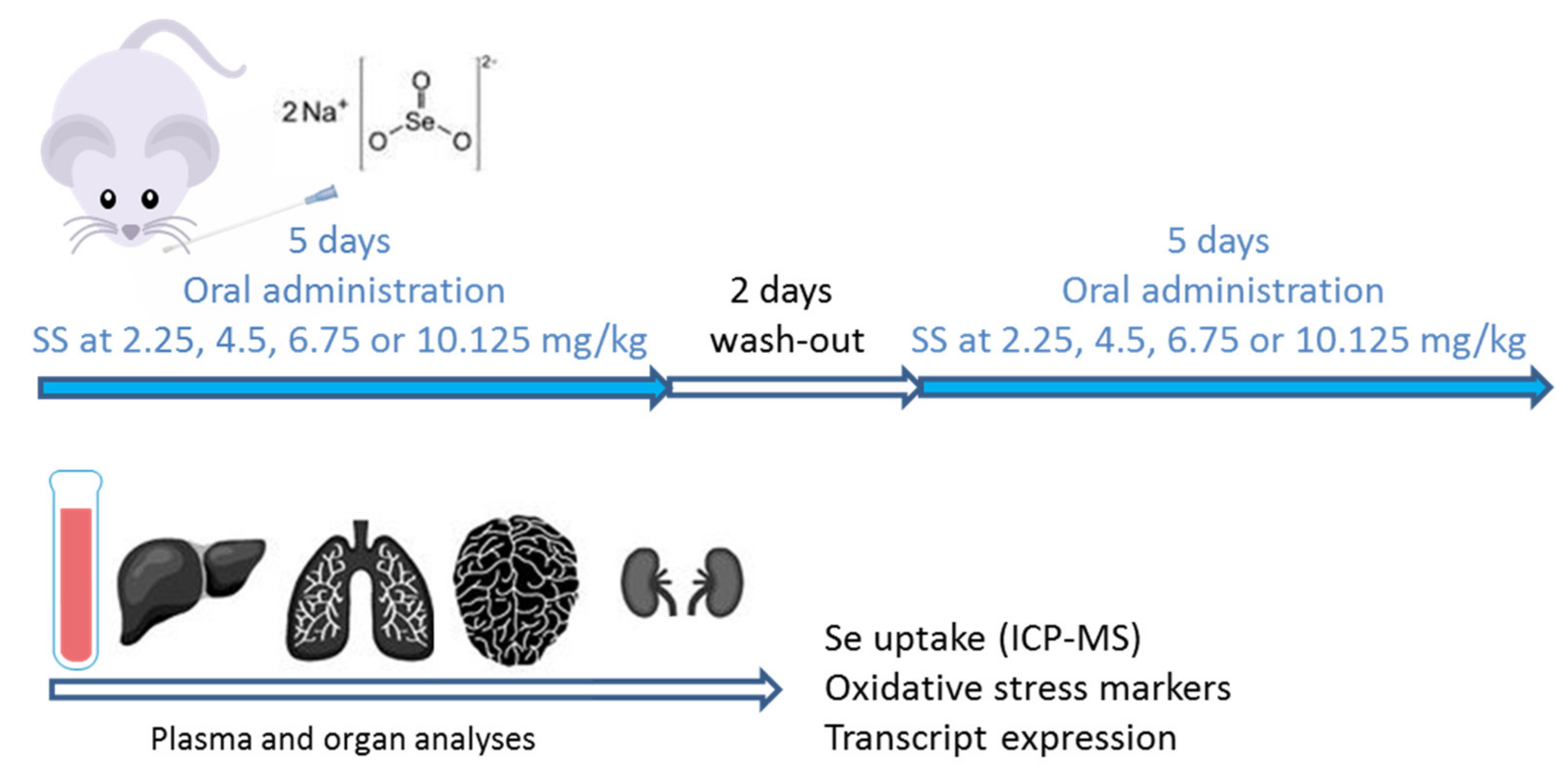
2.2.1. Until 6.75 mg/kg, the Mice Did Not Exhibit Signs of Toxicity
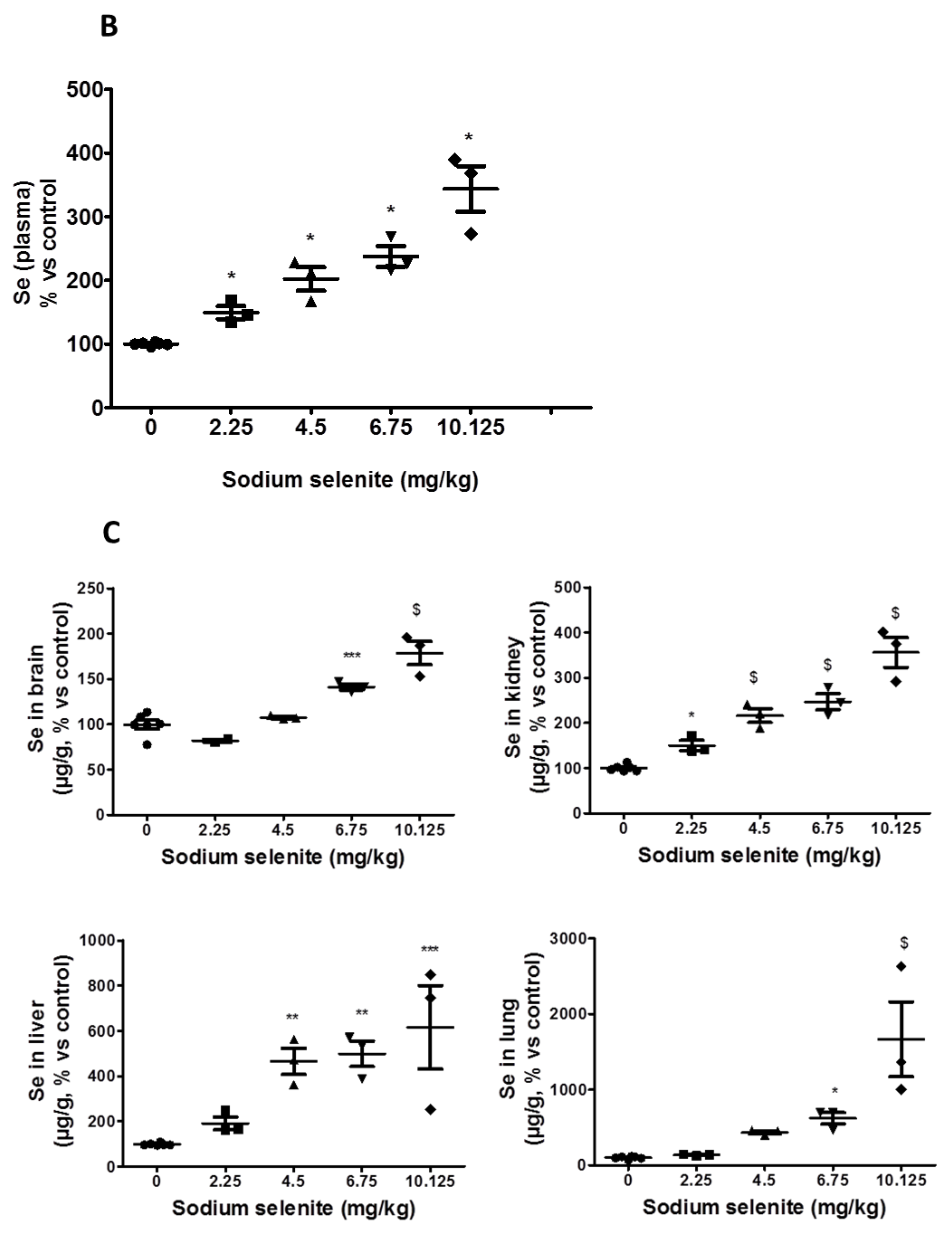
2.2.2. Se in Plasma and Organs Were Found at Different Levels
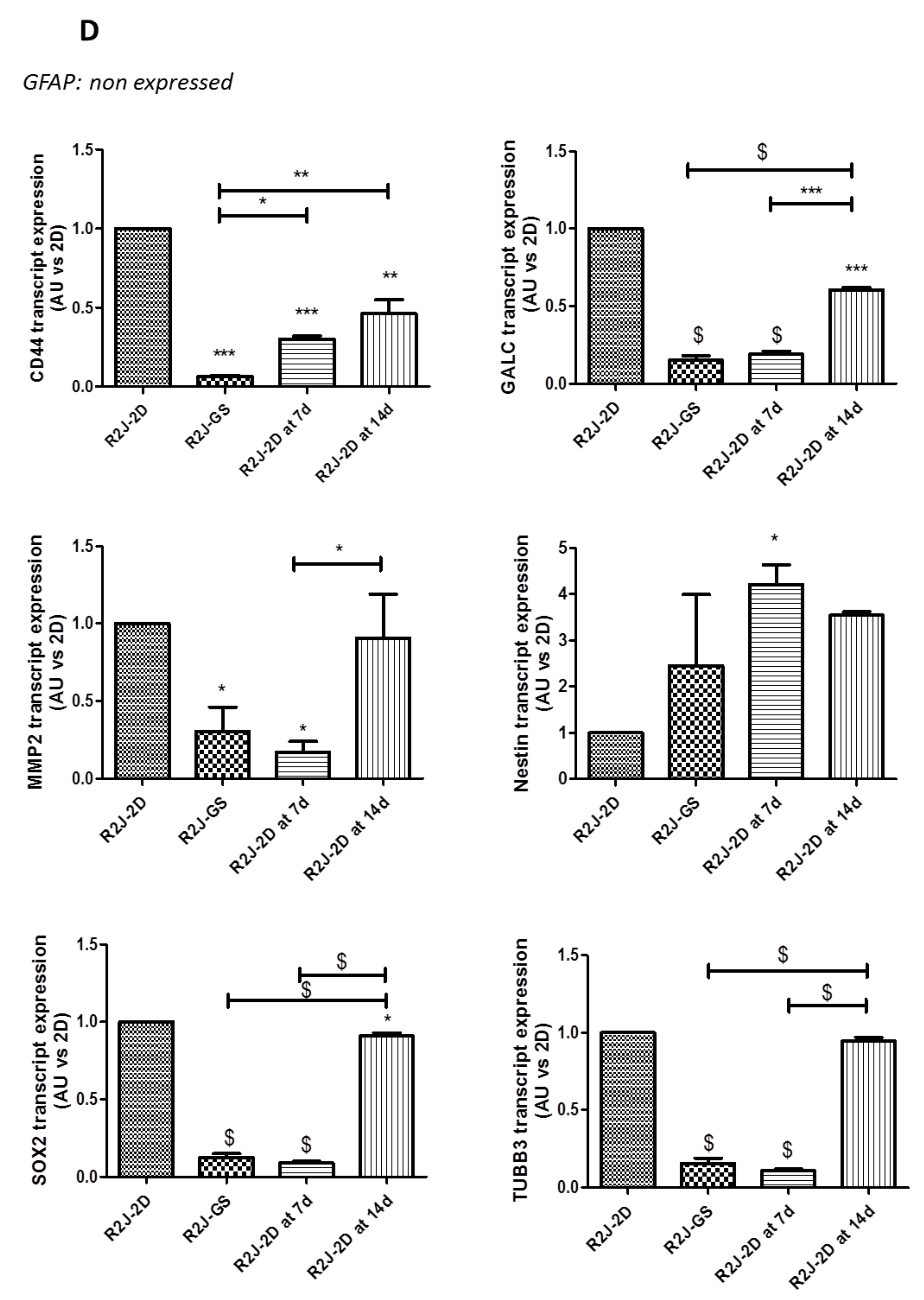
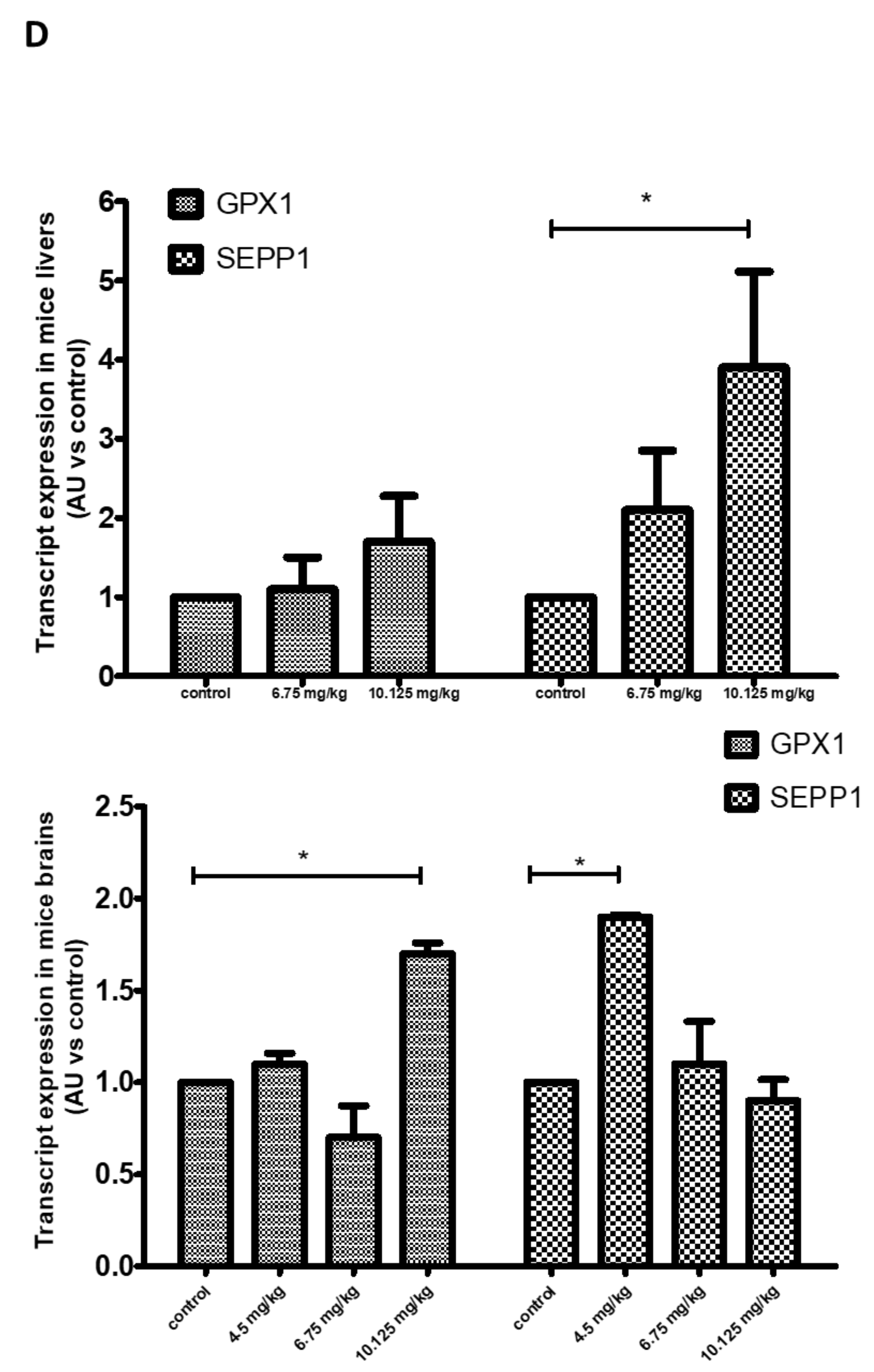
2.2.3. Transcript Expression in SS-Supplemented Mice
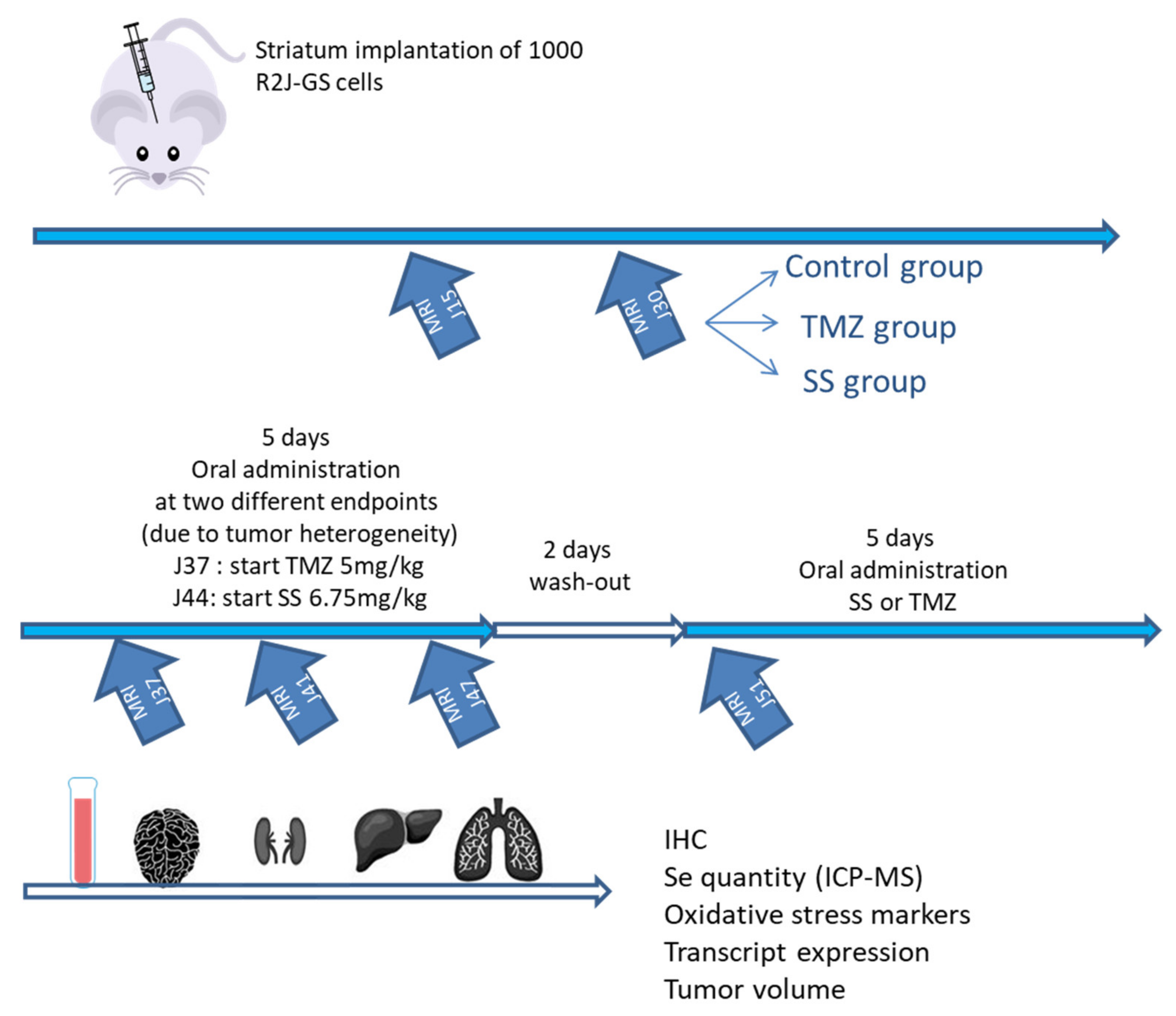
2.3. In Vivo Study: Tumor Regression Assay
2.3.1. Evolution of Neuronal and Stem Cell Markers after SS and TMZ Treatment in Mice Implanted with R2J-GS Cells
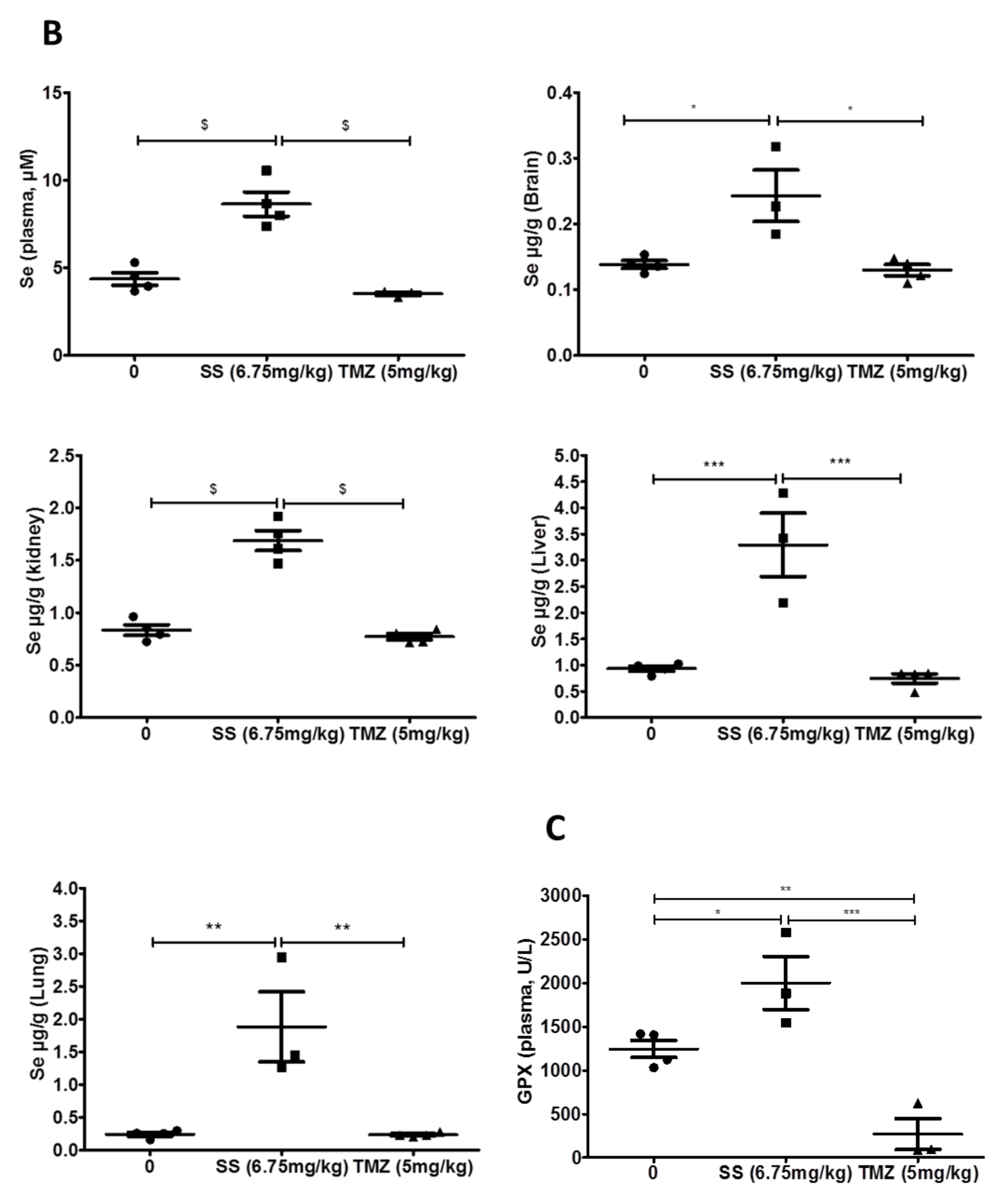
2.3.2. Increase in Se in Plasma and Organs of Mice Receiving SS Orally
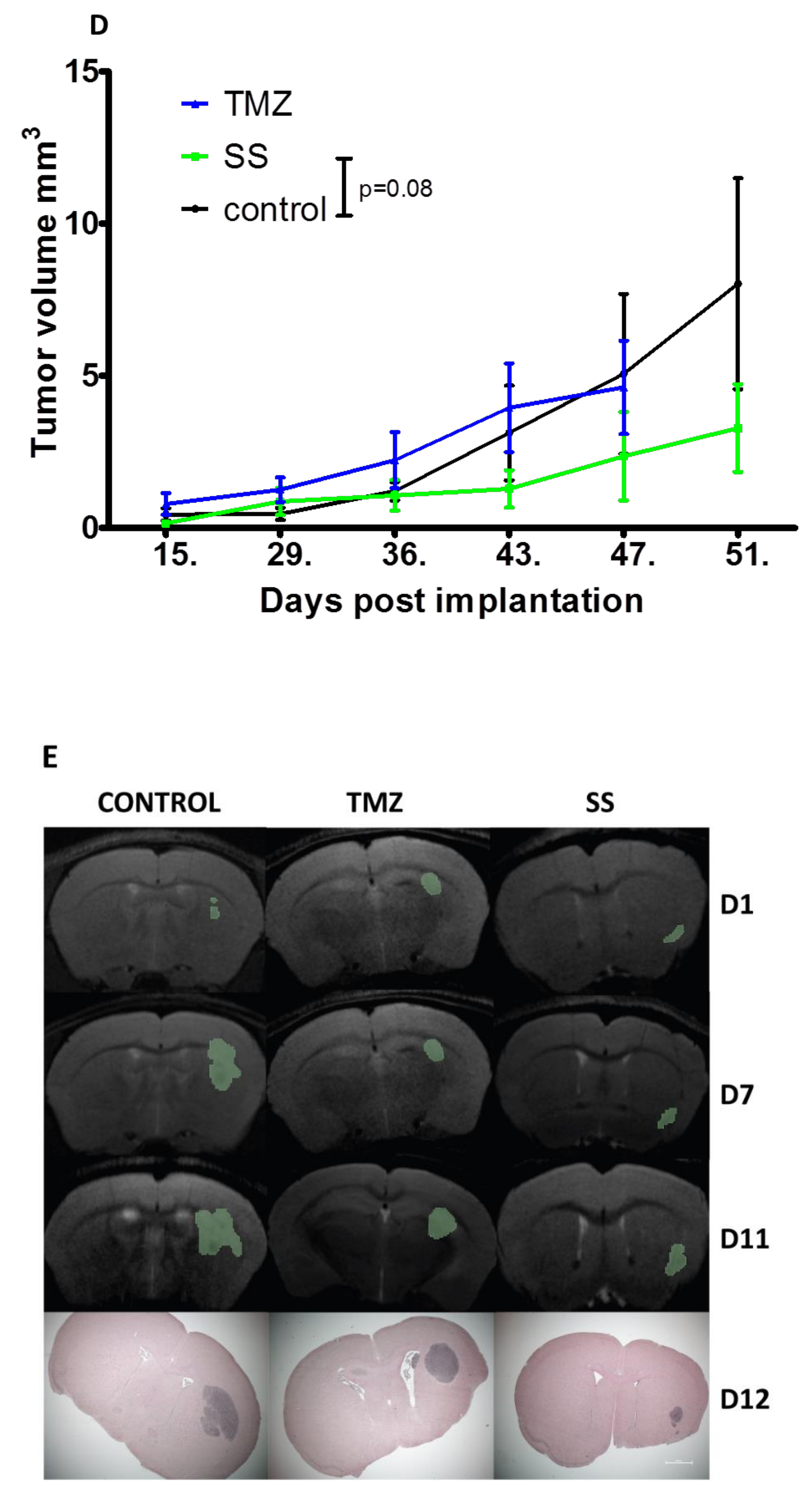
2.3.3. Evolution of the Xenograft Tumor as a Function of the Treatment (SS vs. TMZ): Clinical Responses
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Selenium Uptake
4.3. Dosage of Proteins
4.4. Se(0) Measurement
4.5. Real-Time PCR
4.6. Quantitative Determination of Oxidized (GSSG) and Reduced (GSH) Glutathione Levels
4.7. Immunohistochemistry
4.8. Cellular Invasion Assay
4.9. Flow Cytometry
4.10. Soft Agar Cell Culture to Test Secondary Sphere Formation after SS and TMZ Treatments
4.11. In Vivo Experiments
4.12. Delimitation of SS Toxic Doses
4.13. Tissue Preparations
4.14. Determination of Malondialdehyde (MDA)
4.15. GPX1 Determination
4.16. Thiol Group Evaluation
4.17. Striatum R2J-GS Xenograft into Nude Mice and Treatment with SS or TMZ
4.18. Magnetic Resonance Imaging (MRI)
4.19. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Chow, L.M.L.; Endersby, R.; Zhu, X.; Rankin, S.; Qu, C.; Zhang, J.; Broniscer, A.; Ellison, D.W.; Baker, S.J. Cooperativity within and among Pten, P53, and Rb Pathways Induces High-Grade Astrocytoma in Adult Brain. Cancer Cell 2011, 19, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Fan, X.; Xiong, Y.; Wang, Y. A Reignited Debate over the Cell(s) of Origin for Glioblastoma and Its Clinical Implications. Front. Med. 2019, 13, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Berthier, S.; Larrouquère, L.; Champelovier, P.; Col, E.; Lefebvre, C.; Cottet-Rouselle, C.; Arnaud, J.; Garrel, C.; Laporte, F.; Boutonnat, J.; et al. A New Patient-Derived Metastatic Glioblastoma Cell Line: Characterisation and Response to Sodium Selenite Anticancer Agent. Cancers 2018, 11, E12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Brescia, P.; Richichi, C.; Pelicci, G. Current Strategies for Identification of Glioma Stem Cells: Adequate or Unsatisfactory? J. Oncol. 2012, 2012, 376894. [Google Scholar] [CrossRef] [Green Version]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, X.; Chen, Y.-S.; Chen, F.-R.; Xi, S.-Y.; Chen, Z.-P. Glioblastoma Stem Cell Differentiation into Endothelial Cells Evidenced through Live-Cell Imaging. Neuro-Oncol. 2017, 19, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Neradil, J.; Veselska, R. Nestin as a Marker of Cancer Stem Cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, A.E.; Lee, N.; Matthew-Onabanjo, A.N.; Spears, M.E.; Park, S.J.; Youkana, D.; Doshi, M.B.; Peppers, A.; Li, R.; Joseph, A.B.; et al. Selenium Detoxification Is Required for Cancer-Cell Survival. Nat. Metab. 2020, 2, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Brodin, O.; Eksborg, S.; Wallenberg, M.; Asker-Hagelberg, C.; Larsen, E.H.; Mohlkert, D.; Lenneby-Helleday, C.; Jacobsson, H.; Linder, S.; Misra, S.; et al. Pharmacokinetics and Toxicity of Sodium Selenite in the Treatment of Patients with Carcinoma in a Phase I Clinical Trial: The SECAR Study. Nutrients 2015, 7, 4978–4994. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Yang, Y.; Huang, F.; Li, F.; Jiang, Q.; Shi, K.; Xu, C. Selenite Induces Apoptosis in Colorectal Cancer Cells via AKT-Mediated Inhibition of β-Catenin Survival Axis. Cancer Lett. 2012, 315, 78–85. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Shi, K.J.; Wei, W.; Hua, F.Y.; Ci, Y.L.; Jiang, Q.; Li, F.; Wu, P.; Hui, K.Y.; Yang, Y.; et al. The ROS/JNK/ATF2 Pathway Mediates Selenite-Induced Leukemia NB4 Cell Cycle Arrest and Apoptosis In Vitro and In Vivo. Cell Death Dis. 2013, 4, e973. [Google Scholar] [CrossRef]
- Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT Testing—The Challenges for Biomarker-Based Glioma Treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Lunøe, K.; Gabel-Jensen, C.; Stürup, S.; Andresen, L.; Skov, S.; Gammelgaard, B. Investigation of the Selenium Metabolism in Cancer Cell Lines. Met. Integr. Biomet. Sci. 2011, 3, 162–168. [Google Scholar] [CrossRef]
- Campos, B.; Gal, Z.; Baader, A.; Schneider, T.; Sliwinski, C.; Gassel, K.; Bageritz, J.; Grabe, N.; von Deimling, A.; Beckhove, P.; et al. Aberrant Self-Renewal and Quiescence Contribute to the Aggressiveness of Glioblastoma. J. Pathol. 2014, 234, 23–33. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, H.-J.; Jung, C.-W.; Choi, B.-I.; Lee, D.-H.; Park, M.-J. PARK7 Maintains the Stemness of Glioblastoma Stem Cells by Stabilizing Epidermal Growth Factor Receptor Variant III. Oncogene 2021, 40, 508–521. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Platet, N.; Liu, S.Y.; Atifi, M.E.; Oliver, L.; Vallette, F.M.; Berger, F.; Wion, D. Influence of Oxygen Tension on CD133 Phenotype in Human Glioma Cell Cultures. Cancer Lett. 2007, 258, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor Stem Cells Derived from Glioblastomas Cultured in BFGF and EGF More Closely Mirror the Phenotype and Genotype of Primary Tumors than Do Serum-Cultured Cell Lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Bourseau-Guilmain, E.; Lemaire, L.; Griveau, A.; Hervouet, E.; Vallette, F.; Berger, F.; Menei, P.; Benoit, J.-P.; Wion, D.; Garcion, E. In Vitro Expansion of Human Glioblastoma Cells at Non-Physiological Oxygen Tension Irreversibly Alters Subsequent in Vivo Aggressiveness and AC133 Expression. Int. J. Oncol. 2012, 40, 1220–1229. [Google Scholar] [CrossRef]
- Ludwig, K.; Kornblum, H.I. Molecular Markers in Glioma. J. Neurooncol. 2017, 134, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Nagare, R.P.; Sneha, S.; Priya, S.K.; Ganesan, T.S. Cancer Stem Cells—Are Surface Markers Alone Sufficient? Curr. Stem Cell Res. Ther. 2017, 12, 37–44. [Google Scholar] [CrossRef]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The Transcriptional Network for Mesenchymal Transformation of Brain Tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Biswas, S.; Talukder, G.; Sharma, A. Prevention of Cytotoxic Effects of Arsenic by Short-Term Dietary Supplementation with Selenium in Mice in Vivo. Mutat. Res. 1999, 441, 155–160. [Google Scholar] [CrossRef]
- Liu, Y.-R.; Sun, B.; Zhu, G.-H.; Li, W.-W.; Tian, Y.-X.; Wang, L.-M.; Zong, S.-M.; Sheng, P.-Z.; Li, M.; Chen, S.; et al. Selenium-Lentinan Inhibits Tumor Progression by Regulating Epithelial-Mesenchymal Transition. Toxicol. Appl. Pharmacol. 2018, 360, 1–8. [Google Scholar] [CrossRef]
- Li, F.; Jiang, Q.; Shi, K.J.; Luo, H.; Yang, Y.; Xu, C.M. RhoA Modulates Functional and Physical Interaction between ROCK1 and Erk1/2 in Selenite-Induced Apoptosis of Leukaemia Cells. Cell Death Dis. 2013, 4, e708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalieri, R.R.; Scott, K.G. Sodium Selenite Se 75. A More Specific Agent for Scanning Tumors. JAMA 1968, 206, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Loeschner, K.; Hadrup, N.; Hansen, M.; Pereira, S.A.; Gammelgaard, B.; Møller, L.H.; Mortensen, A.; Lam, H.R.; Larsen, E.H. Absorption, Distribution, Metabolism and Excretion of Selenium Following Oral Administration of Elemental Selenium Nanoparticles or Selenite in Rats. Met. Integr. Biomet. Sci. 2014, 6, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Painter, E.P. The Chemistry and Toxicity of Selenium Compounds, with Special Reference to the Selenium Problem. Chem. Rev. 1941, 28, 179–213. [Google Scholar] [CrossRef]
- Hu, M.L.; Tappel, A.L. Selenium as a Sulfhydryl Redox Catalyst and Survey of Potential Selenium-Dependent Enzymes. J. Inorg. Biochem. 1987, 30, 239–248. [Google Scholar] [CrossRef]
- Krittaphol, W.; McDowell, A.; Thomson, C.D.; Mikov, M.; Fawcett, J.P. Biotransformation of L-Selenomethionine and Selenite in Rat Gut Contents. Biol. Trace Elem. Res. 2011, 139, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Lamana, J.; Abad-Alvaro, I.; Bierla, K.; Laborda, F.; Szpunar, J.; Lobinski, R. Detection and Characterization of Biogenic Selenium Nanoparticles in Selenium-Rich Yeast by Single Particle ICP MS. J. Anal. At. Spectrom. 2018, 33, 452–460. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Goltyaev, M.V.; Mal’tseva, V.N.; Turovsky, E.A.; Sarimov, R.M.; Simakin, A.V.; Gudkov, S.V. Mechanisms of the Cytotoxic Effect of Selenium Nanoparticles in Different Human Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 7798. [Google Scholar] [CrossRef]
- Olm, E.; Fernandes, A.P.; Hebert, C.; Rundlöf, A.-K.; Larsen, E.H.; Danielsson, O.; Björnstedt, M. Extracellular Thiol-Assisted Selenium Uptake Dependent on the x(c)- Cystine Transporter Explains the Cancer-Specific Cytotoxicity of Selenite. Proc. Natl. Acad. Sci. USA 2009, 106, 11400–11405. [Google Scholar] [CrossRef] [Green Version]
- Dokic, I.; Hartmann, C.; Herold-Mende, C.; Régnier-Vigouroux, A. Glutathione Peroxidase 1 Activity Dictates the Sensitivity of Glioblastoma Cells to Oxidative Stress. Glia 2012, 60, 1785–1800. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Selenoprotein P-Expression, Functions, and Roles in Mammals. Biochim. Biophys. Acta 2009, 1790, 1441–1447. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.F.; Hill, K.E. Regulation of Selenium Metabolism and Transport. Annu. Rev. Nutr. 2015, 35, 109–134. [Google Scholar] [CrossRef]
- Seboussi, R.; Faye, B.; Alhadrami, G.; Askar, M.; Ibrahim, W.; Mahjoub, B.; Hassan, K.; Moustafa, T.; Elkhouly, A. Selenium Distribution in Camel Blood and Organs after Different Level of Dietary Selenium Supplementation. Biol. Trace Elem. Res. 2010, 133, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Ning, S.; Knox, S.J. Sodium Selenite Radiosensitizes Hormone-Refractory Prostate Cancer Xenograft Tumors but Not Intestinal Crypt Cells in Vivo. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Thamilselvan, V.; Menon, M.; Thamilselvan, S. Combination of Carmustine and Selenite Effectively Inhibits Tumor Growth by Targeting Androgen Receptor, Androgen Receptor-Variants, and Akt in Preclinical Models: New Hope for Patients with Castration Resistant Prostate Cancer. Int. J. Cancer 2016, 139, 1632–1647. [Google Scholar] [CrossRef] [Green Version]
- McConnell, D.; McGreevy, J.; Williams, M.; Litofsky, N. Do Anti-Oxidants Vitamin D3, Melatonin, and Alpha-Lipoic Acid Have Synergistic Effects with Temozolomide on Cultured Glioblastoma Cells? Medicines 2018, 5, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazane-Puch, F.; Champelovier, P.; Arnaud, J.; Trocmé, C.; Garrel, C.; Faure, P.; Laporte, F. Six-Day Selenium Supplementation Led to Either UVA-Photoprotection or Toxic Effects in Human Fibroblasts Depending on the Chemical Form and Dose of Se. Met. Integr. Biomet. Sci. 2014, 6, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Vacchina, V.; Foix, D.; Menta, M.; Martinez, H.; Séby, F. Optimization of Elemental Selenium (Se(0)) Determination in Yeasts by Anion-Exchange HPLC-ICP-MS. Anal. Bioanal. Chem. 2021, 413, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, A.; Overbergh, L.; Valckx, D.; Decallonne, B.; Bouillon, R.; Mathieu, C. An Overview of Real-Time Quantitative PCR: Applications to Quantify Cytokine Gene Expression. Methods 2001, 25, 386–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerboom, T.P.; Sies, H. Assay of Glutathione, Glutathione Disulfide, and Glutathione Mixed Disulfides in Biological Samples. Methods Enzymol. 1981, 77, 373–382. [Google Scholar] [CrossRef]
- Nygren, H.; Eksborg, S. Stability of Temozolomide in Solutions Aimed for Oral Treatment Prepared from a Commercially Available Powder for Infusion. Pharm. Methods 2012, 3, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Flohé, L.; Günzler, W.A. Assays of Glutathione Peroxidase. Methods Enzymol. 1984, 105, 114–121. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larrouquère, L.; Berthier, S.; Chovelon, B.; Garrel, C.; Vacchina, V.; Paucot, H.; Boutonnat, J.; Faure, P.; Hazane-Puch, F. Preclinical Evaluation of Sodium Selenite in Mice: Toxicological and Tumor Regression Studies after Striatum Implantation of Human Glioblastoma Stem Cells. Int. J. Mol. Sci. 2021, 22, 10646. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910646
Larrouquère L, Berthier S, Chovelon B, Garrel C, Vacchina V, Paucot H, Boutonnat J, Faure P, Hazane-Puch F. Preclinical Evaluation of Sodium Selenite in Mice: Toxicological and Tumor Regression Studies after Striatum Implantation of Human Glioblastoma Stem Cells. International Journal of Molecular Sciences. 2021; 22(19):10646. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910646
Chicago/Turabian StyleLarrouquère, Louis, Sylvie Berthier, Benoit Chovelon, Catherine Garrel, Véronique Vacchina, Hugues Paucot, Jean Boutonnat, Patrice Faure, and Florence Hazane-Puch. 2021. "Preclinical Evaluation of Sodium Selenite in Mice: Toxicological and Tumor Regression Studies after Striatum Implantation of Human Glioblastoma Stem Cells" International Journal of Molecular Sciences 22, no. 19: 10646. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910646