Bivalent Genes Targeting of Glioma Heterogeneity and Plasticity

 , , and
, , and 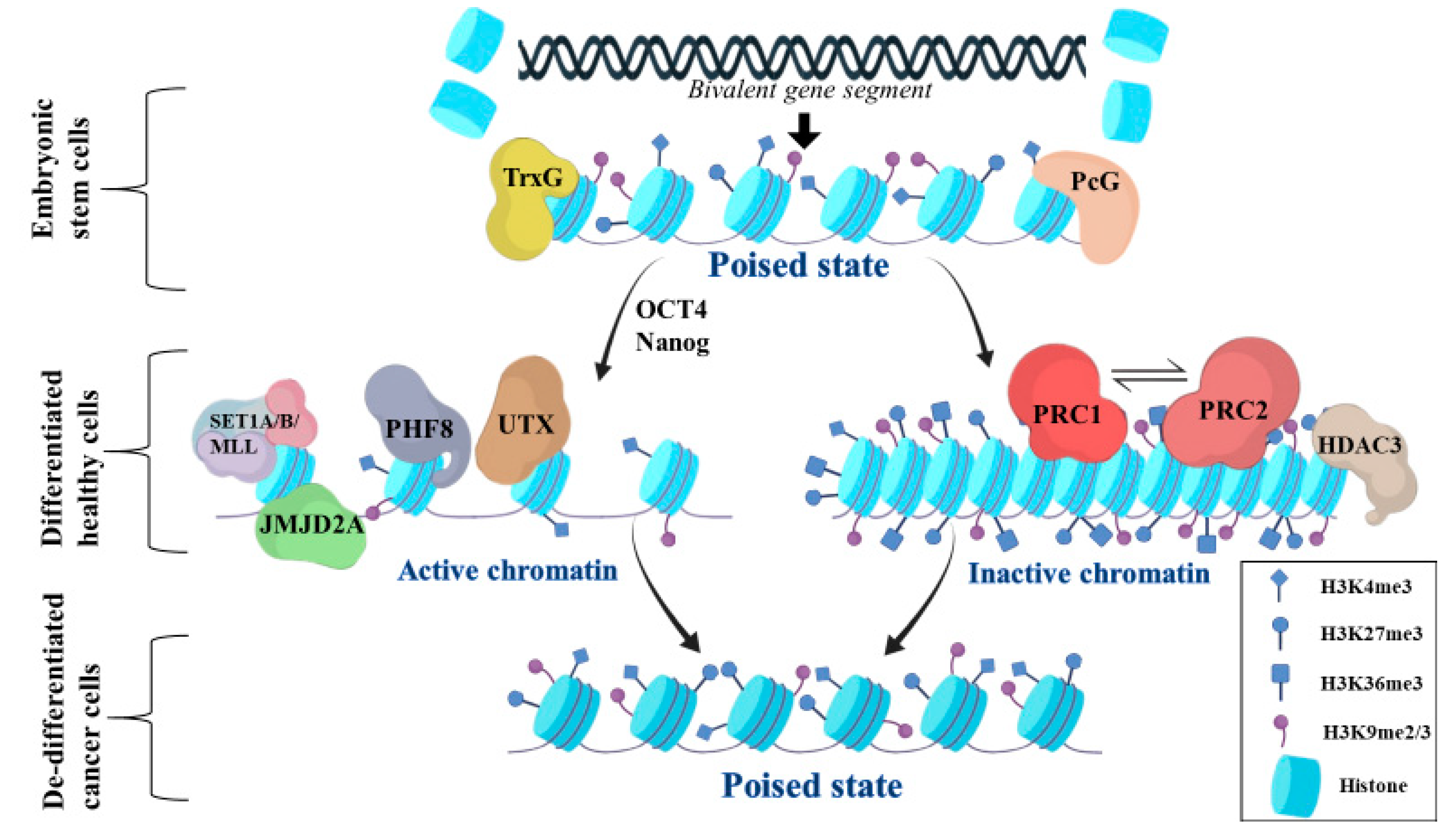{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Aspects of Bivalent Chromatin State
3. Regulation of Bivalent Genes Promoters and Enhancers
3.1. Activation of Bivalent Gene Promoters
3.2. Suppression of Bivalent Gene Promoters
3.3. Regulation of Gene Enhancers
4. Role of Bivalent Genes in Cancer
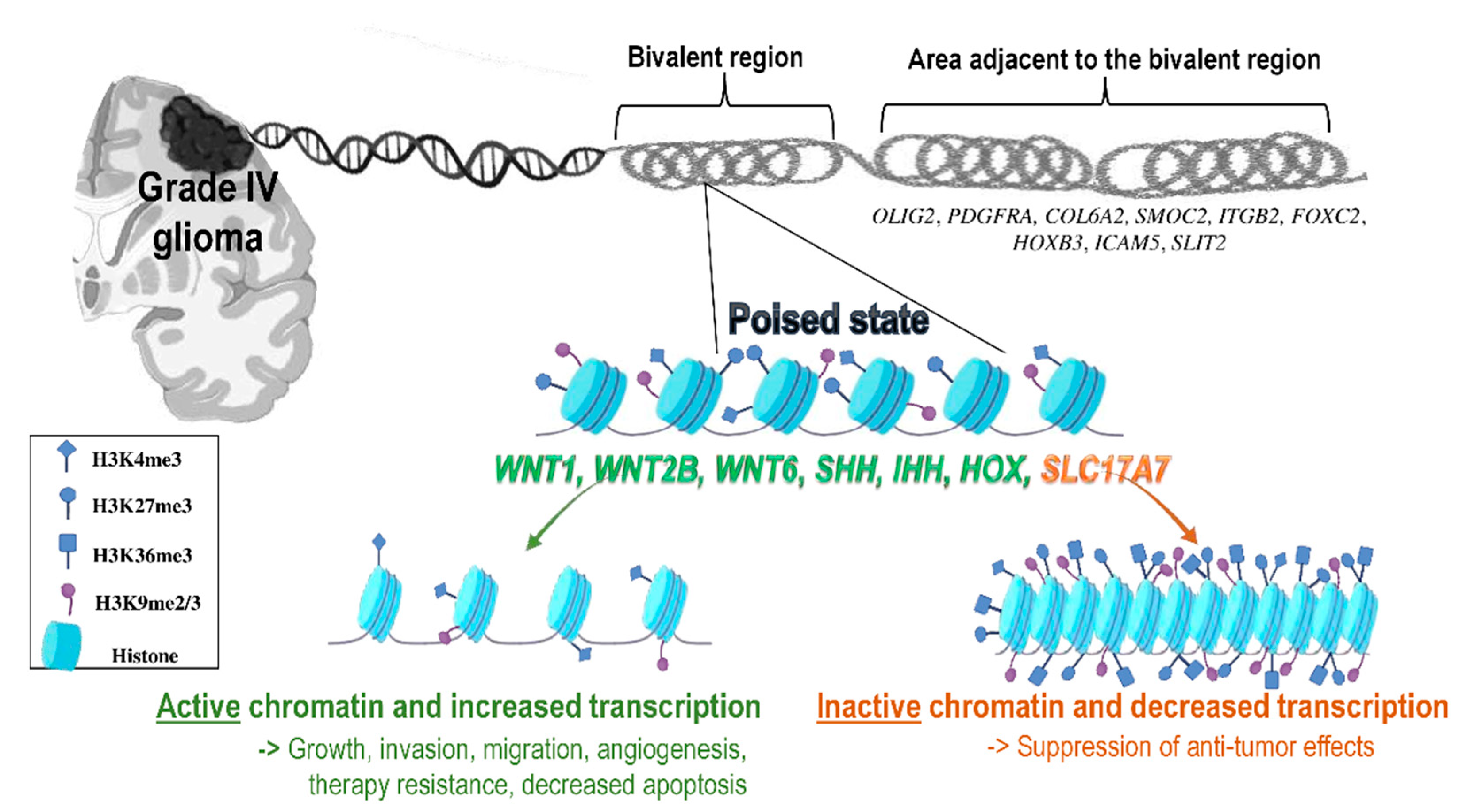
5. Bivalent Genes Regulate Tumor Phenotype in Gliomas
DNA Methylation Confers an Additional Control of Bivalent Gene Expression in Gliomas
6. Targeting Options of Bivalent Genes
7. Conclusions—Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TrxG | Trithorax |
| PcG | Polycomb |
| SET1A/B/MLL | SET Domain Containing 1A, Histone Lysine Methyltransferase/SET Domain Containing 1B |
| PHF8 | PHD Finger Protein 8 |
| JMJD2A | Jumonji Domain Containing 2A |
| UTX | Lysine Demethylase 6A |
| OCT4 | Octamer-binding transcription factor 4 |
| Nanog | Nanog Homeobox |
| PRC2 | Polycomb repressive complex 2 |
| PRC1 | Polycomb repressive complex 1 |
| HDAC3 | Histone deacetylase 3 |
| OLIG2 | Oligodendrocyte Transcription Factor 2 |
| PDGFRA | Platelet Derived Growth Factor Receptor Alpha |
| COL6A2 | Collagen Type VI Alpha 2 Chain |
| SMOC2 | SPARC-related modular calcium-binding protein 2 |
| ITGB2 | Integrin Subunit Beta 2 |
| Forkhead Box C2 | Forkhead Box C2 |
| HOXB3 | Homeobox B3 |
| ICAM5 | Intercellular Adhesion Molecule 5 |
| SLIT2 | Slit Guidance Ligand 2 |
| WNT1 | Wnt Family Member 1 |
| WNT2B | Wnt Family Member 2B |
| WNT6 | Wnt Family Member 6 |
| SHH | Sonic Hedgehog |
| IHH | Indian hedgehog homolog, |
| HOX | Homeobox |
| SLC17A7 | Solute Carrier Family 17 Member 7 |
| H3K4me3 | Histone 3 Lysine 4 trimethylation |
| H3K27me3 | Histone 3 Lysine 27 trimethylation |
| H3K36me3 | Histone 3 Lysine 36 trimethylation |
| H3K9me2/3 | Histone 3 Lysine 9 di- or trimethylation |
References
- Wood, M.D.; Halfpenny, A.M.; Moore, S.R. Applications of molecular neuro-oncology—A review of diffuse glioma integrated diagnosis and emerging molecular entities. Diagn. Pathol. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Yang, W.; Ma, J.; Zhang, J.; Liao, M.; Xu, L.; Xu, M.; Yi, L. Mutant-allele tumor heterogeneity in malignant glioma effectively predicts neoplastic recurrence. Oncol. Lett. 2019, 18, 6108–6116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Cai, J.; Jiang, C. Recent Advances in Targeted Therapy for Glioma. Curr. Med. Chem. 2017, 24, 1365–1381. [Google Scholar] [CrossRef] [PubMed]
- Freese, C.; Takiar, V.; Fouladi, M.; DeWire, M.; Breneman, J.; Pater, L. Radiation and subsequent reirradiation outcomes in the treatment of diffuse intrinsic pontine glioma and a systematic review of the reirradiation literature. Pract. Radiat. Oncol. 2017, 7, 86–92. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro. Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone methyltransferase SETDB1: A common denominator of tumorigenesis with therapeutic potential. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Dabrowski, M.; Wojtas, B. Global DNA Methylation Patterns in Human Gliomas and Their Interplay with Other Epigenetic Modifications. Int. J. Mol. Sci. 2019, 20, 3478. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.F. Epigenomics in cancer management. Cancer Manag. Res. 2010, 2, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batham, J.; Lim, P.S.; Rao, S. SETDB-1: A Potential Epigenetic Regulator in Breast Cancer Metastasis. Cancers 2019, 11, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Fukuda, A.; Matsumoto, Y.; Hanyu, Y.; Sono, M.; Fukunaga, Y.; Masuda, T.; Araki, O.; Nagao, M.; Yoshkawa, T.; et al. SETDB1 Inhibits p53-Mediated Apoptosis and is Required for Formation of Pancreatic Ductal Adenocarcinomas in Mice. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.-F.; Sun, Q.-Y.; Ding, L.-W.; Chien, W.; Liu, X.-Y.; Mayakonda, A.; Jiang, Y.-Y.; Loh, X.-Y.; Ran, X.-B.; Doan, N.B.; et al. The c-MYC–BMI1 axis is essential for SETDB1-mediated breast tumourigenesis. J. Pathol. 2018, 246, 89–102. [Google Scholar] [CrossRef]
- Blanco, E.; González-Ramírez, M.; Alcaine-Colet, A.; Aranda, S.; Di Croce, L. The Bivalent Genome: Characterization, Structure, and Regulation. Trends Genet. 2020, 36, 118–131. [Google Scholar] [CrossRef]
- Lindeman, L.C.; Andersen, I.S.; Reiner, A.H.; Li, N.; Aanes, H.; Østrup, O.; Winata, C.; Mathavan, S.; Müller, F.; Aleström, P.; et al. Prepatterning of developmental gene expression by modified histones before zygotic genome activation. Dev. Cell 2011, 21, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Voigt, P.; Tee, W.-W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fattah, R.; Xiao, A.; Bomgardner, D.; Pease, C.-S.; Lopes, M.-B.S.; Hussaini, I.M. Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J. Pathol. 2006, 209, 15–24. [Google Scholar] [CrossRef]
- Wang, J.-X.; Zeng, Q.; Chen, L.; Du, J.-C.; Yan, X.-L.; Yuan, H.-F.; Zhai, C.; Zhou, J.-N.; Jia, Y.-L.; Yue, W.; et al. SPINDLIN1 Promotes Cancer Cell Proliferation through Activation of WNT/TCF-4 Signaling. Mol. Cancer Res. 2012, 10, 326–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chen, Z.; Mao, Z.; Zhang, H.; Ding, X.; Chen, S.; Zhang, X.; Xu, R.; Zhu, B. Nucleolar protein Spindlin1 recognizes H3K4 methylation and stimulates the expression of rRNA genes. EMBO Rep. 2011, 12, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-W.; Zhao, M.-H.; Liang, S.; Guo, J.; Lin, Z.-L.; Li, Y.-H.; Jo, Y.-J.; Kim, N.-H.; Cui, X.-S. Spindlin 1 is essential for metaphase II stage maintenance and chromosomal stability in porcine oocytes. Mol. Hum. Reprod. 2017, 23, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anatskaya, O.V.; Vinogradov, A.E.; Vainshelbaum, N.M.; Giuliani, A.; Erenpreisa, J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. Int. J. Mol. Sci. 2020, 21, 8759. [Google Scholar] [CrossRef] [PubMed]
- Karamouzis, M.V.; Badra, F.A.; Papavassiliou, A.G. Breast cancer: The upgraded role of HER-3 and HER-4. Int. J. Biochem. Cell Biol. 2007, 39, 851–856. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, B.; Drogaris, P.; Lin, H.-H.S.; Armstrong, H.; Hiragami-Hamada, K.; Imhof, A.; Bonneil, E.; Thibault, P.; Verreault, A.; Festenstein, R.J. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 2011, 7, e1001354. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Liu, Y.; Su, X.; Lee, J.-E.; Song, Y.; Wang, D.; Ge, K.; Gao, J.; Zhang, M.Q.; Li, H. Molecular basis for histone H3 “K4me3-K9me3/2” methylation pattern readout by Spindlin1. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Cai, L.; Rothbart, S.B.; Lu, R.; Xu, B.; Chen, W.-Y.; Tripathy, A.; Rockowitz, S.; Zheng, D.; Patel, D.J.; Allis, C.D.; et al. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 2013, 49, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M.; Lee, J.; Noh, K.-M.; Choi, W.-Y.; Jeon, S.; Oh, G.T.; Kim-Ha, J.; Jin, Y.; Cho, S.-W.; Kim, Y.-J. Intragenic CpG islands play important roles in bivalent chromatin assembly of developmental genes. Proc. Natl. Acad. Sci. USA 2017, 114, E1885–E1894. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.K.; Frietze, S.E.; Gordon, J.A.; Heath, J.L.; Messier, T.; Hong, D.; Boyd, J.R.; Kang, M.; Imbalzano, A.N.; Lian, J.B.; et al. Bivalent Epigenetic Control of Oncofetal Gene Expression in Cancer. Mol. Cell. Biol. 2017, 37, e00352-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-Z.; Tsai, Y.-P.; Yang, M.-H.; Huang, C.-H.; Chang, S.-Y.; Chang, C.-C.; Teng, S.-C.; Wu, K.-J. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Nottke, A.; Colaiácovo, M.P.; Shi, Y. Developmental roles of the histone lysine demethylases. Development 2009, 136, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, Y.; Nakaki, R.; Inagaki, T.; Yoshida, A.; Kano, Y.; Kimura, H.; Tanaka, T.; Tsutsumi, S.; Nakao, M.; Doi, T.; et al. H3K4/H3K9me3 Bivalent Chromatin Domains Targeted by Lineage-Specific DNA Methylation Pauses Adipocyte Differentiation. Mol. Cell 2015, 60, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.D.; Chu, F.; Racki, L.R.; de la Cruz, C.C.; Burlingame, A.L.; Panning, B.; Narlikar, G.J.; Shokat, K.M. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 2007, 128, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Spicuglia, S.; Vanhille, L. Chromatin signatures of active enhancers. Nucleus 2012, 3, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarvey, K.M.; Van Neste, L.; Cope, L.; Ohm, J.E.; Herman, J.G.; Van Criekinge, W.; Schuebel, K.E.; Baylin, S.B. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008, 68, 5753–5759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGarvey, K.M.; Fahrner, J.A.; Greene, E.; Martens, J.; Jenuwein, T.; Baylin, S.B. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006, 66, 3541–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, A.H.; McGarvey, K.M.; Baylin, S.B. The cancer epigenome--components and functional correlates. Genes Dev. 2006, 20, 3215–3231. [Google Scholar] [CrossRef] [Green Version]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, H.P.; Cai, Y.; McGarvey, K.M.; Easwaran, H.; Van Neste, L.; Ohm, J.E.; O’Hagan, H.M.; Baylin, S.B. Polycomb CBX7 promotes initiation of heritable repression of genes frequently silenced with cancer-specific DNA hypermethylation. Cancer Res. 2009, 69, 6322–6330. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Weikert, S.; Christoph, F.; Köllermann, J.; Müller, M.; Schrader, M.; Miller, K.; Krause, H. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int. J. Mol. Med. 2005, 16, 349–353. [Google Scholar] [CrossRef]
- Matsukawa, Y.; Semba, S.; Kato, H.; Ito, A.; Yanagihara, K.; Yokozaki, H. Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 2006, 97, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Croonquist, P.A.; Van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.W.; Battenhouse, A.M.; Shivram, H.; Morris, A.R.; Cowperthwaite, M.C.; Shpak, M.; Iyer, V.R. Bivalent Chromatin Domains in Glioblastoma Reveal a Subtype-Specific Signature of Glioma Stem Cells. Cancer Res. 2018, 78, 2463–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Yu, N.-K.; Kaang, B.-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, C.S.; Le Boiteux, E.; Arnaud, P.; Costa, B.M. HOX gene cluster (de)regulation in brain: From neurodevelopment to malignant glial tumours. Cell. Mol. Life Sci. 2020, 77, 3797–3821. [Google Scholar] [CrossRef] [PubMed]
- Buccoliero, A.M.; Castiglione, F.; Degl’Innocenti, D.R.; Ammanati, F.; Giordano, F.; Sanzo, M.; Mussa, F.; Genitori, L.; Taddei, G.L. Hox-D Genes Expression in Pediatric Low-grade Gliomas: Real-time-PCR Study. Cell. Mol. Neurobiol. 2009, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kurscheid, S.; Bady, P.; Sciuscio, D.; Samarzija, I.; Shay, T.; Vassallo, I.; Criekinge, W.V.; Daniel, R.T.; van den Bent, M.J.; Marosi, C.; et al. Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX-signature in glioblastoma. Genome Biol. 2015, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; Suvà, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An Aberrant Transcription Factor Network Essential for Wnt Signaling and Stem Cell Maintenance in Glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Lee, H.; Yoon, J.-G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369–5381. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-H.; Kim, E.-J.; Hitomi, M.; Oh, S.-Y.; Jin, X.; Jeon, H.-M.; Beck, S.; Jin, X.; Kim, J.-K.; Park, C.G.; et al. The LIM-only transcription factor LMO2 determines tumorigenic and angiogenic traits in glioma stem cells. Cell Death Differ. 2015, 22, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.-B.; Zhang, S.-Y.; Zhao, B.-L.; Ding, R.; Liu, H.; Li, S. Knockdown of MMP11 inhibits proliferation and invasion of gastric cancer cells. Int. J. Immunopathol. Pharmacol. 2013, 26, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Pan, H.; Xia, T.; Xue, J.; Cheng, L.; Fan, P.; Zhang, Y.; Zhu, W.; Xue, Y.; Liu, X.; et al. Up-regulation of S100A16 expression promotes epithelial-mesenchymal transition via Notch1 pathway in breast cancer. J. Biomed. Sci. 2014, 21, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, U.; Luthra, S.; Santana-Santos, L.; Mao, P.; Kim, S.-H.; Minata, M.; Li, J.; Benos, P.V.; De-wang, M.; Hu, B.; et al. Gene expression profiling distinguishes proneural glioma stem cells from mesenchymal glioma stem cells. Genom. Data 2015, 5, 333–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Pannell, R.; Forster, A.; Rabbitts, T.H. The oncogenic LIM-only transcription factor Lmo2 regulates angiogenesis but not vasculogenesis in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Dai, X.; Chen, Y.; Sun, C.; Zhu, Q.; Zhao, H.; Liu, G.; Huang, Q.; Lan, Q. MiR-30a-5p is induced by Wnt/β-catenin pathway and promotes glioma cell invasion by repressing NCAM. Biochem. Biophys. Res. Commun. 2015, 465, 374–380. [Google Scholar] [CrossRef]
- Court, F.; Le Boiteux, E.; Fogli, A.; Müller-Barthélémy, M.; Vaurs-Barrière, C.; Chautard, E.; Pereira, B.; Biau, J.; Kemeny, J.-L.; Khalil, T.; et al. Transcriptional alterations in glioma result primarily from DNA methylation-independent mechanisms. Genome Res. 2019, 29, 1605–1621. [Google Scholar] [CrossRef] [Green Version]
- Bernhart, S.H.; Kretzmer, H.; Holdt, L.M.; Jühling, F.; Ammerpohl, O.; Bergmann, A.K.; Northoff, B.H.; Doose, G.; Siebert, R.; Stadler, P.F.; et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016, 6, 37393. [Google Scholar] [CrossRef]
- Li, Y.; Ren, Y.; Wang, Y.; Tan, Y.; Wang, Q.; Cai, J.; Zhou, J.; Yang, C.; Zhao, K.; Yi, K.; et al. A Compound AC1Q3QWB Selectively Disrupts HOTAIR-Mediated Recruitment of PRC2 and Enhances Cancer Therapy of DZNep. Theranostics 2019, 9, 4608–4623. [Google Scholar] [CrossRef]
- Ciarapica, R.; Carcarino, E.; Adesso, L.; De Salvo, M.; Bracaglia, G.; Leoncini, P.P.; Dall’agnese, A.; Verginelli, F.; Milano, G.M.; Boldrini, R.; et al. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 2014, 14, 139. [Google Scholar] [CrossRef] [Green Version]
- A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects with Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02601937 (accessed on 16 November 2020).
- A Phase II, Multicenter Study of the EZH2 Inhibitor Tazemetostat in Adult Subjects with INI1-Negative Tumors or Relapsed/Refractory Synovial Sarcoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02601950 (accessed on 16 November 2020).
- Williams, M.J.; Singleton, W.G.B.; Lowis, S.P.; Malik, K.; Kurian, K.M. Therapeutic Targeting of Histone Modifications in Adult and Pediatric High-Grade Glioma. Front. Oncol. 2017, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients with Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peereboom, D.M.; Shepard, D.R.; Ahluwalia, M.S.; Brewer, C.J.; Agarwal, N.; Stevens, G.H.J.; Suh, J.H.; Toms, S.A.; Vogelbaum, M.A.; Weil, R.J.; et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J. Neurooncol. 2010, 98, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Chamberlain, M.C.; Grossman, S.A.; Peereboom, D.M.; Lesser, G.J.; Batchelor, T.T.; Desideri, S.; Salazar, A.M.; Ye, X. A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro. Oncol. 2010, 12, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Butowski, N.; Chang, S.M.; Lamborn, K.R.; Polley, M.-Y.; Pieper, R.; Costello, J.F.; Vandenberg, S.; Parvataneni, R.; Nicole, A.; Sneed, P.K.; et al. Phase II and pharmacogenomics study of enzastaurin plus temozolomide during and following radiation therapy in patients with newly diagnosed glioblastoma multiforme and gliosarcoma. Neuro. Oncol. 2011, 13, 1331–1338. [Google Scholar] [CrossRef]
- Weller, M.; Gorlia, T.; Cairncross, J.G.; van den Bent, M.J.; Mason, W.; Belanger, K.; Brandes, A.A.; Bogdahn, U.; Macdonald, D.R.; Forsyth, P.; et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011, 77, 1156–1164. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Vandoros, G.P.; Papavassiliou, A.G. FK228 (depsipeptide): A HDAC inhibitor with pleiotropic antitumor activities. Cancer Chemother. Pharmacol. 2006, 58, 711–715. [Google Scholar] [CrossRef]
- Wu, Y.; Dong, L.; Bao, S.; Wang, M.; Yun, Y.; Zhu, R. FK228 augmented temozolomide sensitivity in human glioma cells by blocking PI3K/AKT/mTOR signal pathways. Biomed. Pharmacother. 2016, 84, 462–469. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.-A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Morales La Madrid, A.; Hashizume, R.; Kieran, M.W. Future Clinical Trials in DIPG: Bringing Epigenetics to the Clinic. Front. Oncol. 2015, 5, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Cheng, J.; Zhang, X.; Wang, R.; Zhang, W.; Lin, H.; Xiao, X.; Cai, S.; Chen, X.; Cheng, H. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2010, 19, 2888–2896. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markouli, M.; Strepkos, D.; Papavassiliou, K.A.; Papavassiliou, A.G.; Piperi, C. Bivalent Genes Targeting of Glioma Heterogeneity and Plasticity. Int. J. Mol. Sci. 2021, 22, 540. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020540
Markouli M, Strepkos D, Papavassiliou KA, Papavassiliou AG, Piperi C. Bivalent Genes Targeting of Glioma Heterogeneity and Plasticity. International Journal of Molecular Sciences. 2021; 22(2):540. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020540
Chicago/Turabian StyleMarkouli, Mariam, Dimitrios Strepkos, Kostas A. Papavassiliou, Athanasios G. Papavassiliou, and Christina Piperi. 2021. "Bivalent Genes Targeting of Glioma Heterogeneity and Plasticity" International Journal of Molecular Sciences 22, no. 2: 540. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020540