Editing and Chemical Modifications on Non-Coding RNAs in Cancer: A New Tale with Clinical Significance

 , , and
, , and
Abstract
:1. Introduction
2. NcRNA Editing in Cancer
2.1. ADAR Dependent Editing
2.2. APOBEC Dependent Editing
2.3. Non-ADAR, Non-APOBEC Editing Mechanisms
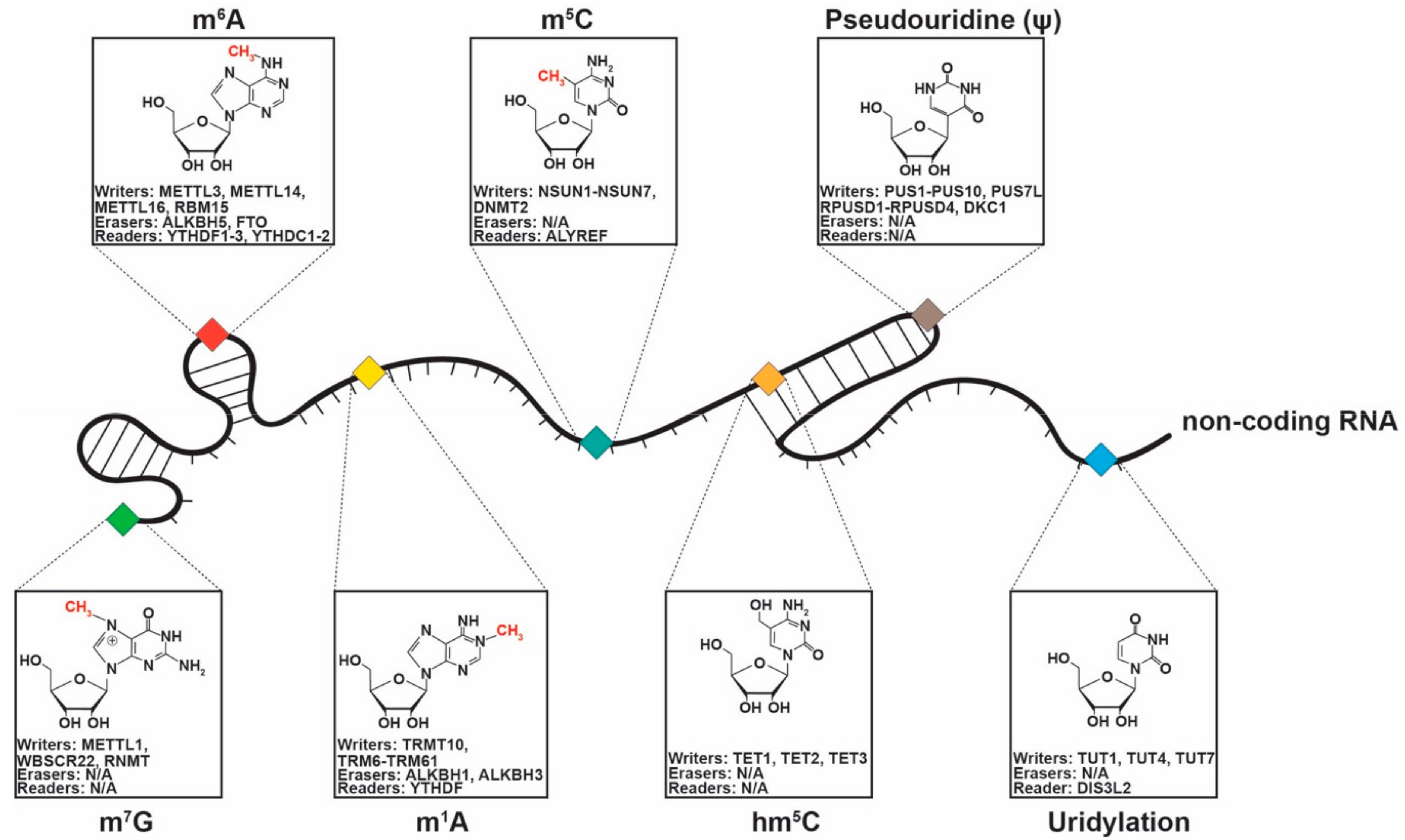
3. NcRNA Chemical Modifications in Cancer
3.1. 5-Methylcytosine
3.2. 5-Hydroxymethylcytosine (hm5C)
3.3. N6-Methyladenosine (m6A)
3.4. N1-Methyladenosine (m1A)
3.5. Pseudouridylation (Ψ)
3.6. Uridylation
3.7. 7-Methylguanosine (m7G)
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A-to-I | adenosine to inosine |
| ACF | Apobec-1 complementation factor |
| ADAR | adenosine deaminase acting on RNA |
| ALKBH5 | alpha-ketoglutarate-dependent dioxygenase alkB homolog 5 |
| AMFR | autocrine motility factor receptor |
| AML | acute myeloid leukemia |
| APOBEC | apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like |
| ASB2 | ankyrin repeat and SOCS box containing 2 |
| BIK | BCL-2-interacting-killer |
| C-to-U | cytidine to uridine |
| CA19-9 | carbohydrate antigen 19-9 |
| CAE | carcinoembryonic antigen |
| cDNA | complementary DNA |
| circRNA | circular RNA |
| CLL | chronic lymphocytic leukemia |
| CSF-1 | colony stimulating factor 1 |
| CXCR4 | CXC motif chemokine receptor 4 |
| DIS3L2 | DIS3-like exonuclease 2 |
| DKC1 | dyskerin pseudouridine synthase 1 |
| DND1 | dead-end protein homolog 1 |
| DNMT | DNA methyltransferases |
| DRAI | DNA-to-RNA allelic imbalance |
| ESCC | esophageal squamous cell carcinoma |
| FOXM1 | forkhead box M1 |
| FTO | fat mass and obesity associated protein |
| GAC | glutaminase isoform C |
| gDNA | genomic DNA |
| GLS | glutaminase |
| hm5C | 5-hydroxymethylcytosine |
| KGA | glutaminase kidney isoform |
| LIFR | leukemia inhibitory factor receptor |
| lncRNA | long non-coding RNA |
| LTR | long terminal repeat |
| m1A | N1-Methyladenosine |
| m5C | 5-methylcytosine |
| m6A | N6-Methyladenosine |
| MALAT1 MALDI-TOF-MS | metastasis associated lung adenocarcinoma transcript 1 matrix-assisted laser desorption/ionization-time of flight- mass spectrometry |
| MDS | myelodysplastic syndrome |
| METTL1 | methyltransferase like 1 |
| miRNA | microRNA |
| MMP2 | matrix metallopeptidase 2 |
| MPN | myeloproliferative neoplasm |
| ncRNA | non-coding RNA |
| NMR | NSUN2 methylated lncRNA |
| NSCLC | non-small-cell lung cancer |
| NSUN | NOP2/Sun RNA methyltransferase |
| PARVA | alpha-parvin |
| PD1 | programmed cell death protein 1 |
| pre-miRNA | precursor miRNA |
| pri-miRNA | primary miRNA |
| PROTAC | proteolysis-targeting chimera |
| PUS | pseudouridine synthase |
| RARA | retinoic acid receptor alpha |
| RARG | retinoic acid receptor gamma |
| RBM15 | RNA-binding motif protein 15 |
| RDD | RNA-DNA difference |
| rRNA RT-PCR | ribosomal RNA reverse transcription-polymerase chain reaction |
| siRNA | small interfering RNA |
| SNHG1 | Small nucleolar RNA host gene 1 |
| SNHG7 snoRNA | Small nucleolar RNA host gene 7 small nucleolar RNA |
| SNP snRNA | single nucleotide polymorphism small nuclear RNA |
| SOX1 | SRY-Box transcription factor 1 |
| SRA1 | steroid receptor RNA activator 1 |
| TERC | telomerase RNA component |
| TETt | en-eleven family demethylases |
| TNFR2 | tumor necrosis factor receptor 2 |
| TRAIL | TNF-related apoptosis inducing ligand |
| TRM10 | tRNA methyltransferase 10 homologue A |
| TRM6 | tRNA methyltransferase non-catalytic subunit 6 |
| TRM61 | tRNA methyltransferase catalytic subunit 61 |
| tRNA | transfer RNA |
| TUT1 | terminal uridylyltransferase 1 |
| XIST | X-inactive specific transcript |
| ZFAS1 | zinc finger antisense 1 |
| Ψ | pseudouridylation |
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.P.; Kopetz, S.; Ajani, J.A.; Calin, G.A. Non-coding RNAs in GI cancers: From cancer hallmarks to clinical utility. Gut 2020, 69, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Bullock, M.; Calin, G. The Clinical Relevance of Long Non-Coding RNAs in Cancer. Cancers 2015, 7, 2169–2182. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Calore, F.; Paone, A.; Galli, R.; Calin, G.A. Epigenetic regulation of miRNAs in cancer. Adv. Exp. Med. Biol. 2013, 754, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Michlewski, G.; Caceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target interactions—Beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.Y.; Kuo, H.C. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019, 26, 29. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoloso, M.S.; Spizzo, R.; Shimizu, M.; Rossi, S.; Calin, G.A. MicroRNAs—The micro steering wheel of tumour metastases. Nat. Rev. Cancer 2009, 9, 293–302. [Google Scholar] [CrossRef]
- Spizzo, R.; Nicoloso, M.S.; Croce, C.M.; Calin, G.A. SnapShot: MicroRNAs in Cancer. Cell 2009, 137, 586–586.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Hansen, T.B.; Veno, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Dragomir, M.; Calin, G.A. Circular RNAs in Cancer—Lessons Learned From microRNAs. Front. Oncol 2018, 8, 179. [Google Scholar] [CrossRef] [Green Version]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—An update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef]
- Pardini, B.; Sabo, A.A.; Birolo, G.; Calin, G.A. Noncoding RNAs in Extracellular Fluids as Cancer Biomarkers: The New Frontier of Liquid Biopsies. Cancers 2019, 11, 1170. [Google Scholar] [CrossRef] [Green Version]
- Morales, S.; Monzo, M.; Navarro, A. Epigenetic regulation mechanisms of microRNA expression. Biomol. Concepts 2017, 8, 203–212. [Google Scholar] [CrossRef]
- Esteller, M.; Pandolfi, P.P. The Epitranscriptome of Noncoding RNAs in Cancer. Cancer Discov. 2017, 7, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Christofi, T.; Zaravinos, A. RNA editing in the forefront of epitranscriptomics and human health. J. Transl. Med. 2019, 17, 319. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; D’Erchia, A.M.; Gallo, A.; Montalvo, A.; Pesole, G. Uncovering RNA Editing Sites in Long Non-Coding RNAs. Front. Bioeng. Biotechnol. 2014, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ji, B.; Song, R.; Wang, S.; Li, T.; Zhang, X.; Chen, K.; Li, T.; Li, J. Accurate detection for a wide range of mutation and editing sites of microRNAs from small RNA high-throughput sequencing profiles. Nucleic Acids Res. 2016, 44, e123. [Google Scholar] [CrossRef]
- Ramaswami, G.; Li, J.B. Identification of human RNA editing sites: A historical perspective. Methods 2016, 107, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Distefano, R.; Nigita, G.; Veneziano, D.; Romano, G.; Croce, C.M.; Acunzo, M. isoTar: Consensus Target Prediction with Enrichment Analysis for MicroRNAs Harboring Editing Sites and Other Variations. Methods Mol. Biol. 2019, 1970, 211–235. [Google Scholar] [CrossRef]
- Nigita, G.; Marceca, G.P.; Tomasello, L.; Distefano, R.; Calore, F.; Veneziano, D.; Romano, G.; Nana-Sinkam, S.P.; Acunzo, M.; Croce, C.M. ncRNA Editing: Functional Characterization and Computational Resources. Methods Mol. Biol. 2019, 1912, 133–174. [Google Scholar] [CrossRef]
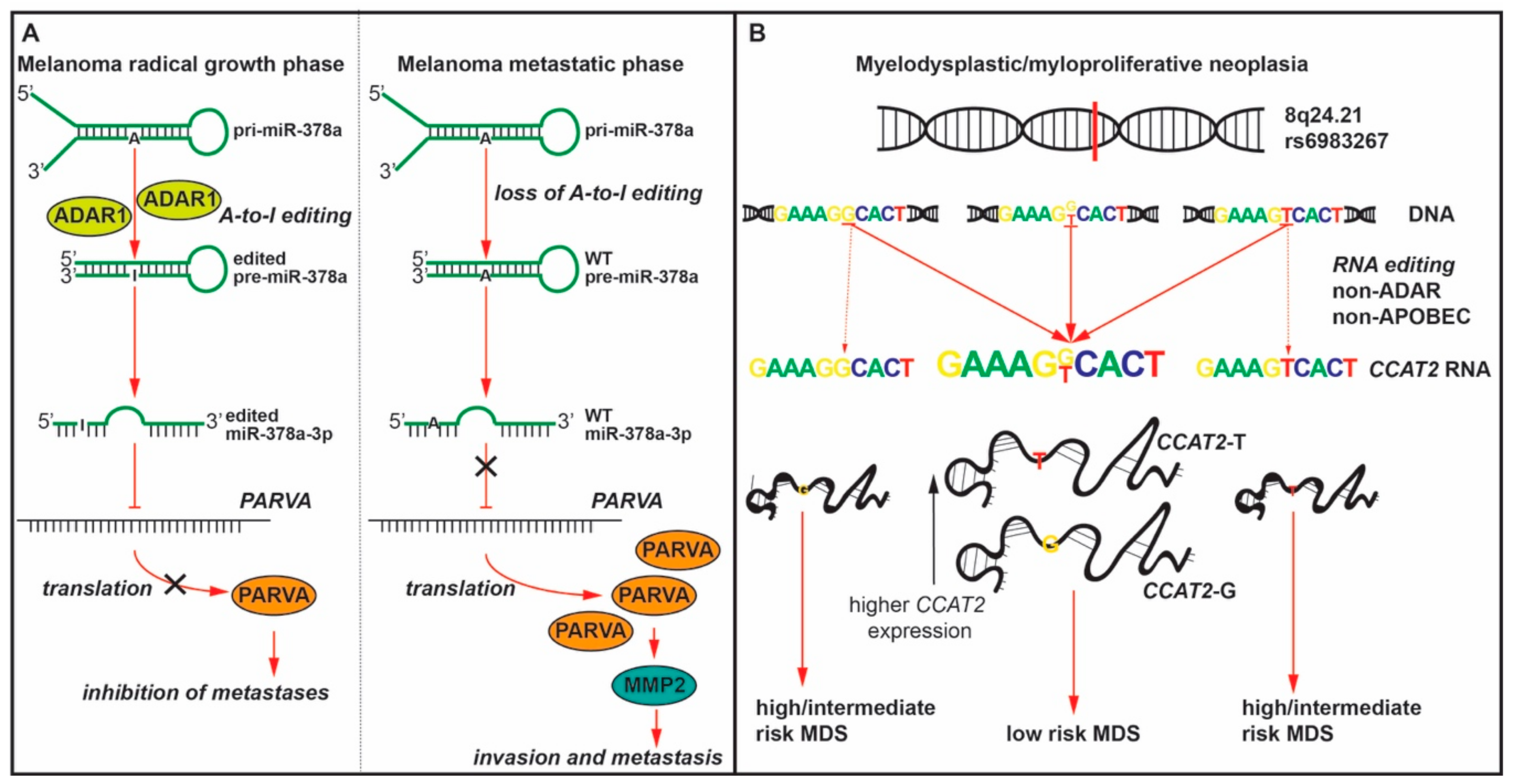
- Shah, M.Y.; Ferracin, M.; Pileczki, V.; Chen, B.; Redis, R.; Fabris, L.; Zhang, X.; Ivan, C.; Shimizu, M.; Rodriguez-Aguayo, C.; et al. Cancer-associated rs6983267 SNP and its accompanying long noncoding RNA CCAT2 induce myeloid malignancies via unique SNP-specific RNA mutations. Genome Res. 2018, 28, 432–447. [Google Scholar] [CrossRef] [Green Version]
- Zinshteyn, B.; Nishikura, K. Adenosine-to-inosine RNA editing. Wiley Interdiscip. Rev. Syst Biol. Med. 2009, 1, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Walkley, C.R.; Li, J.B. Rewriting the transcriptome: Adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heale, B.S.; Keegan, L.P.; O’Connell, M.A. The effect of RNA editing and ADARs on miRNA biogenesis and function. Adv. Exp. Med. Biol. 2010, 700, 76–84. [Google Scholar] [PubMed]
- Kawahara, Y.; Megraw, M.; Kreider, E.; Iizasa, H.; Valente, L.; Hatzigeorgiou, A.G.; Nishikura, K. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008, 36, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Yu, S.; Jeong, K.J.; Zhou, Z.; Han, L.; Tsang, Y.H.; Li, J.; Chen, H.; Mangala, L.S.; et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017, 27, 1112–1125. [Google Scholar] [CrossRef] [Green Version]
- Pinto, Y.; Buchumenski, I.; Levanon, E.Y.; Eisenberg, E. Human cancer tissues exhibit reduced A-to-I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res. 2018, 46, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, Y.; Mojumdar, K.; Zhou, Z.; Jeong, K.J.; Mangala, L.S.; Yu, S.; Tsang, Y.H.; Rodriguez-Aguayo, C.; Lu, Y.; et al. A-to-I-edited miRNA-379-5p inhibits cancer cell proliferation through CD97-induced apoptosis. J. Clin. Invest. 2019, 129, 5343–5356. [Google Scholar] [CrossRef] [Green Version]
- Vaikari, V.P.; Yang, J.; Wu, S.; Alachkar, H. CD97 expression is associated with poor overall survival in acute myeloid leukemia. Exp. Hematol. 2019, 75, 64–73.e64. [Google Scholar] [CrossRef]
- He, Z.; Wu, H.; Jiao, Y.; Zheng, J. Expression and prognostic value of CD97 and its ligand CD55 in pancreatic cancer. Oncol. Lett. 2015, 9, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Li, C.; Trojanowicz, B.; Li, X.; Shi, D.; Zhan, C.; Wang, Z.; Chen, L. CD97 promotion of gastric carcinoma lymphatic metastasis is exosome dependent. Gastric Cancer 2016, 19, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Tomaselli, S.; Galeano, F.; Alon, S.; Raho, S.; Galardi, S.; Polito, V.A.; Presutti, C.; Vincenti, S.; Eisenberg, E.; Locatelli, F.; et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015, 16, 5. [Google Scholar] [CrossRef]
- Choudhury, Y.; Tay, F.C.; Lam, D.H.; Sandanaraj, E.; Tang, C.; Ang, B.T.; Wang, S. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J. Clin. Investig. 2012, 122, 4059–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarini, V.; Silvestris, D.A.; Tassinari, V.; Tomaselli, S.; Alon, S.; Eisenberg, E.; Locatelli, F.; Gallo, A. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids Res. 2018, 46, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, E.; Mobley, A.K.; Braeuer, R.R.; Kamiya, T.; Huang, L.; Vasquez, M.E.; Salameh, A.; Lee, H.J.; Kim, S.J.; Ivan, C.; et al. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat. Cell Biol. 2015, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Torres, G.; Shoshan, E.; Ivan, C.; Huang, L.; Fuentes-Mattei, E.; Paret, H.; Kim, S.J.; Rodriguez-Aguayo, C.; Xie, V.; Brooks, D.; et al. A-to-I miR-378a-3p editing can prevent melanoma progression via regulation of PARVA expression. Nat. Commun. 2018, 9, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.H.; Chen, C.H.; Yeh, K.H.; Li, C.L.; Wu, Y.J.; Chen, D.S.; Chen, P.J.; Yeh, S.H. ADAR2-mediated editing of miR-214 and miR-122 precursor and antisense RNA transcripts in liver cancers. PLoS ONE 2013, 8, e81922. [Google Scholar] [CrossRef] [Green Version]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Anadon, C.; Guil, S.; Simo-Riudalbas, L.; Moutinho, C.; Setien, F.; Martinez-Cardus, A.; Moran, S.; Villanueva, A.; Calaf, M.; Vidal, A.; et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene 2016, 35, 4407–4413. [Google Scholar] [CrossRef]
- Huang, R.S.; Zheng, Y.L.; Zhao, J.; Chun, X. microRNA-381 suppresses the growth and increases cisplatin sensitivity in non-small cell lung cancer cells through inhibition of nuclear factor-kappaB signaling. Biomed. Pharmacother 2018, 98, 538–544. [Google Scholar] [CrossRef]
- Wang, Q.; Hui, H.; Guo, Z.; Zhang, W.; Hu, Y.; He, T.; Tai, Y.; Peng, P.; Wang, L. ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding. RNA 2013, 19, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.T.; Patterson, D.G.; King, V.M.; Amin, S.V.; Polska, C.J.; Houserova, D.; Crucello, A.; Barnhill, E.C.; Miller, M.M.; Sherman, T.D.; et al. ADAR Mediated RNA Editing Modulates MicroRNA Targeting in Human Breast Cancer. Processes 2018, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Song, Y.; Chan, T.H.M.; Yang, H.; Lin, C.H.; Tay, D.J.T.; Hong, H.; Tang, S.J.; Tan, K.T.; Huang, X.X.; et al. An RNA editing/dsRNA binding-independent gene regulatory mechanism of ADARs and its clinical implication in cancer. Nucleic Acids Res. 2017, 45, 10436–10451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemlich, Y.; Greenberg, E.; Ortenberg, R.; Besser, M.J.; Barshack, I.; Jacob-Hirsch, J.; Jacoby, E.; Eyal, E.; Rivkin, L.; Prieto, V.G.; et al. MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J. Clin. Investig. 2013, 123, 2703–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jia, M.; He, Z.; Liu, X.S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 2018, 37, 3924–3936. [Google Scholar] [CrossRef]
- Glaser, A.P.; Fantini, D.; Wang, Y.; Yu, Y.; Rimar, K.J.; Podojil, J.R.; Miller, S.D.; Meeks, J.J. APOBEC-mediated mutagenesis in urothelial carcinoma is associated with improved survival, mutations in DNA damage response genes, and immune response. Oncotarget 2018, 9, 4537–4548. [Google Scholar] [CrossRef] [Green Version]
- Law, E.K.; Sieuwerts, A.M.; LaPara, K.; Leonard, B.; Starrett, G.J.; Molan, A.M.; Temiz, N.A.; Vogel, R.I.; Meijer-van Gelder, M.E.; Sweep, F.C.; et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737. [Google Scholar] [CrossRef] [Green Version]
- Blanc, V.; Park, E.; Schaefer, S.; Miller, M.; Lin, Y.; Kennedy, S.; Billing, A.M.; Ben Hamidane, H.; Graumann, J.; Mortazavi, A.; et al. Genome-wide identification and functional analysis of Apobec-1-mediated C-to-U RNA editing in mouse small intestine and liver. Genome Biol. 2014, 15, R79. [Google Scholar] [CrossRef] [Green Version]
- Salter, J.D.; Smith, H.C. Modeling the Embrace of a Mutator: APOBEC Selection of Nucleic Acid Ligands. Trends Biochem. Sci. 2018, 43, 606–622. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, S.; Balestra, M.E.; Ferrell, L.D.; Fan, J.; Arnold, K.S.; Taylor, S.; Taylor, J.M.; Innerarity, T.L. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc. Natl. Acad. Sci. USA 1995, 92, 8483–8487. [Google Scholar] [CrossRef] [Green Version]
- Valdmanis, P.N.; Roy-Chaudhuri, B.; Kim, H.K.; Sayles, L.C.; Zheng, Y.; Chuang, C.H.; Caswell, D.R.; Chu, K.; Zhang, Y.; Winslow, M.M.; et al. Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene 2015, 34, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koito, A.; Ikeda, T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Chang, C.J.; Xie, X.; Xia, W.; Yang, J.Y.; Wang, S.C.; Wang, Y.; Xia, J.; Chen, L.; Cai, C.; et al. APOBEC3G promotes liver metastasis in an orthotopic mouse model of colorectal cancer and predicts human hepatic metastasis. J. Clin. Investig. 2011, 121, 4526–4536. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Karki, N.; Bhattacharya, C.; Zhu, R.; MacDuff, D.A.; Stenglein, M.D.; Schumacher, A.J.; Demorest, Z.L.; Harris, R.S.; Matin, A.; et al. APOBEC3 inhibits DEAD-END function to regulate microRNA activity. BMC Mol. Biol. 2013, 14, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Chen, J.; Huang, Z. miR-372 promotes breast cancer cell proliferation by directly targeting LATS2. Exp. Ther. Med. 2018, 15, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Shao, Z.M.; He, Q.Z.; Jiang, B.Q.; Wu, Y.; Zhuang, Z.G. Hsa-miR-206 represses the proliferation and invasion of breast cancer cells by targeting Cx43. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2091–2104. [Google Scholar]
- Li, M.; Wang, I.X.; Li, Y.; Bruzel, A.; Richards, A.L.; Toung, J.M.; Cheung, V.G. Widespread RNA and DNA sequence differences in the human transcriptome. Science 2011, 333, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, J.K.; Gilad, Y.; Pritchard, J.K. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302, author reply 1302. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A. Widespread promiscuous genetic information transfer from DNA to RNA. Circ. Res. 2011, 109, 1202–1203. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, C.L.; Majewski, J. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302, author reply 1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.X.; Core, L.J.; Kwak, H.; Brady, L.; Bruzel, A.; McDaniel, L.; Richards, A.L.; Wu, M.; Grunseich, C.; Lis, J.T.; et al. RNA-DNA differences are generated in human cells within seconds after RNA exits polymerase II. Cell Rep. 2014, 6, 906–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafa, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Dragomir, M.P.; Fabris, L.; Bayraktar, R.; Knutsen, E.; Liu, X.; Tang, C.; Li, Y.; Shimura, T.; Ivkovic, T.C.; et al. The Long Noncoding RNA CCAT2 induces chromosomal instability through BOP1—AURKB signaling. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Haiman, C.A.; Le Marchand, L.; Yamamato, J.; Stram, D.O.; Sheng, X.; Kolonel, L.N.; Wu, A.H.; Reich, D.; Henderson, B.E. A common genetic risk factor for colorectal and prostate cancer. Nat. Genet. 2007, 39, 954–956. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.; Webb, E.; Carvajal-Carmona, L.; Broderick, P.; Kemp, Z.; Spain, S.; Penegar, S.; Chandler, I.; Gorman, M.; Wood, W.; et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat. Genet. 2007, 39, 984–988. [Google Scholar] [CrossRef]
- Zanke, B.W.; Greenwood, C.M.; Rangrej, J.; Kustra, R.; Tenesa, A.; Farrington, S.M.; Prendergast, J.; Olschwang, S.; Chiang, T.; Crowdy, E.; et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet. 2007, 39, 989–994. [Google Scholar] [CrossRef]
- Ghoussaini, M.; Song, H.; Koessler, T.; Al Olama, A.A.; Kote-Jarai, Z.; Driver, K.E.; Pooley, K.A.; Ramus, S.J.; Kjaer, S.K.; Hogdall, E.; et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J. Natl. Cancer Inst. 2008, 100, 962–966. [Google Scholar] [CrossRef] [Green Version]
- Redis, R.S.; Vela, L.E.; Lu, W.; Ferreira de Oliveira, J.; Ivan, C.; Rodriguez-Aguayo, C.; Adamoski, D.; Pasculli, B.; Taguchi, A.; Chen, Y.; et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol. Cell 2016, 61, 520–534. [Google Scholar] [CrossRef] [Green Version]
- Ontiveros, R.J.; Stoute, J.; Liu, K.F. The chemical diversity of RNA modifications. Biochem. J. 2019, 476, 1227–1245. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Aleksic, J.; Blanco, S.; Dietmann, S.; Frye, M. Characterizing 5-methylcytosine in the mammalian epitranscriptome. Genome Biol. 2013, 14, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Dinescu, S.; Ignat, S.; Lazar, A.D.; Constantin, C.; Neagu, M.; Costache, M. Epitranscriptomic Signatures in lncRNAs and Their Possible Roles in Cancer. Genes 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, R.; Zander, S.; Gutschner, T. The Dark Side of the Epitranscriptome: Chemical Modifications in Long Non-Coding RNAs. Int. J. Mol. Sci 2017, 18, 2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, M.; Watt, F.M. The RNA Methyltransferase Misu (NSun2) Mediates Myc-Induced Proliferation and Is Upregulated in Tumors. Curr. Biol. 2006, 16, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.; Gao, R.; Chen, Y.; Yang, Z.; Han, P.; Zhang, H.; Dou, Y.; Liu, W.; Wang, W.; Du, G.; et al. Overexpression of NSUN2 by DNA hypomethylation is associated with metastatic progression in human breast cancer. Oncotarget 2017, 8, 20751–20765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trixl, L.; Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip. Rev. RNA 2019, 10, e1510. [Google Scholar] [CrossRef] [Green Version]
- Bantis, A.; Giannopoulos, A.; Gonidi, M.; Liossi, A.; Aggelonidou, E.; Petrakakou, E.; Athanassiades, P.; Athanassiadou, P. Expression of p120, Ki-67 and PCNA as proliferation biomarkers in imprint smears of prostate carcinoma and their prognostic value. Cytopathology 2004, 15, 25–31. [Google Scholar] [CrossRef]
- Saijo, Y.; Sato, G.; Usui, K.; Sato, M.; Sagawa, M.; Kondo, T.; Minami, Y.; Nukiwa, T. Expression of nucleolar protein p120 predicts poor prognosis in patients with stage I lung adenocarcinoma. Ann. Oncol. 2001, 12, 1121–1125. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzelepi, V.; Logotheti, S.; Efstathiou, E.; Troncoso, P.; Aparicio, A.; Sakellakis, M.; Hoang, A.; Perimenis, P.; Melachrinou, M.; Logothetis, C.; et al. Epigenetics and prostate cancer: Defining the timing of DNA methyltransferase deregulation during prostate cancer progression. Pathology 2020, 52, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Guerrero, C.R.; Zhong, N.; Amato, N.J.; Liu, Y.; Liu, S.; Cai, Q.; Ji, D.; Jin, S.-G.; Niedernhofer, L.J.; et al. Tet-Mediated Formation of 5-Hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 2014, 136, 11582–11585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, M.; Koseki, J.; Asai, A.; Yamagata, A.; Shimamura, T.; Motooka, D.; Okuzaki, D.; Kawamoto, K.; Mizushima, T.; Eguchi, H.; et al. Distinct methylation levels of mature microRNAs in gastrointestinal cancers. Nat. Commun. 2019, 10, 3888. [Google Scholar] [CrossRef] [Green Version]
- Cheray, M.; Etcheverry, A.; Jacques, C.; Pacaud, R.; Bougras-Cartron, G.; Aubry, M.; Denoual, F.; Peterlongo, P.; Nadaradjane, A.; Briand, J.; et al. Cytosine methylation of mature microRNAs inhibits their functions and is associated with poor prognosis in glioblastoma multiforme. Mol. Cancer 2020, 19, 36. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Luo, M.; Zhou, C.; Shi, X.; Yang, W.; Lu, Z.; Chen, Z.; Sun, N.; He, J. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett. 2018, 430, 57–66. [Google Scholar] [CrossRef]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S.; et al. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 2016, 351, 282. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Banfai, B.; Jia, H.; Khatun, J.; Wood, E.; Risk, B.; Gundling, W.E., Jr.; Kundaje, A.; Gunawardena, H.P.; Yu, Y.; Xie, L.; et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 2012, 22, 1646–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.F.; Wei, C.Y.; Huang, X.Y.; Peng, R.; Yang, X.; Lu, J.C.; Zhang, C.; Gao, C.; Cai, J.B.; Gao, P.T.; et al. Circular RNA circTRIM33-12 acts as the sponge of MicroRNA-191 to suppress hepatocellular carcinoma progression. Mol. Cancer 2019, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Shu, M.; He, L.; Yu, X.; Liu, X.; Lu, Y.; Chen, Y.; Miao, X.; Chen, X. Epigenomic landscape of 5-hydroxymethylcytosine reveals its transcriptional regulation of lncRNAs in colorectal cancer. Br. J. Cancer 2017, 116, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Pan, Y.; Ma, P.; Liu, Y.; Li, W.; Shu, Y. Multiple functions of m(6)A RNA methylation in cancer. J. Hematol. Oncol. 2018, 11, 48. [Google Scholar] [CrossRef]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.-G.; et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.-T.; Tsang, F.H.-C.; Shen, J.; Cheng, C.L.-H.; Tsang, L.-H.; Ho, D.W.-H.; Chiu, D.K.-C.; Lee, J.M.-F.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835.e814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Ma, L. New Insights into Long Non-Coding RNA MALAT1 in Cancer and Metastasis. Cancers 2019, 11, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.A.; Kinzig, C.G.; DeGregorio, S.J.; Steitz, J.A. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 14013–14018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCown, P.J.; Wang, M.C.; Jaeger, L.; Brown, J.A. Secondary Structural Model of Human MALAT1 Reveals Multiple Structure-Function Relationships. Int. J. Mol. Sci. 2019, 20, 5610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, D.P.; Chen, C.-K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hao, Y.-J.; Zhang, Y.; Li, M.-M.; Wang, M.; Han, W.; Wu, Y.; Lv, Y.; Hao, J.; Wang, L.; et al. m6A RNA Methylation Is Regulated by MicroRNAs and Promotes Reprogramming to Pluripotency. Cell Stem Cell 2015, 16, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.D.; Jaffrey, S.R. Rethinking m(6)A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 2017, 33, 319–342. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Yang, C.; Wu, R.; Huang, L.; Song, S.; Li, W.; Yan, P.; Lin, C.; Li, D.; Zhang, Y. YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Front. Oncol. 2019, 9, 332. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m6 A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148.e6. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, A.; Tanikawa, K.; Tsunetomi, M.; Takai, K.; Ikeda, H.; Konno, J.; Torigoe, T.; Maeda, H.; Kutomi, G.; Okita, K.; et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1alpha mRNA is translated. Cancer Lett. 2016, 376, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Alarcón Claudio, R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie Sohail, F. HNRNPA2B1 Is a Mediator of m6 A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø Cathrine, B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wei, J.; Cui, Y.-H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m6 A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef] [Green Version]
- Saikia, M.; Fu, Y.; Pavon-Eternod, M.; He, C.; Pan, T. Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs. RNA 2010, 16, 1317–1327. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Rauch, S.; Dai, Q.; Cui, X.; Zhang, Z.; Nachtergaele, S.; Sepich, C.; He, C.; Dickinson, B.C. Evolution of a reverse transcriptase to map N(1)-methyladenosine in human messenger RNA. Nat. Methods 2019, 16, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wang, T.; Gonzalez, G.; Wang, Y. Identification of YTH Domain-Containing Proteins as the Readers for N1-Methyladenosine in RNA. Anal. Chem. 2018, 90, 6380–6384. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Li, X.; Yi, C. N1-methyladenosine methylome in messenger RNA and non-coding RNA. Curr. Opin. Chem. Biol. 2018, 45, 179–186. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.-W.; et al. Base-Resolution Mapping Reveals Distinct m1 A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol. Cell 2017, 68, 993–1005.e9. [Google Scholar] [CrossRef] [Green Version]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Liu, F.; Clark, W.; Luo, G.; Wang, X.; Fu, Y.; Wei, J.; Wang, X.; Hao, Z.; Dai, Q.; Zheng, G.; et al. ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell 2016, 167, 816–828.e16. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Qi, M.; Shen, B.; Luo, G.; Wu, Y.; Li, J.; Lu, Z.; Zheng, Z.; Dai, Q.; Wang, H. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019, 47, 2533–2545. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.-H.; Chambers, S.K. Human ALKBH3-induced m1A demethylation increases the CSF-1 mRNA stability in breast and ovarian cancer cells. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2019, 1862, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Yamato, I.; Sho, M.; Shimada, K.; Hotta, K.; Ueda, Y.; Yasuda, S.; Shigi, N.; Konishi, N.; Tsujikawa, K.; Nakajima, Y. PCA-1/ALKBH3 Contributes to Pancreatic Cancer by Supporting Apoptotic Resistance and Angiogenesis. Cancer Res. 2012, 72, 4829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, W.E.; Volkin, E. Nucleoside-5′-Phosphates from Ribonucleic Acid. Nature 1951, 167, 483–484. [Google Scholar] [CrossRef]
- Ge, J.; Yu, Y.-T. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci. 2013, 38, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karijolich, J.; Yi, C.; Yu, Y.-T. Transcriptome-wide dynamics of RNA pseudouridylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, E.; Chandler, J.C.; Varga, M.; Tahoun, M.; Menyhard, D.K.; Schay, G.; Goncalves, T.; Hamar, R.; Legradi, R.; Szekeres, A.; et al. Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc. Natl. Acad. Sci. USA 2020, 117, 15137–15147. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Ludovini, V.; Treré, D.; Siggillino, A.; Vannucci, J.; Bellezza, G.; Crinò, L.; Montanaro, L. Dyskerin and TERC expression may condition survival in lung cancer patients. Oncotarget 2015, 6, 21755–21760. [Google Scholar] [CrossRef]
- Sieron, P.; Hader, C.; Hatina, J.; Engers, R.; Wlazlinski, A.; Müller, M.; Schulz, W.A. DKC1 overexpression associated with prostate cancer progression. Br. J. Cancer 2009, 101, 1410–1416. [Google Scholar] [CrossRef] [Green Version]
- Montanaro, L.; Calienni, M.; Bertoni, S.; Rocchi, L.; Sansone, P.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Carnicelli, D.; et al. Novel Dyskerin-Mediated Mechanism of p53 Inactivation through Defective mRNA Translation. Cancer Res. 2010, 70, 4767. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Weng, H.; Deng, X.; Chen, J. RNA Modifications in Cancer: Functions, Mechanisms, and Therapeutic Implications. Annu. Rev. Cancer Biol. 2020, 4, 221–240. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Patton, J.R.; Davis, S.L.; Florence, B.; Ames, S.J.; Spanjaard, R.A. Regulation of Nuclear Receptor Activity by a Pseudouridine Synthase through Posttranscriptional Modification of Steroid Receptor RNA Activator. Mol. Cell 2004, 15, 549–558. [Google Scholar] [CrossRef]
- Jana, S.; Hsieh, A.C.; Gupta, R. Reciprocal amplification of caspase-3 activity by nuclear export of a putative human RNA-modifying protein, PUS10 during TRAIL-induced apoptosis. Cell Death Dis. 2017, 8, e3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Li, G.; Cheng, B.; Jiang, R. ZFAS1 functions as an oncogenic long non-coding RNA in bladder cancer. Biosci. Rep. 2018, 38, BSR20180475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Gao, H.; Li, S.; Ni, X.; Zhu, Z. Long non-coding RNA ZFAS1 correlates with clinical progression and prognosis in cancer patients. Oncotarget 2017, 8, 61561–61569. [Google Scholar] [CrossRef]
- Grammatikakis, I.; Panda, A.C.; Abdelmohsen, K.; Gorospe, M. Long noncoding RNAs(lncRNAs) and the molecular hallmarks of aging. Aging 2014, 6, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Baena-Del Valle, J.A.; Zheng, Q.; Esopi, D.M.; Rubenstein, M.; Hubbard, G.K.; Moncaliano, M.C.; Hruszkewycz, A.; Vaghasia, A.; Yegnasubramanian, S.; Wheelan, S.J.; et al. MYC drives overexpression of telomerase RNA (hTR/TERC) in prostate cancer. J. Pathol. 2018, 244, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.W.; Liu, J.; Liu, Q.; Xu, Q.H.; Li, T.F.; Jin, S.; Xia, T.S. LncRNA SNHG7 promotes the proliferation and inhibits apoptosis of gastric cancer cells by repressing the P15 and P16 expression. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4613–4622. [Google Scholar]
- Tian, T.; Qiu, R.; Qiu, X. SNHG1 promotes cell proliferation by acting as a sponge of miR-145 in colorectal cancer. Oncotarget 2018, 9, 2128–2139. [Google Scholar] [CrossRef]
- Liu, L.; Shi, Y.; Shi, J.; Wang, H.; Sheng, Y.; Jiang, Q.; Chen, H.; Li, X.; Dong, J. The long non-coding RNA SNHG1 promotes glioma progression by competitively binding to miR-194 to regulate PHLDA1 expression. Cell Death Dis. 2019, 10, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes, M.R.; Balzeau, J.; Hagan, J.P. 3′ RNA Uridylation in Epitranscriptomics, Gene Regulation, and Disease. Front. Mol. Biosci. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.-Q.; Lou, Y.-F.; He, Z.-G.; Ji, M. Nucleotidyl transferase TUT1 inhibits lipogenesis in osteosarcoma cells through regulation of microRNA-24 and microRNA-29a. Tumor Biol. 2014, 35, 11829–11835. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Gong, Y.; Zhou, H.; Wang, M.; Kong, L.; Liu, J.; An, T.; Zhu, H.; Li, Y. Star-PAP, a poly(A) polymerase, functions as a tumor suppressor in an orthotopic human breast cancer model. Cell Death Dis. 2017, 8, e2582. [Google Scholar] [CrossRef] [Green Version]
- Alajez, N.M.; Shi, W.; Wong, D.; Lenarduzzi, M.; Waldron, J.; Weinreb, I.; Liu, F.F. Lin28b promotes head and neck cancer progression via modulation of the insulin-like growth factor survival pathway. Oncotarget 2012, 3, 1641–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.Y.; Wei, T.Z.; Luo, Q.C.; Wu, Q.W.; Liu, Q.F.; Yang, M.; Ye, G.D.; Wu, J.F.; Chen, Y.Y.; Sun, G.B.; et al. The Wnt-beta-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J. Cell Sci. 2013, 126, 2877–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, J.E.; Chang, H.-M.; Piskounova, E.; Gregory, R.I. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA 2012, 18, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.-J.; Park, J.-E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-Uridylation of Pre-MicroRNA as a Key Step in the Biogenesis of Group II let-7 MicroRNAs. Cell 2012, 151, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Ustianenko, D.; Hrossova, D.; Potesil, D.; Chalupnikova, K.; Hrazdilova, K.; Pachernik, J.; Cetkovska, K.; Uldrijan, S.; Zdrahal, Z.; Vanacova, S. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA 2013, 19, 1632–1638. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.W.; Liu, Y.; Manjunath, H.; Acharya, A.; Jones, B.T.; Zhang, H.; Chen, B.; Ramalingam, H.; Hammer, R.E.; Xie, Y.; et al. Loss of Dis3l2 partially phenocopies Perlman syndrome in mice and results in up-regulation of Igf2 in nephron progenitor cells. Genes Dev. 2018, 32, 903–908. [Google Scholar] [CrossRef] [Green Version]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol. Cell 2019, 74, 1278–1290.e1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Fujiwara, M.; Hori, M.; Okada, K.; Yazama, F.; Konishi, H.; Xiao, Y.; Qi, G.; Shimamoto, F.; Ota, T.; et al. tRNA modifying enzymes, NSUN2 and METTL1, determine sensitivity to 5-fluorouracil in HeLa cells. PLoS Genet. 2014, 10, e1004639. [Google Scholar] [CrossRef]
- Gustavsson, M.; Ronne, H. Evidence that tRNA modifying enzymes are important in vivo targets for 5-fluorouracil in yeast. RNA 2008, 14, 666–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowling, V.H. Enhanced mRNA cap methylation increases cyclin D1 expression and promotes cell transformation. Oncogene 2010, 29, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilescu, C.; Dragomir, M.; Tanase, M.; Giza, D.; Purnichescu-Purtan, R.; Chen, M.; Yeung, S.J.; Calin, G.A. Circulating miRNAs in sepsis-A network under attack: An in-silico prediction of the potential existence of miRNA sponges in sepsis. PLoS ONE 2017, 12, e0183334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.; Mafra, A.C.P.; Dias, S.M.G.; Vasilescu, C.; Calin, G.A. Using microRNA Networks to Understand Cancer. Int. J. Mol. Sci. 2018, 19, 1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roosbroeck, K.; Bayraktar, R.; Calin, S.; Bloehdorn, J.; Dragomir, M.P.; Okubo, K.; Bertilaccio, M.T.S.; Zupo, S.; You, M.J.; Gaidano, G.; et al. The involvement of microRNA in the pathogenesis of Richter syndrome. Haematologica 2019, 104, 1004–1015. [Google Scholar] [CrossRef]
- Fuentes-Mattei, E.; Bayraktar, R.; Manshouri, T.; Silva, A.M.; Ivan, C.; Gulei, D.; Fabris, L.; Soares do Amaral, N.; Mur, P.; Perez, C.; et al. miR-543 regulates the epigenetic landscape of myelofibrosis by targeting TET1 and TET2. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Volinia, S.; Galasso, M.; Costinean, S.; Tagliavini, L.; Gamberoni, G.; Drusco, A.; Marchesini, J.; Mascellani, N.; Sana, M.E.; Abu Jarour, R.; et al. Reprogramming of miRNA networks in cancer and leukemia. Genome Res. 2010, 20, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Sieuwerts, A.M.; Willis, S.; Burns, M.B.; Look, M.P.; Meijer-Van Gelder, M.E.; Schlicker, A.; Heideman, M.R.; Jacobs, H.; Wessels, L.; Leyland-Jones, B.; et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Horm. Cancer 2014, 5, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Okugawa, Y.; Toiyama, Y.; Shigeyasu, K.; Yamamoto, A.; Shigemori, T.; Yin, C.; Ichikawa, T.; Yasuda, H.; Fujikawa, H.; Yoshiyama, S.; et al. Enhanced AZIN1 RNA editing and overexpression of its regulatory enzyme ADAR1 are important prognostic biomarkers in gastric cancer. J. Transl. Med. 2018, 16, 366. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.H.; Qamra, A.; Tan, K.T.; Guo, J.; Yang, H.; Qi, L.; Lin, J.S.; Ng, V.H.; Song, Y.; Hong, H.; et al. ADAR-Mediated RNA Editing Predicts Progression and Prognosis of Gastric Cancer. Gastroenterology 2016, 151, 637–650.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.M.; Zeng, H.D.; Zhang, C.Y.; Xu, J.W. Identification of METTL3 as an Adverse Prognostic Biomarker in Hepatocellular Carcinoma. Dig. Dis. Sci. 2020. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.F.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, W.; Yao, S.; Zhou, Y.; Liu, Y.; Huang, P.; Zhou, A.; Liu, J.; Che, L.; Li, J. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m6 A reader YTHDF3. Mol. Cancer 2019, 18, 143. [Google Scholar] [CrossRef]
- Petrescu, G.E.D.; Sabo, A.A.; Torsin, L.I.; Calin, G.A.; Dragomir, M.P. MicroRNA based theranostics for brain cancer: Basic principles. J. Exp. Clin. Cancer Res. 2019, 38, 231. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Pichler, M.; Rodriguez-Aguayo, C.; Nam, S.Y.; Dragomir, M.P.; Bayraktar, R.; Anfossi, S.; Knutsen, E.; Ivan, C.; Fuentes-Mattei, E.; Lee, S.K.; et al. Therapeutic potential of FLANC, a novel primate-specific long non-coding RNA in colorectal cancer. Gut 2020. [Google Scholar] [CrossRef]
- Selvam, C.; Mutisya, D.; Prakash, S.; Ranganna, K.; Thilagavathi, R. Therapeutic potential of chemically modified siRNA: Recent trends. Chem. Biol. Drug Des. 2017, 90, 665–678. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Ribich, S.; Copeland, R.A. RNA-modifying proteins as anticancer drug targets. Nat. Rev. Drug Discov. 2018, 17, 435–453. [Google Scholar] [CrossRef]
- Li, T.; Hu, P.S.; Zuo, Z.; Lin, J.F.; Li, X.; Wu, Q.N.; Chen, Z.H.; Zeng, Z.L.; Wang, F.; Zheng, J.; et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer 2019, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Bedi, R.K.; Huang, D.; Eberle, S.A.; Wiedmer, L.; Sledz, P.; Caflisch, A. Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. Chem. Med. Chem. 2020, 15, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Li, X.; Mu, Z.; Zhou, J.; Zhou, P.; Xie, C.; Jiang, S. FTO Inhibition Enhances the Antitumor Effect of Temozolomide by Targeting MYC-miR-155/23a Cluster-MXI1 Feedback Circuit in Glioma. Cancer Res. 2020, 80, 3945–3958. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e10. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Cancer | ncRNA | Enzyme | Editing | Effects | Consequence | Ref |
|---|---|---|---|---|---|---|
| Breast, lung, ovarian, renal cancer cell lines | miR-379-5p | ADAR2 | ↓ | ↑ ADGRE5 | cell proliferation and inhibited apoptosis | [36] |
| Breast, ovarian, renal cancer cell lines | mir-200b-3p | ADAR1/2 | ↑ | ↑ ZEB1/2 ↓ LIFR | cell invasion and migration | [34] |
| Glioblastoma | miR-589-3p | ADAR2 | ↓ | ↓ PCDH2 ↑ ADAM12 | cell proliferation, invasion and migration | [42] |
| Glioblastoma | miR-221/222 miR-21 | ADAR2 | ↓ | ↓ p27Kip1 | cell proliferation | [40] |
| Glioblastoma | miR-376a cluster | ADAR1 | ↓ | ↓ RAP2A ↑ AMFR | cell invasion and migration | [41] |
| Hepatocellular carcinoma | miR-214 | ADAR2 | ↑ | ↑ RAB15 | cell proliferation, invasion, angiogenesis | [45] |
| Leukemia | let-7 | ADAR1 | ↑ | ↑ LIN28B | enhanced self-renewal | [46] |
| Melanoma | miR-378a-3p | ADAR1 | ↓ | ↑ PARVA | metastasis | [44] |
| Melanoma | miR-455-5p | ADAR1 | ↓ | ↓ CPEB1 | tumor growth and metastasis | [43] |
| NSCLC | miR-381-3p | ADAR1 | ↑ | N/A | cell proliferation, invasion | [47] |
| MDS/ MPN | CCAT2 at the rs6983267 SNP | N/A | ↑ | Homozygous GG/TT -> heterozygous G/T | low risk MDS | [29] |
| Chem. Mod. | Cancer | ncRNA | Enzyme | Mod | Effects | Consequence | Ref |
|---|---|---|---|---|---|---|---|
| m5C | Pancreatic cancer and colorectal cancer | miR-200c-3p, miR-21-3p | NSUN2 | ↑ | interaction between miRNA and AGO is modified | N/A | [95] |
| GBM | miR-16-5p, miR-181a-5p, miR-181b-5p, miR-181d-5p, miR-210-3p | DNMT3A/ AGO4 | ↑ | Cytosine methylated miR-181a-5p loses its capacity to suppress BIM (apoptosis regulator) | Decreases apoptosis and increases invasion and proliferation rate. | [96] | |
| ESCC | NMR | NSUN2 | ↑ | Attenuates the methylation of PLOD3, COL4A5, LAMB1, HSPG2 | Increases migration and invasion | [97] | |
| m6A | Pancreatic cancer and colorectal cancer | let-7a-5p, miR-17-5p | METTL3 and METTL4 | ↑ | interaction between miRNA and AGO is modified | m6A modified miRNAs have a reduced ability to inhibit mRNAs | [95] |
| Ψ | Lung cancer | TERC | N/A | N/A | N/A | Telomere shortening, pro-oncogenic | [146] |
| Uridylation | Osteosarcoma | miR-24, miR-29a | TUT1 | N/A | ↑ PPARgamma ↑ SREBP-1c | Stimulates lipogenesis, tumor progression | [163] |
| Breast cancer | let-7a, let-7f | LIN28 | ↑ | ↑ HRAS, ↑ HMGA2, | Expansion of cancer stem cells | [166] | |
| Head and Neck cancer | let-7 family | LIN28B | ↑ | ↑ HMGA2, ↑ CCND2, ↑ IGF1R, ↑ IGF2BP2 | Oncogenesis and cancer progression | [165] | |
| m7G | Lung and colon cancer cells | let-7 family (let-7-5p seed), hsa-miR-125a-5p, hsa-miR-92b-3p | METTL1 | ↑ | ↓ HMGA2 | Cell migration | [171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torsin, L.I.; Petrescu, G.E.D.; Sabo, A.A.; Chen, B.; Brehar, F.M.; Dragomir, M.P.; Calin, G.A. Editing and Chemical Modifications on Non-Coding RNAs in Cancer: A New Tale with Clinical Significance. Int. J. Mol. Sci. 2021, 22, 581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020581
Torsin LI, Petrescu GED, Sabo AA, Chen B, Brehar FM, Dragomir MP, Calin GA. Editing and Chemical Modifications on Non-Coding RNAs in Cancer: A New Tale with Clinical Significance. International Journal of Molecular Sciences. 2021; 22(2):581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020581
Chicago/Turabian StyleTorsin, Ligia I., George E. D. Petrescu, Alexandru A. Sabo, Baoqing Chen, Felix M. Brehar, Mihnea P. Dragomir, and George A. Calin. 2021. "Editing and Chemical Modifications on Non-Coding RNAs in Cancer: A New Tale with Clinical Significance" International Journal of Molecular Sciences 22, no. 2: 581. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020581