Elevated Expression of Cathepsin K in Periodontal Ligament Fibroblast by Inflammatory Cytokines Accelerates Osteoclastogenesis via Paracrine Mechanism in Periodontal Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
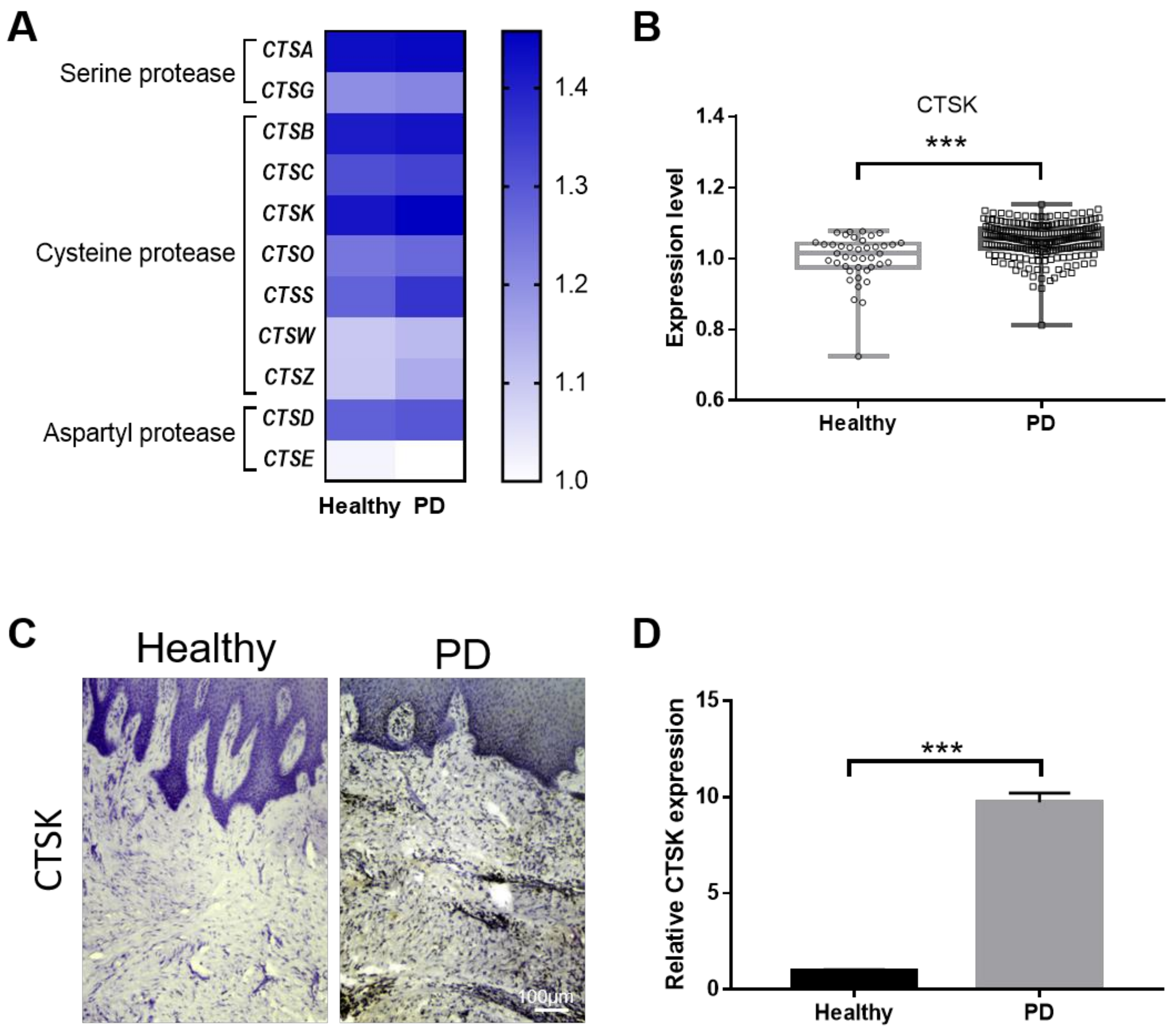
2.1. Gingival Tissue from PD Patients Has Increased Levels of CTSK
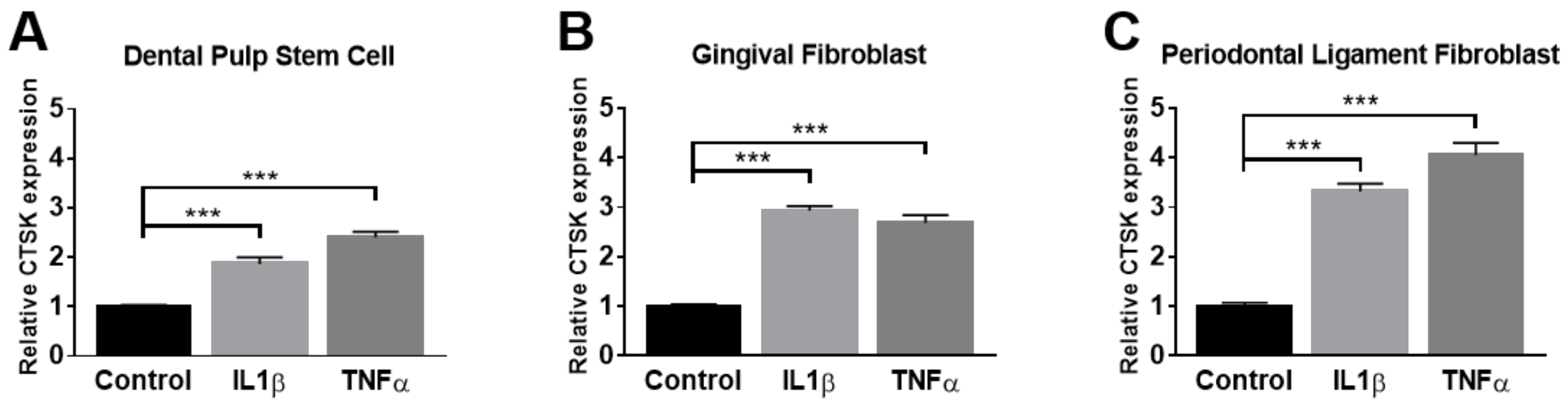
2.2. Administration of TNFα and IL-1β Elevates the Expression of CTSK in Osteoclastogenesis-Supporting Cells
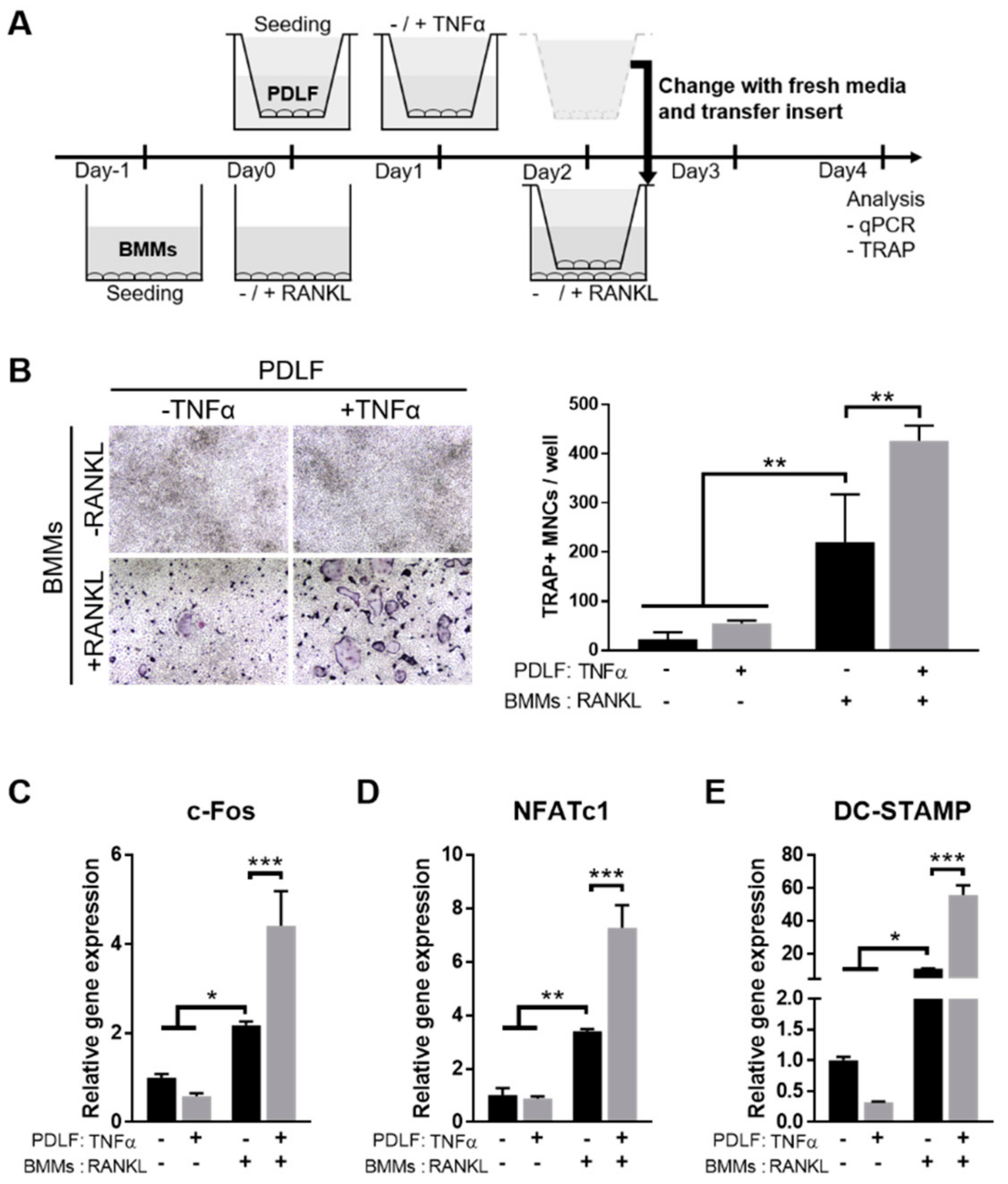
2.3. TNFα Enhances the Osteoclastogenesis-Supporting Activity of PDLFs
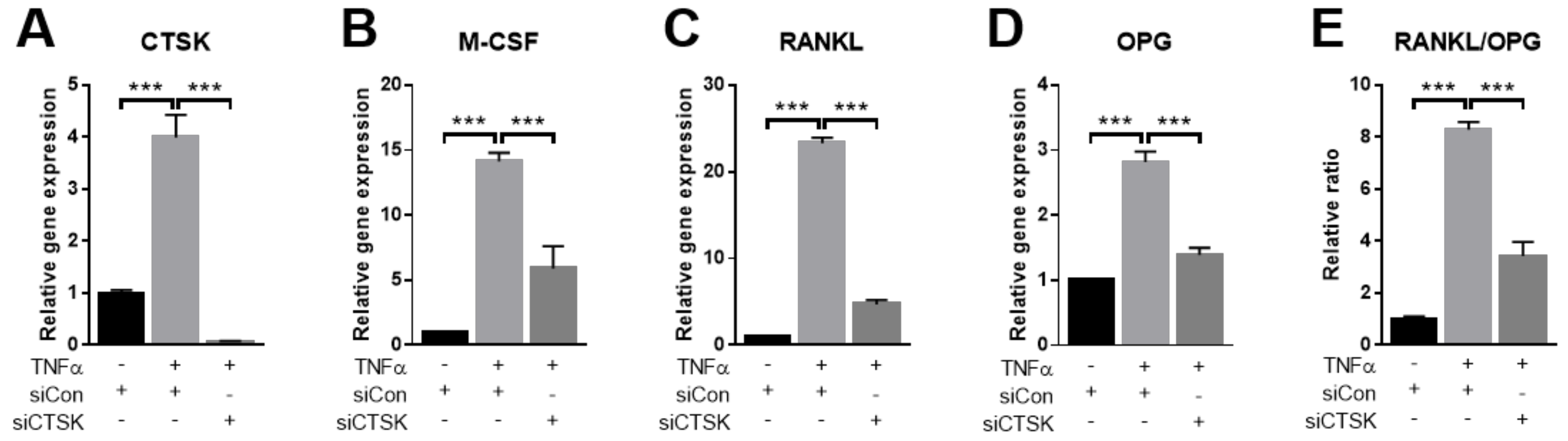
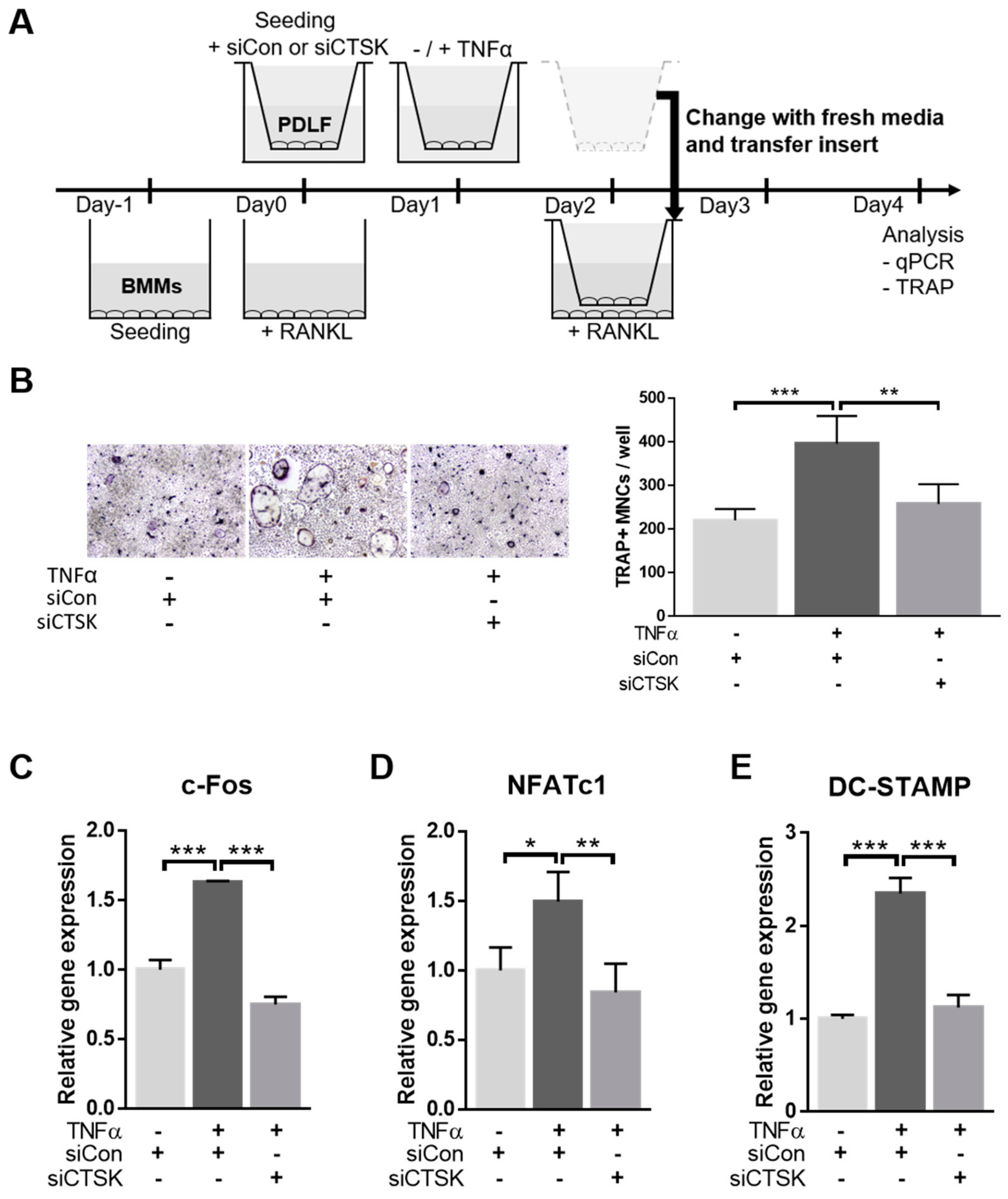
2.4. TNFα-Stimulated Expression of Osteoclastogenesis-Modulating Factors Were Ameliorated by CTSK-Silencing in PDLFs
2.5. CTSK Is Responsible for TNFα-Induced Paracrine Function of PDLFs in Osteoclastogenesis
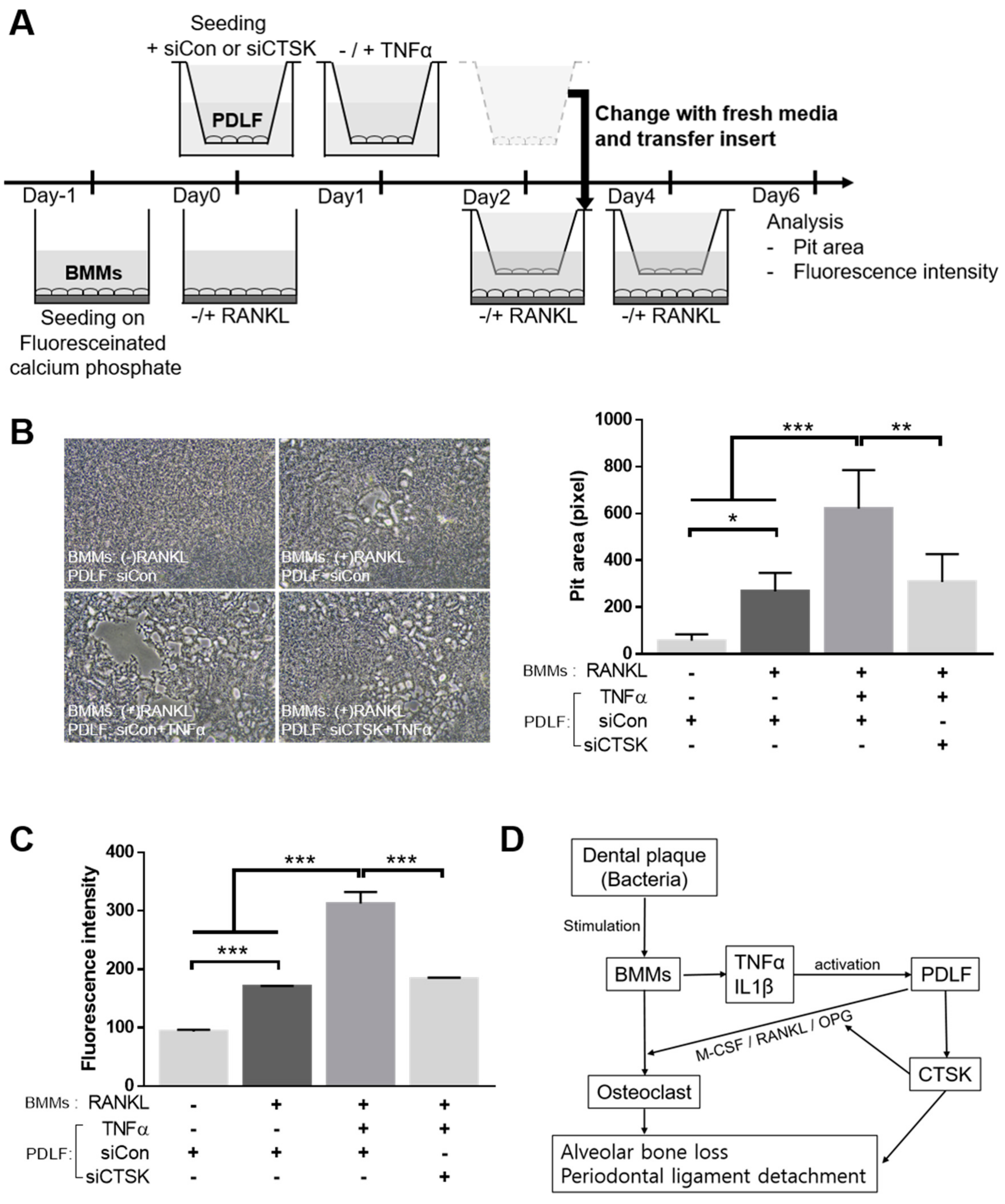
2.6. CTSK Is Involved in TNFα-Activated Paracrine Function of PDLF in Bone Resorption
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Cytokine Treatment
4.3. BMM Isolation and Osteoclastogenic Cultures
4.4. Public Data Acquisition and Processing
4.5. Immunohistochemistry
4.6. qPCR
4.7. Transwell Co-Culture Osteoclastogenesis System
4.8. Transfection with siRNA
4.9. Resorption Assay
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CTSK | Cathepsin K |
| PD | Periodontal disease |
| TNFα | Tumor necrosis factor-alpha |
| PDLF | Periodontal ligament fibroblast |
| DPSC | Dental pulp stem cell |
| GF | Gingival fibroblast |
| RANKL | Receptor activator of nuclear factor kappa-B ligand |
| M-CSF | Macrophage-colony stimulating factor |
| OPG | Osteoprotegerin |
| BMM | Bone marrow-derived macrophage |
| TRAP | Tartrate-resistant acid phosphatase |
References
- Williams, R.C. Periodontal disease. N. Engl. J. Med. 1990, 322, 373–382. [Google Scholar] [CrossRef]
- Isola, G.; Polizzi, A.; Patini, R.; Ferlito, S.; Alibrandi, A.; Palazzo, G. Association among serum and salivary A. actinomycetemcomitans specific immunoglobulin antibodies and periodontitis. BMC Oral Health 2020, 20, 283. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. The bacterial etiology of destructive periodontal disease: Current concepts. J. Periodontol. 1992, 63, 322–331. [Google Scholar] [CrossRef]
- Ebersole, J.L.; Kirakodu, S.; Novak, M.J.; Stromberg, A.J.; Shen, S.; Orraca, L.; Gonzalez-Martinez, J.; Burgos, A.; Gonzalez, O.A. Cytokine gene expression profiles during initiation, progression and resolution of periodontitis. J. Clin. Periodontol. 2014, 41, 853–861. [Google Scholar] [CrossRef]
- Gorska, R.; Gregorek, H.; Kowalski, J.; Laskus-Perendyk, A.; Syczewska, M.; Madalinski, K. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. J. Clin. Periodontol. 2003, 30, 1046–1052. [Google Scholar] [CrossRef]
- Isola, G.; Lo Giudice, A.; Polizzi, A.; Alibrandi, A.; Murabito, P.; Indelicato, F. Identification of the different salivary Interleukin-6 profiles in patients with periodontitis: A cross-sectional study. Arch. Oral Biol. 2020, 122, 104997. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Sterrett, J.D. The osteoclast and periodontitis. J. Clin. Periodontol. 1986, 13, 258–269. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Taubman, M.A.; Valverde, P.; Han, X.; Kawai, T. Immune Response: The Key to Bone Resorption in Periodontal Disease. J. Periodontol. 2005, 76 (Suppl. S11), 2033–2041. [Google Scholar] [CrossRef]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Takayanagi, H. Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol. Rev. 2009, 231, 241–256. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Kukita, T.; Wada, N.; Kukita, A.; Kakimoto, T.; Sandra, F.; Toh, K.; Nagata, K.; Iijima, T.; Horiuchi, M.; Matsusaki, H.; et al. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J. Exp. Med. 2004, 200, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Drake, F.H.; Dodds, R.A.; James, I.E.; Connor, J.R.; Debouck, C.; Richardson, S.; Lee-Rykaczewski, E.; Coleman, L.; Rieman, D.; Barthlow, R.; et al. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J. Biol. Chem. 1996, 271, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Drake, M.T.; Clarke, B.L.; Oursler, M.J.; Khosla, S. Cathepsin K Inhibitors for Osteoporosis: Biology, Potential Clinical Utility, and Lessons Learned. Endocr. Rev. 2017, 38, 325–350. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; He, X.W.; Shi, Y.H.; Liu, Y.S.; Liu, F.D.; Hu, Y.; Zhuang, M.T.; Feng, X.Y.; Zhao, L.; Zhao, B.Q.; et al. Cathepsin K Knockout Exacerbates Haemorrhagic Transformation Induced by Recombinant Tissue Plasminogen Activator After Focal Cerebral Ischaemia in Mice. Cell Mol. Neurobiol. 2019, 39, 823–831. [Google Scholar] [CrossRef]
- Cheng, X.W.; Kikuchi, R.; Ishii, H.; Yoshikawa, D.; Hu, L.; Takahashi, R.; Shibata, R.; Ikeda, N.; Kuzuya, M.; Okumura, K.; et al. Circulating cathepsin K as a potential novel biomarker of coronary artery disease. Atherosclerosis 2013, 228, 211–216. [Google Scholar] [CrossRef]
- Lutgens, E.; Lutgens, S.P.; Faber, B.C.; Heeneman, S.; Gijbels, M.M.; de Winther, M.P.; Frederik, P.; van der Made, I.; Daugherty, A.; Sijbers, A.M.; et al. Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation 2006, 113, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Hirai, T.; Kunigami, T.; Kamano, S.; Gober, H.J.; Okamoto, K.; Nishikawa, K.; Latz, E.; Golenbock, D.T.; Aoki, K.; et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science 2008, 319, 624–627. [Google Scholar] [CrossRef]
- Oshiro, T.; Shibasaki, Y.; Martin, T.J.; Sasaki, T. Immunolocalization of vacuolar-type H+-ATPase, cathepsin K, matrix metalloproteinase-9, and receptor activator of NFkappaB ligand in odontoclasts during physiological root resorption of human deciduous teeth. Anat. Rec. 2001, 264, 305–311. [Google Scholar] [CrossRef]
- Mogi, M.; Otogoto, J. Expression of cathepsin-K in gingival crevicular fluid of patients with periodontitis. Arch. Oral Biol. 2007, 52, 894–898. [Google Scholar] [CrossRef]
- Yongchaitrakul, T.; Lertsirirangson, K.; Pavasant, P. Human periodontal ligament cells secrete macrophage colony-stimulating factor in response to tumor necrosis factor-alpha in vitro. J. Periodontol. 2006, 77, 955–962. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Bostanci, N.; Hashim, A.; Johansson, A.; Aduse-Opoku, J.; Curtis, M.A.; Hughes, F.J. Regulation of RANKL and OPG gene expression in human gingival fibroblasts and periodontal ligament cells by Porphyromonas gingivalis: A putative role of the Arg-gingipains. Microb. Pathog. 2007, 43, 46–53. [Google Scholar] [CrossRef]
- Hummel, K.M.; Petrow, P.K.; Franz, J.K.; Muller-Ladner, U.; Aicher, W.K.; Gay, R.E.; Bromme, D.; Gay, S. Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction. J. Rheumatol. 1998, 25, 1887–1894. [Google Scholar]
- Sukhova, G.K.; Shi, G.P.; Simon, D.I.; Chapman, H.A.; Libby, P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Investig. 1998, 102, 576–583. [Google Scholar] [CrossRef] [Green Version]
- Konttinen, Y.T.; Mandelin, J.; Li, T.F.; Salo, J.; Lassus, J.; Liljestrom, M.; Hukkanen, M.; Takagi, M.; Virtanen, I.; Santavirta, S. Acidic cysteine endoproteinase cathepsin K in the degeneration of the superficial articular hyaline cartilage in osteoarthritis. Arthritis Rheum. 2002, 46, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Otogoto, J.; Ota, N.; Inagaki, H.; Minami, M.; Kojima, K. Interleukin 1 beta, interleukin 6, beta 2-microglobulin, and transforming growth factor-alpha in gingival crevicular fluid from human periodontal disease. Arch. Oral Biol. 1999, 44, 535–539. [Google Scholar] [CrossRef]
- Offenbacher, S.; Odle, B.M.; Van Dyke, T.E. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J. Periodontal. Res. 1986, 21, 101–112. [Google Scholar] [CrossRef]
- Beklen, A.; Al-Samadi, A.; Konttinen, Y.T. Expression of cathepsin K in periodontitis and in gingival fibroblasts. Oral Dis. 2015, 21, 163–169. [Google Scholar] [CrossRef]
- Leblebicioglu, B.; Lim, J.S.; Cario, A.C.; Beck, F.M.; Walters, J.D. pH changes observed in the inflamed gingival crevice modulate human polymorphonuclear leukocyte activation in vitro. J. Periodontol. 1996, 67, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Newman, H.N. Focal infection. J. Dent. Res. 1996, 75, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Mattila, K.J.; Nieminen, M.S.; Valtonen, V.V.; Rasi, V.P.; Kesaniemi, Y.A.; Syrjala, S.L.; Jungell, P.S.; Isoluoma, M.; Hietaniemi, K.; Jokinen, M.J. Association between dental health and acute myocardial infarction. BMJ 1989, 298, 779–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonetti, M.S.; Van Dyke, T.E.; Working Group 1 of the Joint EFP/AAP Workshop. Periodontitis and atherosclerotic cardiovascular disease: Consensus report of the Joint EFP/AAPWorkshop on Periodontitis and Systemic Diseases. J. Periodontol. 2013, 84 (Suppl. S4), S24–S29. [Google Scholar] [CrossRef]
- Isola, G.; Polizzi, A.; Alibrandi, A.; Williams, R.C.; Leonardi, R. Independent impact of periodontitis and cardiovascular disease on elevated soluble urokinase-type plasminogen activator receptor (suPAR) levels. J. Periodontol. 2020. [Google Scholar] [CrossRef]
- Chapple, I.L.; Genco, R.; Working Group 2 of Joint EFP/AAP Workshop. Diabetes and periodontal diseases: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J. Clin. Periodontol. 2013, 40 (Suppl. S14), S106–S112. [Google Scholar] [CrossRef]
- Linden, G.J.; Herzberg, M.C.; Working Group 4 of Joint EFP/AAP Workshop. Periodontitis and systemic diseases: A record of discussions of working group 4 of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J. Clin. Periodontol. 2013, 40 (Suppl. S14), S20–S23. [Google Scholar] [CrossRef]
- Hao, L.; Zhu, G.; Lu, Y.; Wang, M.; Jules, J.; Zhou, X.; Chen, W. Deficiency of cathepsin K prevents inflammation and bone erosion in rheumatoid arthritis and periodontitis and reveals its shared osteoimmune role. FEBS Lett. 2015, 589, 1331–1339. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Yin, W.; Yang, L.; Xue, L.; Ren, J.; Wei, W.; Lu, Q.; Ding, H.; Liu, Z.; Nabar, N.R.; et al. Inhibition of Ctsk alleviates periodontitis and comorbid rheumatoid arthritis via downregulation of the TLR9 signalling pathway. J. Clin. Periodontol. 2019, 46, 286–296. [Google Scholar] [CrossRef]
- Krajewski, A.C.; Biessei, J.; Kunze, M.; Maersch, S.; Perabo, L.; Noack, M.J. Influence of lipopolysaccharide and interleukin-6 on RANKL and OPG expression and release in human periodontal ligament cells. APMIS 2009, 117, 746–754. [Google Scholar] [CrossRef]
- Lekic, P.; McCulloch, C.A. Periodontal ligament cell population: The central role of fibroblasts in creating a unique tissue. Anat. Rec. 1996, 245, 327–341. [Google Scholar] [CrossRef]
- Nahm, D.S.; Kim, H.J.; Mah, J.; Baek, S.H. In vitro expression of matrix metalloproteinase-1, tissue inhibitor of metalloproteinase-1 and transforming growth factor-beta1 in human periodontal ligament fibroblasts. Eur. J. Orthod. 2004, 26, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Van der Pauw, M.T.; Van den Bos, T.; Everts, V.; Beertsen, W. Enamel matrix-derived protein stimulates attachment of periodontal ligament fibroblasts and enhances alkaline phosphatase activity and transforming growth factor beta1 release of periodontal ligament and gingival fibroblasts. J. Periodontol. 2000, 71, 31–43. [Google Scholar] [CrossRef]
- Karst, M.; Gorny, G.; Galvin, R.J.; Oursler, M.J. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J. Cell Physiol. 2004, 200, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Van den Brule, S.; Misson, P.; Buhling, F.; Lison, D.; Huaux, F. Overexpression of cathepsin K during silica-induced lung fibrosis and control by TGF-beta. Respir. Res. 2005, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Yoo, J.H.; Lee, J.H.; Lee, Y.; Bae, M.K.; Kim, Y.D.; Kim, H.J. Connective tissue growth factor (CTGF) regulates the fusion of osteoclast precursors by inhibiting Bcl6 in periodontitis. Int. J. Med. Sci. 2020, 17, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Torrente, A.; Lukk, M.; Xue, V.; Parkinson, H.; Rung, J.; Brazma, A. Identification of Cancer Related Genes Using a Comprehensive Map of Human Gene Expression. PLoS ONE 2016, 11, e0157484. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, S.C.; Kim, Y.N.; Choi, Y.; Joo, J.-Y.; Hwang, J.J.; Bae, M.-K.; Kim, H.J. Elevated Expression of Cathepsin K in Periodontal Ligament Fibroblast by Inflammatory Cytokines Accelerates Osteoclastogenesis via Paracrine Mechanism in Periodontal Disease. Int. J. Mol. Sci. 2021, 22, 695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020695
Heo SC, Kim YN, Choi Y, Joo J-Y, Hwang JJ, Bae M-K, Kim HJ. Elevated Expression of Cathepsin K in Periodontal Ligament Fibroblast by Inflammatory Cytokines Accelerates Osteoclastogenesis via Paracrine Mechanism in Periodontal Disease. International Journal of Molecular Sciences. 2021; 22(2):695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020695
Chicago/Turabian StyleHeo, Soon Chul, Yu Na Kim, YunJeong Choi, Ji-Young Joo, Jae Joon Hwang, Moon-Kyoung Bae, and Hyung Joon Kim. 2021. "Elevated Expression of Cathepsin K in Periodontal Ligament Fibroblast by Inflammatory Cytokines Accelerates Osteoclastogenesis via Paracrine Mechanism in Periodontal Disease" International Journal of Molecular Sciences 22, no. 2: 695. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020695