Trauma, a Matter of the Heart—Molecular Mechanism of Post-Traumatic Cardiac Dysfunction

{kind=link}
Abstract
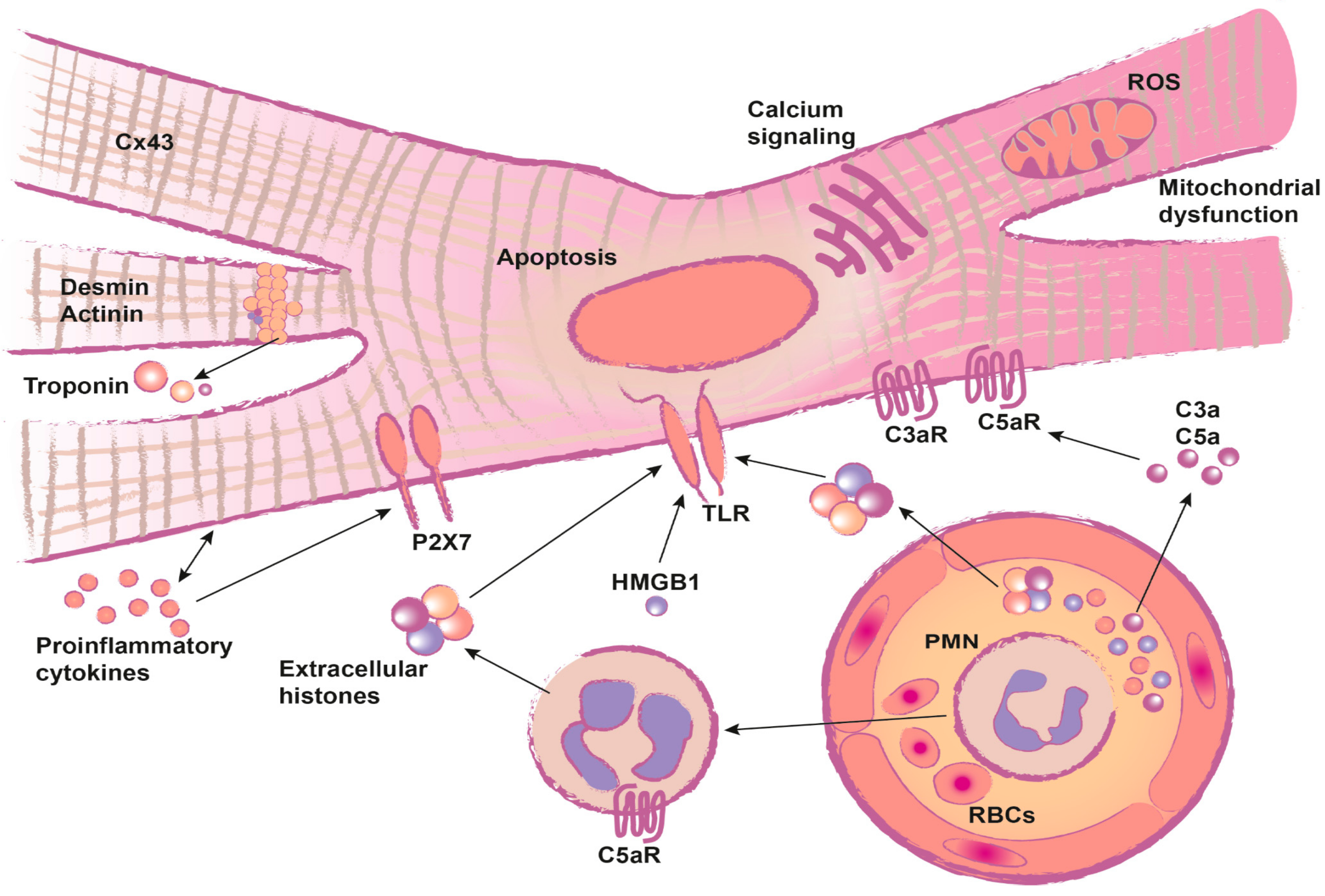
:1. Introduction
2. Review
2.1. Troponin and Heart Fatty Acid Binding Protein (HFABP): Markers for Post-Traumatic Cardiac Injury
2.2. Functional Impairment after Traumatic Heart Injury
2.2.1. Inflammation
2.2.2. Systemic Release of DAMPs and the Impact on the Heart
2.2.3. Complement Activation after Trauma and the Impact on the Heart
2.2.4. Structural Alterations
2.2.5. Cardiac Metabolism after Trauma
2.3. Secondary/Indirect Cardiac Damage
2.4. Treatment Strategies
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | atrial fibrillation |
| AIS | abbreviated injury scale |
| Akt | protein kinase B |
| C5a/R | complement factor 5a/receptor |
| CINC | cytokine-induced neutrophil chemoattractant |
| CM | cardiomyocyte |
| DAMPs | danger-associated molecular patterns |
| ECG | electrocardiogram |
| ECHO | echocardiography |
| EF | ejection fraction |
| ERK | extracellular signal-regulated kinase |
| GLUT | glucose transporter |
| HF | heart failure |
| HFABP | heart type fatty acid binding protein |
| HMGB-1 | high mobility group box-1 protein |
| ICAM | intercellular adhesion molecule |
| ICU | intensive care unit |
| IL | interleukin |
| iNOs | inducible nitric oxide synthase |
| ISS | injury severity score |
| JAM | junctional adhesion molecule |
| MAPK | mitogen-activated protein kinase |
| MI | myocardial infarction |
| MMP | matrix metalloprotease |
| MPO | myeloperoxidase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NFkB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | nitric oxide |
| NT-proBNP | N-terminal B-type natriuretic peptide |
| PAMPs | pathogen-associated molecular patterns |
| PI3K | phosphoinositide 3-kinase |
| ROS | reactive oxygen species |
| SAH | subarachnoid hemorrhage |
| SAPS II | simplified acute physiology score |
| SERCA | sarco-/endoplasmic reticulum calcium ATPase |
| SIRS | systemic inflammatory response syndrome |
| TBI | traumatic brain injury |
| TISCI | trauma-induced secondary cardiac injury |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
| TnI/T | troponin I/T |
| TUNEL | terminal deoxynucleatidyl transferase dUTP nick end labeling |
| ZO-1 | Zonula occludens-1 |
References
- German Trauma Society (DGU). Committee on Emergency Medicine, Intensive Care and Trauma Management (Sektion NIS) and AUC—Academy for Trauma Surgery. In Annual Report 2015; German Trauma Society: Berlin, Germany, 2015. [Google Scholar]
- Rady, M.Y.; Edwards, J.D.; Nightingale, P. Early cardiorespiratory findings after severe blunt thoracic trauma and their relation to outcome. Br. J. Surg. 1992, 79, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Hanschen, M.; Kanz, K.-G.; Kirchhoff, C.; Khalil, P.N.; Wierer, M.; van Griensven, M.; Laugwitz, K.-L.; Biberthaler, P.; Lefering, R.; Huber-Wagner, S. Blunt cardiac injury in the severely injured-a retrospective multicentre study. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Nirgiotis, J.G.; Colon, R.; Sweeney, M.S. Blunt trauma to the heart: The pathophysiology of injury. J. Emerg. Med. 1990, 8, 617–623. [Google Scholar] [CrossRef]
- Crown, L.A.; Hawkins, W. Commotio cordis: Clinical implications of blunt cardiac trauma. Am. Fam. Physician 1997, 55, 2467–2470. [Google Scholar]
- Huber, S.; Biberthaler, P.; Delhey, P.; Trentzsch, H.; Winter, H.; van Griensven, M.; Lefering, R.; Huber-Wagner, S. Predictors of poor outcomes after significant chest trauma in multiply injured patients: A retrospective analysis from the German Trauma Registry (Trauma Register DGU®). Scand. J. Trauma Resusc. Emerg. Med. 2014, 22, 52. [Google Scholar] [CrossRef] [Green Version]
- Seguin, P.; Laviolle, B.; Maurice, A.; Leclercq, C.; Malledant, Y. Atrial fibrillation in trauma patients requiring intensive care. Intensive Care Med. 2006, 32, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Hadjizacharia, P.; O’Keeffe, T.; Brown, C.V.R.; Inaba, K.; Salim, A.; Chan, L.S.; Demetriades, D.; Rhee, P. Incidence, risk factors, and outcomes for atrial arrhythmias in trauma patients. Am. Surg. 2011, 77, 634–639. [Google Scholar] [CrossRef]
- Kalbitz, M.; Pressmar, J.; Stecher, J.; Weber, B.; Weiss, M.; Schwarz, S.; Miltner, E.; Gebhard, F.; Huber-Lang, M. The role of troponin in blunt cardiac injury after multiple trauma in humans. World J. Surg. 2017, 41, 162–169. [Google Scholar] [CrossRef]
- Vasile, V.C.; Chai, H.-S.; Abdeldayem, D.; Afessa, B.; Jaffe, A.S. Elevated cardiac troponin T levels in critically ill patients with sepsis. Am. J. Med. 2013, 126, 1114–1121. [Google Scholar] [CrossRef]
- Korff, S.; Katus, H.A.; Giannitsis, E. Differential diagnosis of elevated troponins. Heart 2006, 92, 987–993. [Google Scholar] [CrossRef] [Green Version]
- Bock, J.S.; Benitez, R.M. Blunt cardiac injury. Cardiol. Clin. 2012, 30, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.K.; Kalbitz, M.; Halbgebauer, R.; Eisele, P.; Messerer, D.A.C.; Weckbach, S.; Schultze, A.; Braumuller, S.; Gebhard, F.; Huber-Lang, M.S.; et al. Early structural changes of the heart after experimental polytrauma and hemorrhagic shock. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, B.; Mendler, M.R.; Lackner, I.; Pressmar, J.; Haffner-Luntzer, M.; Hofler, S.; Braun, C.K.; Hummler, H.; Schwarz, S.; Kalbitz, M.; et al. Tissue damage in the heart after cardiac arrest induced by asphyxia and hemorrhage in newborn pigs. Pediatr. Res. 2019, 86. [Google Scholar] [CrossRef] [PubMed]
- Kalbitz, M.; Schwarz, S.; Weber, B.; Bosch, B.; Pressmar, J.; Hoenes, F.M.; Braun, C.K.; Horst, K.; Simon, T.P.; Pfeifer, R.; et al. Cardiac depression in pigs after multiple trauma-characterization of posttraumatic structural and functional alterations. Sci. Rep. 2017, 7, 17861. [Google Scholar] [CrossRef]
- Friden, V.; Starnberg, K.; Muslimovic, A.; Ricksten, S.-E.; Bjurman, C.; Forsgard, N.; Wickman, A.; Hammarsten, O. Clearance of cardiac troponin T with and without kidney function. Clin. Biochem. 2017, 50, 468–474. [Google Scholar] [CrossRef]
- Mair, J.; Lindahl, B.; Hammarsten, O.; Muller, C.; Giannitsis, E.; Huber, K.; Mockel, M.; Plebani, M.; Thygesen, K.; Jaffe, A.S.; et al. How is cardiac troponin released from injured myocardium? Eur. Heart J. Acute Cardiovasc. Care 2017, 7, 553–560. [Google Scholar] [CrossRef]
- Mair, J. Tissue release of cardiac markers: From physiology to clinical applications. Clin. Chem. Lab. Med. 1999, 37, 1077–1084. [Google Scholar] [CrossRef]
- Piper, H.M.; Schwartz, P.; Spahr, R.; Hutter, J.F.; Spieckermann, P.G. Early enzyme release from myocardial cells is not due to irreversible cell damage. J. Mol. Cell. Cardiol. 1984, 16, 385–388. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; McNally, E.M. Plasma membrane repair in health and disease. Curr. Top. Membr. 2016, 77, 67–96. [Google Scholar] [CrossRef] [Green Version]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- Hessel, M.H.M.; Atsma, D.E.; van der Valk, E.J.M.; Bax, W.H.; Schalij, M.J.; van der Laarse, A. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Eur. J. Physiol. 2008, 455, 979–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streng, A.S.; Jacobs, L.H.J.; Schwenk, R.W.; Cardinaels, E.P.M.; Meex, S.J.R.; Glatz, J.F.C.; Wodzig, W.K.W.H.; van Dieijen-Visser, M.P. Cardiac troponin in ischemic cardiomyocytes: Intracellular decrease before onset of cell death. Exp. Mol. Pathol. 2014, 96, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Kalbitz, M.; Grailer, J.J.; Fattahi, F.; Jajou, L.; Herron, T.J.; Campbell, K.F.; Zetoune, F.S.; Bosmann, M.; Sarma, J.V.; Huber-Lang, M.; et al. Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J. 2015, 29, 2185–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Communal, C.; Sumandea, M.; de Tombe, P.; Narula, J.; Solaro, R.J.; Hajjar, R.J. Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 6252–6256. [Google Scholar] [CrossRef] [Green Version]
- Hessel, M.H.M.; Michielsen, E.C.H.J.; Atsma, D.E.; Schalij, M.J.; van der Valk, E.J.M.; Bax, W.H.; Hermens, W.T.; van Dieijen-Visser, M.P.; van der Laarse, A. Release kinetics of intact and degraded troponin I and T after irreversible cell damage. Exp. Mol. Pathol. 2008, 85, 90–95. [Google Scholar] [CrossRef]
- Bangert, A.; Andrassy, M.; Muller, A.-M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Goser, S.; Korkmaz-Icoz, S.; Zittrich, S.; et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164. [Google Scholar] [CrossRef] [Green Version]
- Gami, B.N.; Patel, D.S.; Haridas, N.; Chauhan, K.P.; Shah, H.; Trivedi, A. Utility of heart-type fatty acid binding protein as a new biochemical marker for the early diagnosis of acute coronary syndrome. J. Clin. Diagn. Res. 2015, 9, BC22–BC24. [Google Scholar] [CrossRef]
- McMahon, C.G.; Lamont, J.V.; Curtin, E.; McConnell, R.I.; Crockard, M.; Kurth, M.J.; Crean, P.; Fitzgerald, S.P. Diagnostic accuracy of heart-type fatty acid-binding protein for the early diagnosis of acute myocardial infarction. Am. J. Emerg. Med. 2012, 30, 267–274. [Google Scholar] [CrossRef]
- Motz, B.M.; Baimas-George, M.; Barnes, T.E.; Ragunanthan, B.V.; Symanski, J.D.; Christmas, A.B.; Sing, R.F.; Ross, S.W. Mitigating clinical waste in the trauma intensive care unit: Limited clinical utility of cardiac troponin testing for trauma patients with atrial fibrillation. Am. J. Surg. 2019, 219. [Google Scholar] [CrossRef]
- Maron, B.J.; Boren, S.D.; Estes, N.M. Early descriptions of sudden cardiac death due to commotio cordis occurring in baseball. Heart Rhythm 2010, 7, 992–993. [Google Scholar] [CrossRef]
- Maron, B.J.; Ahluwalia, A.; Haas, T.S.; Semsarian, C.; Link, M.S.; Estes, N.M. Global epidemiology and demographics of commotio cordis. Heart Rhythm 2011, 8, 1969–1971. [Google Scholar] [CrossRef] [PubMed]
- Madias, C.; Maron, B.J.; Supron, S.; Estes, N.M.; Link, M.S. Cell membrane stretch and chest blow-induced ventricular fibrillation: Commotio cordis. J. Cardiovasc. Electrophysiol. 2008, 19, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Mubayed, L.; Romme, A.; Nguyen, H.H. QT Prolongation after minor head trauma in a pediatric patient. Pediatr. Cardiol. 2019, 41. [Google Scholar] [CrossRef] [PubMed]
- Saric, P.; Ravaee, B.D.; Patel, T.R.; Hoit, B.D. Acute severe mitral regurgitation after blunt chest trauma. Echocardiography 2018, 35, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, H.; Arslan, Y.; Alper, A.; Osmonov, D.; Guvenc, T.S.; Poyraz, E.; Akyuz, S.; Yildiz, M. Severe tricuspid regurgitation and atrioventicular block caused by blunt thoracic trauma in an elderly woman. J. Emerg. Med. 2012, 43, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Tutun, U.; Aksoyek, A.; Parlar, A.I.; Cobanoglu, A. Post-traumatic tricuspid insufficiency: A case report. Ulus Travma Acil Cerrahi Derg. 2011, 17, 563–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driessen, R.; Doodeman, I.; Bogaard, K.; Reichert, S. Contusio cordis, not an innocent diagnosis. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ni, Y.; Chen, X.; Ma, L. Aortic valve tear with severe aortic regurgitation following blunt chest trauma. J. Cardiothorac. Surg. 2011, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Simmers, T.A.; Meijburg, H.W.; de la Riviere, A.B. Traumatic papillary muscle rupture. Ann. Thorac. Surg. 2001, 72, 257–259. [Google Scholar] [CrossRef]
- Kumar, A.; Thota, V.; Dee, L.; Olson, J.; Uretz, E.; Parrillo, J.E. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J. Exp. Med. 1996, 183, 949–958. [Google Scholar] [CrossRef]
- Horst, K.; Simon, T.P.; Pfeifer, R.; Teuben, M.; Almahmoud, K.; Zhi, Q.; Santos, S.A.; Wembers, C.C.; Leonhardt, S.; Heussen, N.; et al. Characterization of blunt chest trauma in a long-term porcine model of severe multiple trauma. Sci. Rep. 2016, 6, 39659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remick, D.G.; Bolgos, G.R.; Siddiqui, J.; Shin, J.; Nemzek, J.A. Six at six: Interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 2002, 17, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zheng, R.; Hu, S.; Ma, Y.; Choudhry, M.A.; Messina, J.L.; Rue, L.W., III; Bland, K.I.; Chaudry, I.H. Mechanism of cardiac depression after trauma-hemorrhage: Increased cardiomyocyte IL-6 and effect of sex steroids on IL-6 regulation and cardiac function. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2183–H2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Hu, S.; Hsieh, Y.-C.; Choudhry, M.A.; Rue, L.W., III; Bland, K.I.; Chaudry, I.H. Mechanism of IL-6-mediated cardiac dysfunction following trauma-hemorrhage. J. Mol. Cell. Cardiol. 2006, 40, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tao, L.; Jiao, X.; Liu, H.; Cao, Y.; Lopez, B.; Luan, R.-H.; Christopher, T.; Ma, X.L. TNFalpha-initiated oxidative/nitrative stress mediates cardiomyocyte apoptosis in traumatic animals. Apoptosis 2007, 12, 1795–1802. [Google Scholar] [CrossRef]
- Kalbitz, M.; Amann, E.M.; Bosch, B.; Palmer, A.; Schultze, A.; Pressmar, J.; Weber, B.; Wepler, M.; Gebhard, F.; Schrezenmeier, H.; et al. Experimental blunt chest trauma-induced myocardial inflammation and alteration of gap-junction protein connexin 43. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, T.; Akashi, K.; Keira, N.; Matoba, S.; Mano, A.; Shiraishi, J.; Yamanaka, S.; Kobara, M.; Hibino, N.; Hosokawa, S.; et al. Cytokine-induced nitric oxide inhibits mitochondrial energy production and induces myocardial dysfunction in endotoxin-treated rat hearts. J. Mol. Cell. Cardiol. 2004, 37, 775–784. [Google Scholar] [CrossRef]
- Jin, H.; Fujita, T.; Jin, M.; Kurotani, R.; Hidaka, Y.; Cai, W.; Suita, K.; Prajapati, R.; Liang, C.; Ohnuki, Y.; et al. Epac activation inhibits IL-6-induced cardiac myocyte dysfunction. J. Physiol. Sci. 2018, 68, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Duncan, D.J.; Yang, Z.; Hopkins, P.M.; Steele, D.S.; Harrison, S.M. TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium 2010, 47, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Monnerat, G.; Alarcon, M.L.; Vasconcellos, L.R.; Hochman-Mendez, C.; Brasil, G.; Bassani, R.A.; Casis, O.; Malan, D.; Travassos, L.H.; Sepulveda, M.; et al. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016, 7, 13344. [Google Scholar] [CrossRef] [Green Version]
- Remels, A.H.V.; Derks, W.J.A.; Cillero-Pastor, B.; Verhees, K.J.P.; Kelders, M.C.; Heggermont, W.; Carai, P.; Summer, G.; Ellis, S.R.; de Theije, C.C.; et al. NF-kappaB-mediated metabolic remodelling in the inflamed heart in acute viral myocarditis. Biochim. Biophys. Acta 2018, 1864, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma Emerg. Surg. 2019, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.-F. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Crit. Care 2009, 13, R174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horst, K.; Hildebrand, F.; Pfeifer, R.; Hubenthal, S.; Almahmoud, K.; Sassen, M.; Steinfeldt, T.; Wulf, H.; Ruchholtz, S.; Pape, H.C.; et al. Impact of haemorrhagic shock intensity on the dynamic of alarmins release in porcine poly-trauma animal model. Eur. J. Trauma Emerg. Surg. 2016, 42, 67–75. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.; Jiang, H.; Yu, Y.; Yu, P.; Tong, R.; Wu, J.; Zhang, S.; Yao, K.; Zou, Y.; et al. Extracellular high-mobility group box 1 mediates pressure overload-induced cardiac hypertrophy and heart failure. J. Cell. Mol. Med. 2016, 20, 459–470. [Google Scholar] [CrossRef]
- Tian, Y.; Charles, E.J.; Yan, Z.; Wu, D.; French, B.A.; Kron, I.L.; Yang, Z. The myocardial infarct-exacerbating effect of cell-free DNA is mediated by the high-mobility group box 1-receptor for advanced glycation end products-Toll-like receptor 9 pathway. J. Thorac. Cardiovasc. Surg. 2018, 157. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.A.; Grailer, J.J. Acute lung injury and the role of histones. Transl. Respir. Med. 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Alhamdi, Y.; Abrams, S.T.; Cheng, Z.; Jing, S.; Su, D.; Liu, Z.; Lane, S.; Welters, I.; Wang, G.; Toh, C.-H. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit. Care Med. 2015, 43, 2094–2103. [Google Scholar] [CrossRef]
- Kawai, C.; Kotani, H.; Miyao, M.; Ishida, T.; Jemail, L.; Abiru, H.; Tamaki, K. Circulating extracellular histones are clinically relevant mediators of multiple organ injury. Am. J. Pathol. 2016, 186, 829–843. [Google Scholar] [CrossRef] [Green Version]
- Asavarut, P.; Zhao, H.; Gu, J.; Ma, D. The role of HMGB1 in inflammation-mediated organ injury. Acta Anaesthesiol. Taiwanica 2013, 51, 28–33. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Buurman, W.A.; Griffioen, A.W. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 2008, 11, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Y.; Wu, L.; Zhou, Y.-H.; Liu, T.; Han, Q.-F.; Zhang, D.-Y.; Wang, L.-H.; Yao, H.-C. High mobility group box 1 might be a novel therapeutic target in ischemia heart disease. Int. J. Cardiol. 2016, 206, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Kalbitz, M.; Fattahi, F.; Herron, T.J.; Grailer, J.J.; Jajou, L.; Lu, H.; Huber-Lang, M.; Zetoune, F.S.; Sarma, J.V.; Day, S.M.; et al. Complement destabilizes cardiomyocyte function In Vivo after polymicrobial sepsis and In Vitro. J. Immunol. 2016, 197, 2353–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lu, C.; Gao, M.; Cao, X.; Ha, T.; Kalbfleisch, J.H.; Williams, D.L.; Li, C.; Kao, R.L. Toll-like receptor 4 plays a central role in cardiac dysfunction during trauma hemorrhage shock. Shock 2014, 42, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, G.P.; Zellweger, R. Cardiac troponin I as a predictor of arrhythmia and ventricular dysfunction in trauma patients with myocardial contusion. J. Trauma 2004, 57, 801–808. [Google Scholar] [CrossRef]
- Lackner, I.; Weber, B.; Baur, M.; Haffner-Luntzer, M.; Eiseler, T.; Fois, G.; Gebhard, F.; Relja, B.; Marzi, I.; Pfeifer, R.; et al. Midkine is elevated after multiple trauma and acts directly on human cardiomyocytes by altering their functionality and metabolism. Front. Immunol. 2019, 10, 1920. [Google Scholar] [CrossRef]
- Iwashita, N.; Muramatsu, H.; Toriyama, K.; Torii, S.; Muramatsu, T. Expression of midkine in normal and burn sites of rat skin. Burns 1999, 25, 119–124. [Google Scholar] [CrossRef]
- Fischer, V.; Kalbitz, M.; Muller-Graf, F.; Gebhard, F.; Ignatius, A.; Liedert, A.; Haffner-Luntzer, M. Influence of menopause on inflammatory cytokines during murine and human bone fracture healing. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Sakakima, H.; Yoshida, Y.; Muramatsu, T.; Yone, K.; Goto, M.; Ijiri, K.; Izumo, S. Traumatic injury-induced midkine expression in the adult rat spinal cord during the early stage. J. Neurotrauma 2004, 21, 471–477. [Google Scholar] [CrossRef]
- Fukui, S.; Kitagawa-Sakakida, S.; Kawamata, S.; Matsumiya, G.; Kawaguchi, N.; Matsuura, N.; Sawa, Y. Therapeutic effect of midkine on cardiac remodeling in infarcted rat hearts. Ann. Thorac. Surg. 2008, 85, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Netsu, S.; Shishido, T.; Kitahara, T.; Honda, Y.; Funayama, A.; Narumi, T.; Kadowaki, S.; Takahashi, H.; Miyamoto, T.; Arimoto, T.; et al. Midkine exacerbates pressure overload-induced cardiac remodeling. Biochem. Biophys. Res. Commun. 2014, 443, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, Y.; Muramatsu, T.; Hirai, M.; Inui, T.; Kimura, T.; Saito, H.; McCormick, L.M.; Bu, G.; Kadomatsu, K. Nuclear targeting by the growth factor midkine. Mol. Cell. Biol. 2002, 22, 6788–6796. [Google Scholar] [CrossRef] [Green Version]
- Sumida, A.; Horiba, M.; Ishiguro, H.; Takenaka, H.; Ueda, N.; Ooboshi, H.; Opthof, T.; Kadomatsu, K.; Kodama, I. Midkine gene transfer after myocardial infarction in rats prevents remodelling and ameliorates cardiac dysfunction. Cardiovasc. Res. 2010, 86, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiba, M.; Kadomatsu, K.; Yasui, K.; Lee, J.-K.; Takenaka, H.; Sumida, A.; Kamiya, K.; Chen, S.; Sakuma, S.; Muramatsu, T.; et al. Midkine plays a protective role against cardiac ischemia/reperfusion injury through a reduction of apoptotic reaction. Circulation 2006, 114, 1713–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederbichler, A.D.; Hoesel, L.M.; Westfall, M.V.; Gao, H.; Ipaktchi, K.R.; Sun, L.; Zetoune, F.S.; Su, G.L.; Arbabi, S.; Sarma, J.V.; et al. An essential role for complement C5a in the pathogenesis of septic cardiac dysfunction. J. Exp. Med. 2006, 203, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, L.M.; Niederbichler, A.D.; Schaefer, J.; Ipaktchi, K.R.; Gao, H.; Rittirsch, D.; Pianko, M.J.; Vogt, P.M.; Sarma, J.V.; Su, G.L.; et al. C5a-blockade improves burn-induced cardiac dysfunction. J. Immunol. 2007, 178, 7902–7910. [Google Scholar] [CrossRef]
- Mueller, M.; Herzog, C.; Larmann, J.; Schmitz, M.; Hilfiker-Kleiner, D.; Gessner, J.E.; Theilmeier, G. The receptor for activated complement factor 5 (C5aR) conveys myocardial ischemic damage by mediating neutrophil transmigration. Immunobiology 2013, 218, 1131–1138. [Google Scholar] [CrossRef]
- Burk, A.-M.; Martin, M.; Flierl, M.A.; Rittirsch, D.; Helm, M.; Lampl, L.; Bruckner, U.; Stahl, G.L.; Blom, A.M.; Perl, M.; et al. Early complementopathy after multiple injuries in humans. Shock 2012, 37, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Tennenberg, S.D.; Jacobs, M.P.; Solomkin, J.S. Complement-mediated neutrophil activation in sepsis- and trauma-related adult respiratory distress syndrome: Clarification with radioaerosol lung scans. Arch. Surg. 1987, 122, 26–32. [Google Scholar] [CrossRef]
- Zimmermann, T.; Laszik, Z.; Nagy, S.; Kaszaki, J.; Joo, F. The role of the complement system in the pathogenesis of multiple organ failure in shock. Prog. Clin. Biol. Res. 1989, 308, 291–297. [Google Scholar] [PubMed]
- Fosse, E.; Pillgram-Larsen, J.; Svennevig, J.L.; Nordby, C.; Skulberg, A.; Mollnes, T.E.; Abdelnoor, M. Complement activation in injured patients occurs immediately and is dependent on the severity of the trauma. Injury 1998, 29, 509–514. [Google Scholar] [CrossRef]
- Zilow, G.; Joka, T.; Obertacke, U.; Rother, U.; Kirschfink, M. Generation of anaphylatoxin C3a in plasma and bronchoalveolar lavage fluid in trauma patients at risk for the adult respiratory distress syndrome. Crit. Care Med. 1992, 20, 468–473. [Google Scholar] [CrossRef]
- Hoth, J.J.; Wells, J.D.; Jones, S.E.; Yoza, B.K.; McCall, C.E. Complement mediates a primed inflammatory response after traumatic lung injury. J. Trauma Acute Care Surg. 2014, 76, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flierl, M.A.; Perl, M.; Rittirsch, D.; Bartl, C.; Schreiber, H.; Fleig, V.; Schlaf, G.; Liener, U.; Brueckner, U.B.; Gebhard, F.; et al. The role of C5a in the innate immune response after experimental blunt chest trauma. Shock 2008, 29, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Lackner, I.; Weber, B.; Baur, M.; Fois, G.; Gebhard, F.; Pfeifer, R.; Cinelli, P.; Halvachizadeh, S.; Lipiski, M.; Cesarovic, N.; et al. Complement activation and organ damage after trauma—differential immune response based on surgical treatment strategy. Front. Immunol. 2020, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berg, C.W.; Tambourgi, D.V.; Clark, H.W.; Hoong, S.J.; Spiller, O.B.; McGreal, E.P. Mechanism of neutrophil dysfunction: Neutrophil serine proteases cleave and inactivate the C5a receptor. J. Immunol. 2014, 192, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.A. Role of the complement in experimental sepsis. J. Leukoc. Biol. 2008, 83, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Flierl, M.A.; Nadeau, B.A.; Day, D.E.; Huber-Lang, M.; Mackay, C.R.; Zetoune, F.S.; Gerard, N.P.; Cianflone, K.; Kohl, J.; et al. Functional roles for C5a receptors in sepsis. Nat. Med. 2008, 14, 551–557. [Google Scholar] [CrossRef]
- Fattahi, F.; Ward, P.A. Complement and sepsis-induced heart dysfunction. Mol. Immunol. 2017, 84, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Kalbitz, M.; Fattahi, F.; Grailer, J.J.; Jajou, L.; Malan, E.A.; Zetoune, F.S.; Huber-Lang, M.; Russell, M.W.; Ward, P.A. Complement-induced activation of the cardiac NLRP3 inflammasome in sepsis. FASEB J. 2016, 30, 3997–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattahi, F.; Kalbitz, M.; Malan, E.A.; Abe, E.; Jajou, L.; Huber-Lang, M.S.; Bosmann, M.; Russell, M.W.; Zetoune, F.S.; Ward, P.A.; et al. Complement-induced activation of MAPKs and Akt during sepsis: Role in cardiac dysfunction. FASEB J. 2017, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinus, R.D.; Cook, C.J. The effect of complement C5a on mitochondrial functions of PC12 cells. Neuroreport 2011, 22, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef]
- Del Balzo, U.H.; Levi, R.; Polley, M.J. Cardiac dysfunction caused by purified human C3a anaphylatoxin. Proc. Natl. Acad. Sci. USA 1985, 82, 886–890. [Google Scholar] [CrossRef] [Green Version]
- Mueller, K.A.L.; Patzelt, J.; Sauter, M.; Maier, P.; Gekeler, S.; Klingel, K.; Kandolf, R.; Seizer, P.; Gawaz, M.; Geisler, T.; et al. Myocardial expression of the anaphylatoxin receptor C3aR is associated with cardiac inflammation and prognosis in patients with non-ischaemic heart failure. ESC Heart Fail. 2018, 5, 846–857. [Google Scholar] [CrossRef]
- Giannoudis, P.V.; Smith, R.M.; Bellamy, M.C.; Morrison, J.F.; Dickson, R.A.; Guillou, P.J. Stimulation of the inflammatory system by reamed and unreamed nailing of femoral fractures: An analysis of the second hit. J. Bone Joint Surg. Br. 1999, 81, 356–361. [Google Scholar] [CrossRef]
- Napier, B.A.; Brubaker, S.W.; Sweeney, T.E.; Monette, P.; Rothmeier, G.H.; Gertsvolf, N.A.; Puschnik, A.; Carette, J.E.; Khatri, P.; Monack, D.M.; et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J. Exp. Med. 2016, 213, 2365–2382. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.K.; Schuessler, R.B.; Saffitz, J.E. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ. Res. 2003, 92, 1138–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glukhov, A.V.; Fedorov, V.V.; Kalish, P.W.; Ravikumar, V.K.; Lou, Q.; Janks, D.; Schuessler, R.B.; Moazami, N.; Efimov, I.R. Conduction remodeling in human end-stage nonischemic left ventricular cardiomyopathy. Circulation 2012, 125, 1835–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, C.; Zweifel, M.; Zuppinger, C.; Carrel, T.; Martin, D.; Haefliger, J.-A.; Delacretaz, E. Connexin 43 expression in human hypertrophied heart due to pressure and volume overload. Physiol. Res. 2010, 59, 35–42. [Google Scholar] [PubMed]
- Foertsch, S.; Lackner, I.; Weber, B.; Fuchsl, A.M.; Langgartner, D.; Wirkert, E.; Peters, S.; Fois, G.; Pressmar, J.; Fegert, J.M.; et al. Sensory contact to the stressor prevents recovery from structural and functional heart damage following psychosocial trauma. Brain Behav. Immun. 2019, 80, 667–677. [Google Scholar] [CrossRef]
- Gutstein, D.E.; Morley, G.E.; Tamaddon, H.; Vaidya, D.; Schneider, M.D.; Chen, J.; Chien, K.R.; Stuhlmann, H.; Fishman, G.I. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 2001, 88, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Agullo-Pascual, E.; Cerrone, M.; Delmar, M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. FEBS Lett. 2014, 588, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Yasui, K.; Kada, K.; Hojo, M.; Lee, J.K.; Kamiya, K.; Toyama, J.; Opthof, T.; Kodama, I. Cell-to-cell interaction prevents cell death in cultured neonatal rat ventricular myocytes. Cardiovasc. Res. 2000, 48, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Barker, R.J.; Price, R.L.; Gourdie, R.G. Increased association of ZO-1 with connexin43 during remodeling of cardiac gap junctions. Circ. Res. 2002, 90, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Pyle, W.G.; Solaro, R.J. At the crossroads of myocardial signaling: The role of Z-discs in intracellular signaling and cardiac function. Circ. Res. 2004, 94, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Ramspacher, C.; Steed, E.; Boselli, F.; Ferreira, R.; Faggianelli, N.; Roth, S.; Spiegelhalter, C.; Messaddeq, N.; Liebling, M.; Vermot, J.; et al. Developmental alterations in heart biomechanics and skeletal muscle function in desmin mutants suggest an early pathological root for desminopathies. Cell Rep. 2015, 11, 1564–1576. [Google Scholar] [CrossRef] [Green Version]
- Van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.H.; de Walle, H.E.K.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef]
- Panagopoulou, P.; Davos, C.H.; Milner, D.J.; Varela, E.; Cameron, J.; Mann, D.L.; Capetanaki, Y. Desmin mediates TNF-alpha-induced aggregate formation and intercalated disk reorganization in heart failure. J. Cell Biol. 2008, 181, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Gard, J.J.; Yamada, K.; Green, K.G.; Eloff, B.C.; Rosenbaum, D.S.; Wang, X.; Robbins, J.; Schuessler, R.B.; Yamada, K.A.; Saffitz, J.E.; et al. Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc. Res. 2005, 67, 539–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, F.; Campbell, S.E.; Gerdes, A.M. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: II. cytoskeletal remodeling. J. Mol. Cell. Cardiol. 1999, 31, 319–331. [Google Scholar] [CrossRef]
- Sheng, J.-J.; Feng, H.-Z.; Pinto, J.R.; Wei, H.; Jin, J.-P. Increases of desmin and alpha-actinin in mouse cardiac myofibrils as a response to diastolic dysfunction. J. Mol. Cell. Cardiol. 2016, 99, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Lackner, I.; Weber, B.; Knecht, D.; Horst, K.; Relja, B.; Gebhard, F.; Pape, H.-C.; Huber-Lang, M.; Hildebrand, F.; Kalbitz, M.; et al. Cardiac glucose and fatty acid transport after experimental mono- and polytrauma. Shock 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Jensen, G.L. Inflammation as the key interface of the medical and nutrition universes: A provocative examination of the future of clinical nutrition and medicine. JPEN J. Parenter. Enteral Nutr. 2006, 30, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Preiser, J.-C.; van Zanten, A.R.H.; Berger, M.M.; Biolo, G.; Casaer, M.P.; Doig, G.S.; Griffiths, R.D.; Heyland, D.K.; Hiesmayr, M.; Iapichino, G.; et al. Metabolic and nutritional support of critically ill patients: Consensus and controversies. Crit. Care 2015, 19, 35. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, F.M.; Diczku, V.; Sherry, A.D.; Malloy, C.R. Substrate selection in the isolated working rat heart: Effects of reperfusion, afterload, and concentration. Basic Res. Cardiol. 1995, 90, 388–396. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Taegtmeyer, H. Improved energy homeostasis of the heart in the metabolic state of exercise. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1490–H1501. [Google Scholar] [CrossRef]
- Wentz, A.E.; d’Avignon, D.A.; Weber, M.L.; Cotter, D.G.; Doherty, J.M.; Kerns, R.; Nagarajan, R.; Reddy, N.; Sambandam, N.; Crawford, P.A.; et al. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J. Biol. Chem. 2010, 285, 24447–24456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonekess, B.O. Competition between lactate and fatty acids as sources of ATP in the isolated working rat heart. J. Mol. Cell. Cardiol. 1997, 29, 2725–2733. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.J.; Piel, D.A.; Acton, P.D.; Zhou, R.; Ferrari, V.A.; Karp, J.S.; Deutschman, C.S. Evidence of myocardial hibernation in the septic heart. Crit. Care Med. 2005, 33, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Miura, T.; Miki, T.; Sakamoto, J.; Nakamura, Y.; Ikeda, Y.; Kobayashi, H.; Shimamoto, K. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc. Res. 2004, 61, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.; Wende, A.R.; Abel, E.D.; Moreno, A.P.; Sachse, F.B.; Punske, B.B. Absence of glucose transporter 4 diminishes electrical activity of mouse hearts during hypoxia. Exp. Physiol. 2013, 98, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Papp, A.; Uusaro, A.; Parviainen, I.; Hartikainen, J.; Ruokonen, E. Myocardial function and haemodynamics in extensive burn trauma: Evaluation by clinical signs, invasive monitoring, echocardiography and cytokine concentrations: A prospective clinical study. Acta Anaesthesiol. Scand. 2003, 47, 1257–1263. [Google Scholar] [CrossRef]
- Horton, J.W.; Garcia, N.M.; White, D.J.; Keffer, J. Postburn cardiac contractile function and biochemical markers of postburn cardiac injury. J. Am. Coll. Surg. 1995, 181, 289–298. [Google Scholar]
- Mlcak, R.P.; Suman, O.E.; Murphy, K.; Herndon, D.N. Effects of growth hormone on anthropometric measurements and cardiac function in children with thermal injury. Burns 2005, 31, 60–66. [Google Scholar] [CrossRef]
- Guillory, A.N.; Clayton, R.P.; Herndon, D.N.; Finnerty, C.C. Cardiovascular dysfunction following burn injury: What we have learned from rat and mouse models. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [Green Version]
- Ballard-Croft, C.; Carlson, D.; Maass, D.L.; Horton, J.W. Burn trauma alters calcium transporter protein expression in the heart. J. Appl. Physiol. 2004, 97, 1470–1476. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.D.; Chen, Z.R.; Li, R.; Lou, S.F. Nitric oxide synthesis in myocardium following burn injury in rats. Burns 1998, 24, 455–459. [Google Scholar] [CrossRef]
- Cao, W.; Xie, Y.-H.; Li, X.-Q.; Zhang, X.-K.; Chen, Y.-T.; Kang, R.; Chen, X.; Miao, S.; Wang, S.-W. Burn-induced apoptosis of cardiomyocytes is survivin dependent and regulated by PI3K/Akt, p38 MAPK and ERK pathways. Basic Res. Cardiol. 2011, 106, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Dutton, R.P.; Stansbury, L.G.; Leone, S.; Kramer, E.; Hess, J.R.; Scalea, T.M. Trauma mortality in mature trauma systems: Are we doing better? An analysis of trauma mortality patterns, 1997–2008. J. Trauma 2010, 69, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Bahloul, M.; Chaari, A.N.; Kallel, H.; Khabir, A.; Ayadi, A.; Charfeddine, H.; Hergafi, L.; Chaari, A.D.; Chelly, H.E.; Ben Hamida, C.; et al. Neurogenic pulmonary edema due to traumatic brain injury: Evidence of cardiac dysfunction. Am. J. Crit. Care 2006, 15, 462–470. [Google Scholar] [CrossRef]
- Prathep, S.; Sharma, D.; Hallman, M.; Joffe, A.; Krishnamoorthy, V.; Mackensen, G.B.; Vavilala, M.S. Preliminary report on cardiac dysfunction after isolated traumatic brain injury. Crit. Care Med. 2014, 42, 142–147. [Google Scholar] [CrossRef] [Green Version]
- James, P.; Ellis, C.J.; Whitlock, R.M.; McNeil, A.R.; Henley, J.; Anderson, N.E. Relation between troponin T concentration and mortality in patients presenting with an acute stroke: Observational study. BMJ 2000, 320, 1502–1504. [Google Scholar] [CrossRef] [Green Version]
- Wittstein, I.S.; Thiemann, D.R.; Lima, J.A.C.; Baughman, K.L.; Schulman, S.P.; Gerstenblith, G.; Wu, K.C.; Rade, J.J.; Bivalacqua, T.J.; Champion, H.C.; et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N. Engl. J. Med. 2005, 352, 539–548. [Google Scholar] [CrossRef]
- Kono, T.; Morita, H.; Kuroiwa, T.; Onaka, H.; Takatsuka, H.; Fujiwara, A. Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: Neurogenic stunned myocardium. J. Am. Coll. Cardiol. 1994, 24, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Mashaly, H.A.; Provencio, J.J. Inflammation as a link between brain injury and heart damage: The model of subarachnoid hemorrhage. Cleve. Clin. J. Med. 2008, 75, S26–S30. [Google Scholar] [CrossRef] [Green Version]
- Shivalkar, B.; van Loon, J.; Wieland, W.; Tjandra-Maga, T.B.; Borgers, M.; Plets, C.; Flameng, W. Variable effects of explosive or gradual increase of intracranial pressure on myocardial structure and function. Circulation 1993, 87, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, M.; Ali, A.; Ashley, E.; Freed, D.; Clarke, K.; Tsui, S.; Parameshwar, J.; Large, S. Is stress cardiomyopathy the underlying cause of ventricular dysfunction associated with brain death? J. Heart Lung Transplant. 2010, 29, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Brandspiegel, H.Z.; Marinchak, R.A.; Rials, S.J.; Kowey, P.R. A broken heart. Circulation 1998, 98, 1349. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Recalde, A.; Costero, O.; Oliver, J.M.; Iborra, C.; Ruiz, E.; Sobrino, J.A. Pheochromocytoma-related cardiomyopathy: Inverted Takotsubo contractile pattern. Circulation 2006, 113, e738–e739. [Google Scholar] [CrossRef] [PubMed]
- Akashi, Y.J.; Nakazawa, K.; Sakakibara, M.; Miyake, F.; Sasaka, K. Reversible left ventricular dysfunction “takotsubo” cardiomyopathy related to catecholamine cardiotoxicity. J. Electrocardiol. 2002, 35, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Meune, C.; Bertherat, J.; Dousset, B.; Jude, N.; Bertagna, X.; Duboc, D.; Weber, S. Reduced myocardial contractility assessed by tissue Doppler echocardiography is associated with increased risk during adrenal surgery of patients with pheochromocytoma: Report of a preliminary study. J. Am. Soc. Echocardiogr. 2006, 19, 1466–1470. [Google Scholar] [CrossRef]
- Mori, H.; Ishikawa, S.; Kojima, S.; Hayashi, J.; Watanabe, Y.; Hoffman, J.I.; Okino, H. Increased responsiveness of left ventricular apical myocardium to adrenergic stimuli. Cardiovasc. Res. 1993, 27, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Richard, C. Stress-related cardiomyopathies. Ann. Intensive Care 2011, 1, 39. [Google Scholar] [CrossRef] [Green Version]
- Friesenecker, B.E.; Tsai, A.G.; Martini, J.; Ulmer, H.; Wenzel, V.; Hasibeder, W.R.; Intaglietta, M.; Dünser, M.W. Arteriolar vasoconstrictive response: Comparing the effects of arginine vasopressin and norepinephrine. Crit. Care 2006, 10, R75. [Google Scholar] [CrossRef] [Green Version]
- Bolli, R.; Marban, E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999, 79, 609–634. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. High circulating adrenaline levels at admission predict increased mortality after trauma. J. Trauma Acute Care Surg. 2012, 72, 428–436. [Google Scholar] [CrossRef]
- Ostrowski, S.R.; Pedersen, S.H.; Jensen, J.S.; Mogelvang, R.; Johansson, P.I. Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Crit. Care 2013, 17, R32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostadal, B.; Parizek, A.; Ostadalova, I.; Kolar, F. Cardiotoxicity of β-mimetic catecholamines during ontogenetic development—Possible risks of antenatal therapy. Can. J. Physiol. Pharmacol. 2018, 96, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Rona, G.; Chappel, C.I.; Balazs, T.; Gaudry, R. An infarct-like myocardial lesion and other toxic manifestations produced by isoproterenol in the rat. AMA. Arch. Pathol. 1959, 67, 443–455. [Google Scholar]
- Fleckenstein, A. Specific Inhibitors and Promoters of Calcium Action in the Excitation Contraction Coupling of Heart Muscle and their Role in the Prevention or Production of Myocardial Lesions. In Calcium and the Heart; Academic Press: Cambridge, MA, USA, 1971; pp. 135–185. [Google Scholar]
- Bybee, K.A.; Prasad, A. Stress-related cardiomyopathy syndromes. Circulation 2008, 118, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Banki, N.; Kopelnik, A.; Tung, P.; Lawton, M.T.; Gress, D.; Drew, B.; Dae, M.; Foster, E.; Parmley, W.; Zaroff, J.; et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J. Neurosurg. 2006, 105, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, M.B.; Willet, D.; Keffer, J. The use of cardiac troponin-I (cTnI) to determine the incidence of myocardial ischemia and injury in patients with aneurysmal and presumed aneurysmal subarachnoid hemorrhage. Acta Neurochir. 1998, 140, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Scheitz, J.F.; Erdur, H.; Haeusler, K.G.; Audebert, H.J.; Roser, M.; Laufs, U.; Endres, M.; Nolte, C.H. Insular cortex lesions, cardiac troponin, and detection of previously unknown atrial fibrillation in acute ischemic stroke: Insights from the troponin elevation in acute ischemic stroke study. Stroke 2015, 46, 1196–1201. [Google Scholar] [CrossRef] [Green Version]
- Cannon, W.B. “Voodoo” death: American anthropologist. Am. J. Public Health 2002, 92, 1593–1596. [Google Scholar] [CrossRef]
- Kim, B.S.; Kim, T.-H.; Oh, J.-H.; Kwon, C.H.; Kim, S.H.; Kim, H.-J.; Hwang, H.K.; Chung, S.-M. Association between preoperative high sensitive troponin I levels and cardiovascular events after hip fracture surgery in the elderly. J. Geriatr. Cardiol. 2018, 15, 215–221. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Lin, C.-L.; Hsu, H.-C.; Chung, W.-S. Increased risk of coronary heart disease in patients with hip fracture: A nationwide cohort study. Osteoporos. Int. 2015, 26, 1849–1855. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Liu, C.-J.; Chen, P.-J.; Huang, C.-C.; Hsu, C.-Y.; Chen, Z.-Y.; Chan, W.-L.; Huang, P.-H.; Chen, T.-J.; Chung, C.-M.; et al. Hip fracture and risk of acute myocardial infarction: A nationwide study. J. Bone Miner. Res. 2013, 28, 404–411. [Google Scholar] [CrossRef] [PubMed]
- De’Ath, H.D.; Manson, J.; Davenport, R.; Glasgow, S.; Renfrew, I.; Davies, L.C.; Uppal, R.; Brohi, K. Trauma-induced secondary cardiac injury is associated with hyperacute elevations in inflammatory cytokines. Shock 2013, 39, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Lackner, I.; Knecht, D.; Braun, C.K.; Gebhard, F.; Huber-Lang, M.; Hildebrand, F.; Horst, K.; Pape, H.-C.; Ignatius, A.; et al. Systemic and cardiac alterations after long bone fracture. Shock 2020, 54. [Google Scholar] [CrossRef] [PubMed]
- Batsis, J.A.; Huddleston, J.M.; Melton, L.J., III; Huddleston, P.M.; Lopez-Jimenez, F.; Larson, D.R.; Gullerud, R.E.; McMahon, M.M. Body mass index and risk of adverse cardiac events in elderly patients with hip fracture: A population-based study. J. Am. Geriatr. Soc. 2009, 57, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Giganti, M.G.; Liuni, F.; Celi, M.; Gasbarra, E.; Zenobi, R.; Tresoldi, I.; Modesti, A.; Bei, R.; Tarantino, U. Changes in serum levels of TNF-alpha, IL-6, OPG, RANKL and their correlation with radiographic and clinical assessment in fragility fractures and high energy fractures. J. Biol. Regul. Homeost. Agents 2012, 26, 671–680. [Google Scholar]
- Yu, M.-D.; Su, B.-H.; Zhang, X.-X. Morphologic and molecular alteration during tibia fracture healing in rat. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, G.; Li, S.; Zhang, M.; Li, H.; Wang, K. TNF-alpha- mediated-p38-dependent signaling pathway contributes to myocyte apoptosis in rats subjected to surgical trauma. Cell. Physiol. Biochem. 2015, 35, 1454–1466. [Google Scholar] [CrossRef]
- Natanson, C.; Eichenholz, P.W.; Danner, R.L.; Eichacker, P.Q.; Hoffman, W.D.; Kuo, G.C.; Banks, S.M.; MacVittie, T.J.; Parrillo, J.E. Endotoxin and tumor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock. J. Exp. Med. 1989, 169, 823–832. [Google Scholar] [CrossRef]
- Bryant, D.; Becker, L.; Richardson, J.; Shelton, J.; Franco, F.; Peshock, R.; Thompson, M.; Giroir, B. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1998, 97, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; Chen, W.P.; Kuo, Y.L.; Ho, Y.J.; Lee, S.S.; Su, M.J. Thaliporphine preserves cardiac function of endotoxemic rabbits by both directly and indirectly attenuating NFkappaB signaling pathway. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Li, Y.; Mu, T. HMGB1 neutralizing antibody attenuates cardiac injury and apoptosis induced by hemorrhagic shock/resuscitation in rats. Biol. Pharm. Bull. 2015, 38, 1150–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadfield, D.; Hopkins, P.; Hart, N.; Finney, C.; Penhaligon, B.; Molai, J.; Rafferty, G. ESICM LIVES 2018. Intensive Care Med. Exp. 2018, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Gruda, M.C.; Ruggeberg, K.-G.; O’Sullivan, P.; Guliashvili, T.; Scheirer, A.R.; Golobish, T.D.; Capponi, V.J.; Chan, P.P. Broad adsorption of sepsis-related PAMP and DAMP molecules, mycotoxins, and cytokines from whole blood using CytoSorb(R) sorbent porous polymer beads. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Bosmann, M.; Ward, P.A. Protein-based therapies for acute lung injury: Targeting neutrophil extracellular traps. Expert Opin. Ther. Targets 2014, 18, 703–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T.; et al. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Z.; Wang, Z.; Li, X.; Li, X.; Cao, T.; Bi, Y.; Zhou, J.; Chen, X.; Yu, D.; Zhu, L.; et al. Protective effect of quercetin on posttraumatic cardiac injury. Sci. Rep. 2016, 6, 30812. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-C.; Hwang, T.-L.; Liu, F.-W.; Yu, H.-P. Tropisetron attenuates cardiac injury in a rat trauma-hemorrhage model. Shock 2012, 38, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Yang, S.; Champattanachai, V.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-{kappa}B signaling. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H515–H523. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, B.; Lackner, I.; Gebhard, F.; Miclau, T.; Kalbitz, M. Trauma, a Matter of the Heart—Molecular Mechanism of Post-Traumatic Cardiac Dysfunction. Int. J. Mol. Sci. 2021, 22, 737. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020737
Weber B, Lackner I, Gebhard F, Miclau T, Kalbitz M. Trauma, a Matter of the Heart—Molecular Mechanism of Post-Traumatic Cardiac Dysfunction. International Journal of Molecular Sciences. 2021; 22(2):737. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020737
Chicago/Turabian StyleWeber, Birte, Ina Lackner, Florian Gebhard, Theodore Miclau, and Miriam Kalbitz. 2021. "Trauma, a Matter of the Heart—Molecular Mechanism of Post-Traumatic Cardiac Dysfunction" International Journal of Molecular Sciences 22, no. 2: 737. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020737