Carboxypeptidase U (CPU, TAFIa, CPB2) in Thromboembolic Disease: What Do We Know Three Decades after Its Discovery?


Abstract
:1. Introduction
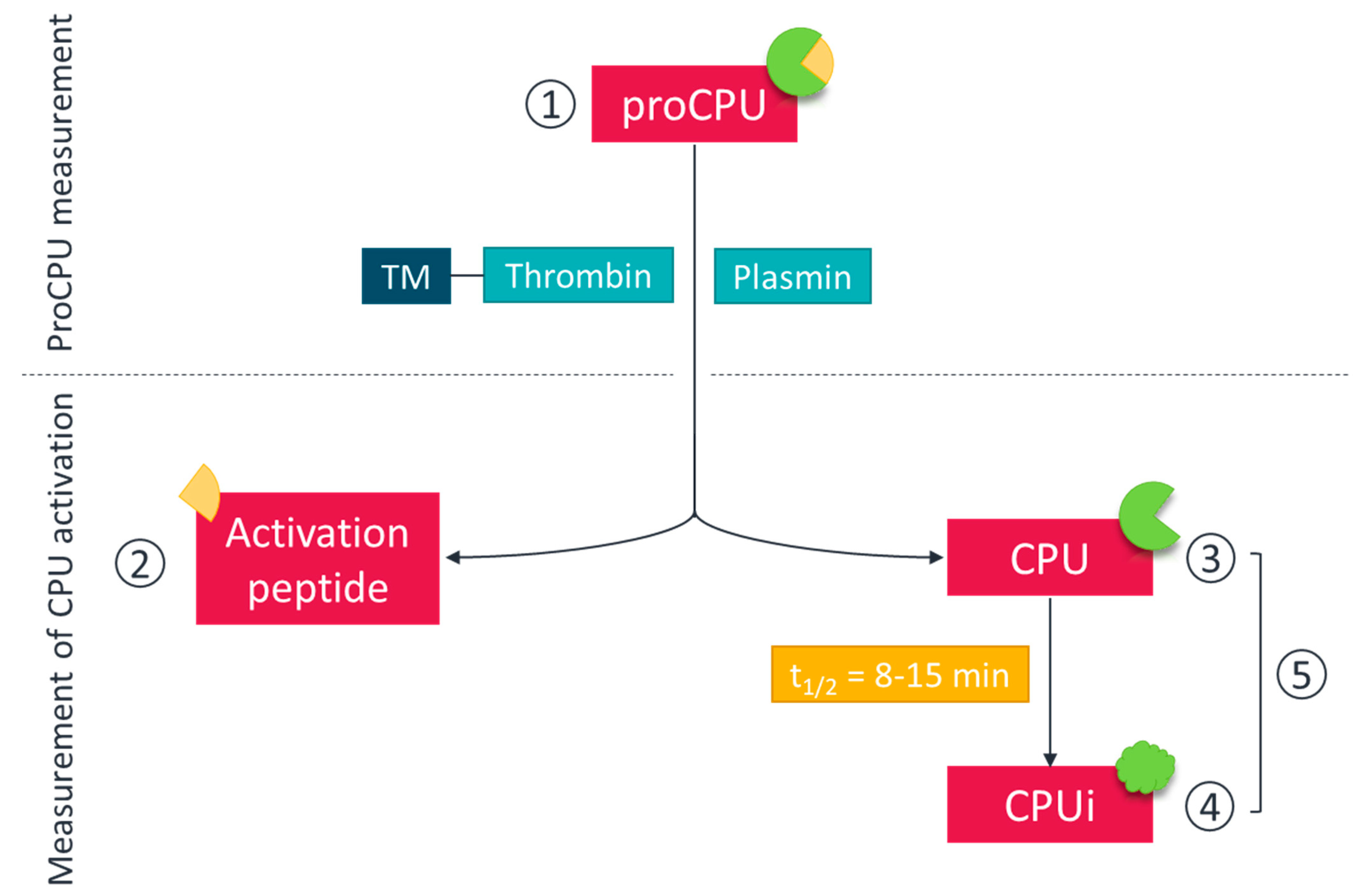
2. Measurement of ProCPU, CPU and CPUi: Methods, Challenges and Pitfalls
2.1. ProCPU
2.2. CPU
2.3. Assessment of Overall ProCPU Activation
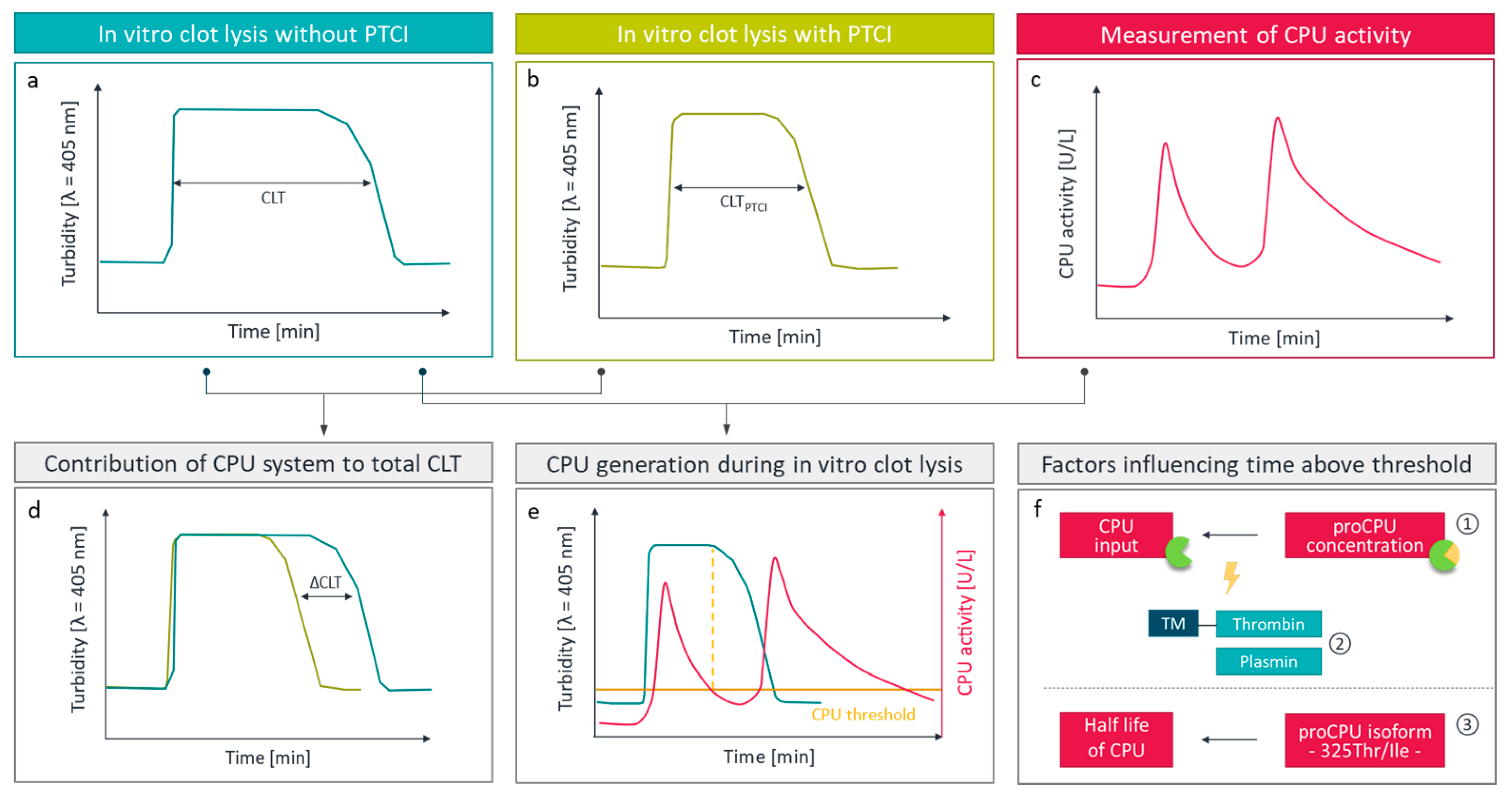
2.4. Assessment of CPU by Functional Fibrinolysis Assays
3. Plasma ProCPU Concentration and ProCPU Polymorphisms as Risk Factors?
3.1. Venous Thrombosis
3.2. Arterial Thrombosis
3.3. Cardiovascular Risk Factors
3.3.1. Hypertension
3.3.2. Hyperlipidemia
3.3.3. Diabetes Mellitus
4. Can ProCPU, CPU or CPUi Serve as Diagnostic or Prognostic Biomarkers for Thromboembolic Disease?
4.1. Venous Thrombosis
4.2. Arterial Thrombosis
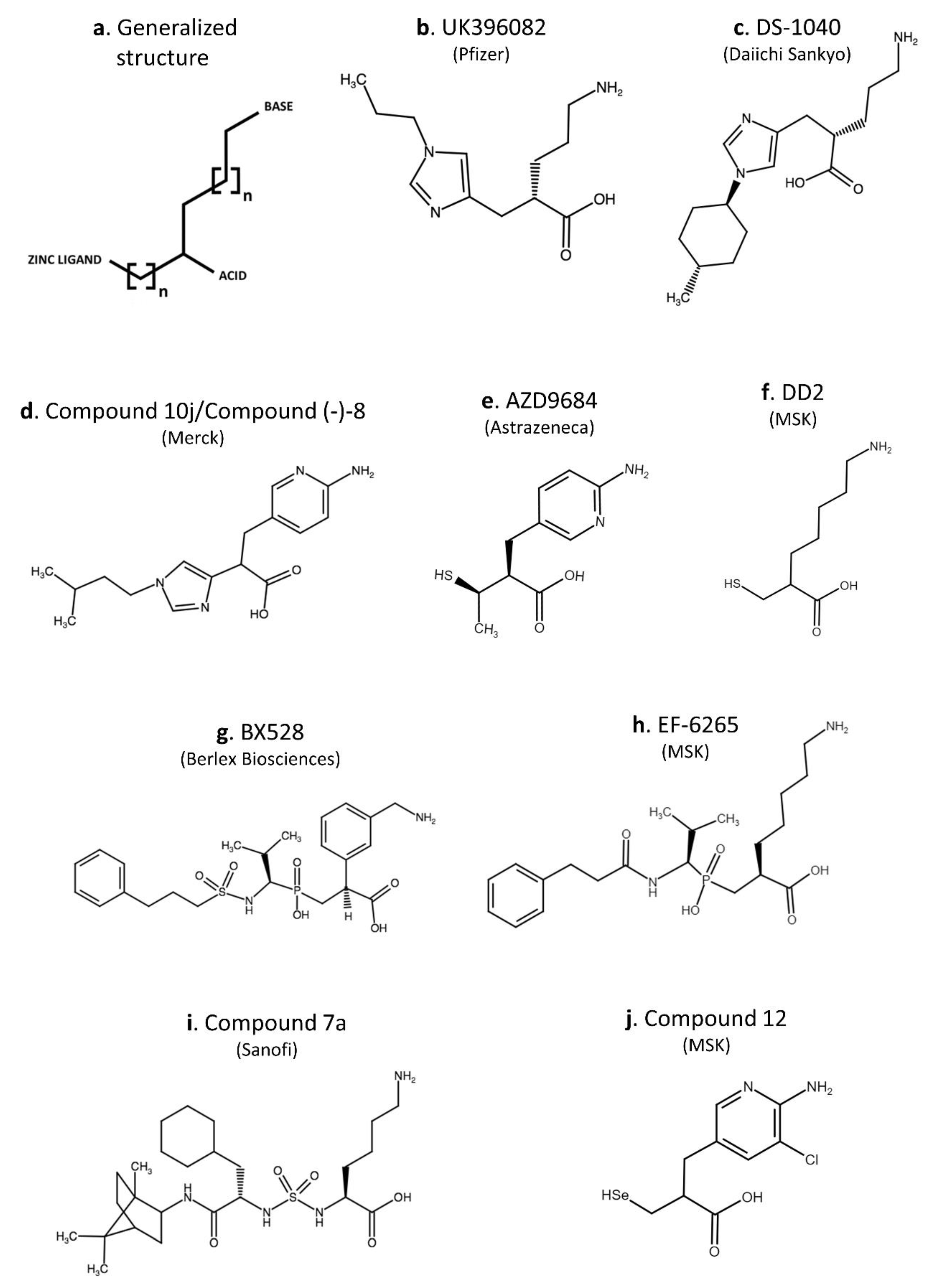
5. Potential Benefit of the Use of CPU Inhibitors—Overview on Inhibitors Anno 2020
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hendriks, D.; Wang, W.; Scharpé, S.; Lommaert, M.P.; van Sande, M. Purification and characterization of a new arginine carboxypeptidase in human serum. BBA Gen. Subj. 1990, 1034, 86–92. [Google Scholar] [CrossRef]
- Eaton, D.L.; Malloy, B.E.; Tsai, S.P.; Henzel, W.; Drayna, D. Isolation, molecular cloning, and partial characterization of a novel carboxypeptidase B from human plasma. J. Biol. Chem. 1991, 266, 21833–21838. [Google Scholar] [CrossRef]
- Bajzar, L.; Manuel, R.; Nesheim, M.E. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 1995, 270, 14477–14484. [Google Scholar] [CrossRef] [Green Version]
- Campbell, W.; Okada, H. An arginine specific carboxypeptidase generated in blood during coagulation or inflammation which is unrelated to carboxypeptidase N or its subunits. Biochem. Biophys. Res. Commun. 1989, 162, 933–939. [Google Scholar] [CrossRef]
- Heylen, E. An update on the role of carboxypeptidase U (TAFIa) in fibrinolysis. Front. Biosci. 2011, 16, 2427. [Google Scholar] [CrossRef] [Green Version]
- Plug, T.; Meijers, J.C.M. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor. J. Thromb. Haemost. 2016, 14, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Marx, P.F.; Brondijk, T.H.C.; Plug, T.; Romijn, R.A.; Hemrika, W.; Meijers, J.C.M.; Huizinga, E.G. Crystal structures of TAFI elucidate the inactivation mechanism of activated TAFI: A novel mechanism for enzyme autoregulation. Blood 2008, 112, 2803–2809. [Google Scholar] [CrossRef] [Green Version]
- Leurs, J.; Hendriks, D. Carboxypeptidase U (TAFIa): A metallocarboxypeptidase with a distinct role in haemostasis and a possible risk factor for thrombotic disease. Thromb. Haemost. 2005, 94, 471–487. [Google Scholar] [CrossRef]
- Heylen, E.; Van Goethem, S.; Willemse, J.; Olsson, T.; Augustyns, K.; Hendriks, D. Development of a sensitive and selective assay for the determination of procarboxypeptidase U (thrombin-activatable fibrinolysis inhibitor) in plasma. Anal. Biochem. 2010, 396, 152–154. [Google Scholar] [CrossRef]
- Guo, X.; Morioka, A.; Kaneko, Y.; Okada, N.; Obata, K.; Nomura, T.; Campbell, W.; Okada, H. Arginine carboxypeptidase (CPR) in human plasma determined with sandwich ELISA. Microbiol. Immunol. 1999, 43, 691–698. [Google Scholar] [CrossRef]
- Mosnier, L.O.; Von Dem Borne, P.A.K.; Meijers, J.C.M.; Bouma, B.N. Plasma TAFI levels influence the clot lysis time in healthy individuals in the presence of an intact intrinsic pathway of coagulation. Thromb. Haemost. 1998, 80, 829–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceresa, E.; Brouwers, E.; Peeters, M.; Jern, C.; Declerck, P.J.; Gils, A. Development of ELISAs measuring the extent of TAFI activation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strömqvist, M.; Schatteman, K.; Leurs, J.; Verkerk, R.; Andersson, J.O.; Johansson, T.; Scharpé, S.; Hendriks, D. Immunological assay for the determination of procarboxypeptidase U antigen levels in human plasma. Thromb. Haemost. 2001, 85, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Willemse, J.L.; Hendriks, D.F. Measurement of procarboxypeptidase U (TAFI) in human plasma: A laboratory challenge. Clin. Chem. 2006, 52, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Hillmayer, K.; Brouwers, E.; León-Tamariz, F.; Meijers, J.C.M.; Marx, P.F.; Declerck, P.J.; Gils, A. Development of sandwich-type ELISAs for the quantification of rat and murine thrombin activatable fibrinolysis inhibitor in plasma. J. Thromb. Haemost. 2008, 6, 132–138. [Google Scholar] [CrossRef]
- Gils, A.; Alessi, M.C.; Brouwers, E.; Peeters, M.; Marx, P.; Leurs, J.; Bouma, B.; Hendriks, D.; Juhan-Vague, I.; Declerck, P.J. Development of a genotype 325-specific proCPU/TAFI ELISA. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1122–1127. [Google Scholar] [CrossRef]
- Guimarães, A.H.C.; Van Tilburg, N.H.; Vos, H.L.; Bertina, R.M.; Rijken, D.C. Association between thrombin activatable fibrinolysis inhibitor genotype and levels in plasma: Comparison of different assays. Br. J. Haematol. 2004, 124, 659–665. [Google Scholar] [CrossRef]
- Heylen, E.; Willemse, J.L.; Hendriks, D.F. Comparative study of commercially available procarboxypeptidase U (thrombin-activatable fibrinolysis inhibitor) assays. J. Thromb. Haemost. 2011, 9, 1407–1409. [Google Scholar] [CrossRef]
- Boffa, M.B.; Wang, W.; Bajzar, L.; Nesheim, M.E. Plasma and recombinant thrombin-activable fibrinolysis inhibitor (TAFI) and activated TAFI compared with respect to glycosylation, thrombin/thrombomodulin-dependent activation, thermal stability, and enzymatic properties. J. Biol. Chem. 1998, 273, 2127–2135. [Google Scholar] [CrossRef] [Green Version]
- Willemse, J.; Leurs, J.; Verkerk, R.; Hendriks, D. Development of a fast kinetic method for the determination of carboxypeptidase U (TAFIa) using C-terminal arginine containing peptides as substrate. Anal. Biochem. 2005, 340, 106–112. [Google Scholar] [CrossRef]
- Mock, W.L.; Stanford, D.J. Anisylazoformylarginine: A superior assay substrate for carboxypeptidase B type enzymes. Bioorganic Med. Chem. Lett. 2002, 12, 1193–1194. [Google Scholar] [CrossRef]
- Willemse, J.L.; Polla, M.; Olsson, T.; Hendriks, D.F. Comparative substrate specificity study of carboxypeptidase U (TAFIa) and carboxypeptidase N: Development of highly selective CPU substrates as useful tools for assay development. Clin. Chim. Acta 2008, 387, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.H.; Kim, P.; Nesheim, M.E. Thrombin-activable fibrinolysis inhibitor zymogen does not play a significant role in the attenuation of fibrinolysis. J. Biol. Chem. 2008, 283, 8863–8867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemse, J.L.; Polla, M.; Hendriks, D.F. The intrinsic enzymatic activity of plasma procarboxypeptidase U (TAFI) can interfere with plasma carboxypeptidase N assays. Anal. Biochem. 2006, 356, 157–159. [Google Scholar] [CrossRef]
- Willemse, J.L.; Matus, V.; Heylen, E.; Mezzano, D.; Hendriks, D.F. Influence of the Thr325Ile polymorphism on procarboxypeptidase U (thrombin-activable fibrinolysis inhibitor) activity-based assays [7]. J. Thromb. Haemost. 2007, 5, 872–875. [Google Scholar] [CrossRef]
- Wang, W.; Hendriks, D.F.; Scharpé, S.S. Carboxypeptidase U, a plasma carboxypeptidase with high affinity for plasminogen. J. Biol. Chem. 1994, 269, 15937–15944. [Google Scholar] [CrossRef]
- Santamaría, A.; Borrell, M.; Oliver, A.; Ortín, R.; Forner, R.; Coll, I.; Mateo, J.; Souto, J.C.; Fontcuberta, J. Association of functional thrombin-activatable fibrinolysis inhibitor (TAFI) with conventional cardiovascular risk factors and its correlation with other hemostatic factors in a Spanish population. Am. J. Hematol. 2004, 76, 348–352. [Google Scholar] [CrossRef]
- Santamaría, A.; Oliver, A.; Borrell, M.; Mateo, J.; Belvis, R.; Martí-Fábregas, J.; Ortín, R.; Tirado, I.; Souto, J.C.; Fontcuberta, J. Risk of ischemic stroke associated with functional thrombin-activatable fibrinolysis inhibitor plasma levels. Stroke 2003, 34, 2387–2391. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.F.; Fagundes, M.G.; Meijers, J.C.M.; Reitsma, P.H.; Lourenço, D.M.; Morelli, V.M.; Maffei, F.H.; Ferrari, I.C.; Piccinato, C.E.; Silva-Jr, W.A.; et al. Identification of polymorphisms in the 5’-untranslated region of the TAFI gene: Relationship with plasma TAFI levels and risk of venous thrombosis. Haematologica 2001, 86, 510–517. [Google Scholar]
- Schatteman, K.A.; Goossens, F.J.; Scharpé, S.S.; Neels, H.M.; Hendriks, D.F. Assay of procarboxypeptidase U, a novel determinant of the fibrinolytic cascade, in human plasma. Clin. Chem. 1999, 45, 807–813. [Google Scholar] [CrossRef]
- Silveira, A.; Schatteman, K.; Goossens, F.; Moor, E.; Scharpe, S.; Stromqvist, M.; Hendriks, D.; Hamsten, A. Plasma procarboxypeptidase U in men with symptomatic coronary artery disease. Thromb. Haemost. 2000, 84, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, L.; Bruni, F.; Pasqui, A.L.; Pastorelli, M.; Bova, G.; Cercignani, M.; Palazzuoli, A.; Auteri, A. Dyslipidemias and fibrinolysis. Ital. Hear. J. 2002, 3, 579–586. [Google Scholar]
- Brouns, R.; Heylen, E.; Sheorajpanday, R.; Willemse, J.L.; Kunnen, J.; De Surgeloose, D.; Hendriks, D.F.; De Deyn, P.P. Carboxypeptidase U (TAFIa) decreases the efficacy of thrombolytic therapy in ischemic stroke patients. Clin. Neurol. Neurosurg. 2009, 111, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Schatteman, K.A.; Goossens, F.J.; Leurs, J.; Kasahara, Y.; Scharpé, S.S.; Hendriks, D.F. Fast homogeneous assay for plasma procarboxypeptidase U. Clin. Chem. Lab. Med. 2001, 39, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; Heylen, E.; Willemse, J.L.; Sheorajpanday, R.; De Surgeloose, D.; Verkerk, R.; De Deyn, P.P.; Hendriks, D.F. The decrease in procarboxypeptidase U (TAFI) concentration in acute ischemic stroke correlates with stroke severity, evolution and outcome. J. Thromb. Haemost. 2010, 8, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.C.; Leenaerts, D.; Brouns, R.; Engelborghs, S.; Ieven, M.; De Deyn, P.P.; Lambeir, A.M.; Hendriks, D. Procarboxypeptidase U (proCPU, TAFI, proCPB2) in cerebrospinal fluid during ischemic stroke is associated with stroke progression, outcome and blood–brain barrier dysfunction. J. Thromb. Haemost. 2018, 16, 342–348. [Google Scholar] [CrossRef]
- Alessi, M.; Gaudin, C.; Grosjean, P.; Martin, V.; Timsit, S.; Brouns, R.; Montaner, J.; Castellanos, M.; Donazzola, Y.; Cho, T.-H.; et al. Changes in Activated Thrombin-Activatable Fibrinolysis Inhibitor Levels Following Thrombolytic Therapy in Ischemic Stroke Patients Correlate with Clinical Outcome. Cerebrovasc. Dis. 2016, 42, 404–414. [Google Scholar] [CrossRef]
- Cruden, N.L.M.; Graham, C.; Harding, S.A.; Ludlam, C.A.; Fox, K.A.A.; Newby, D.E. Plasma TAFI and soluble CD40 ligand do not predict reperfusion following thrombolysis for acute myocardial infarction. Thromb. Res. 2006, 118, 189–197. [Google Scholar] [CrossRef]
- Zorio, E.; Castelló, R.; Falcó, C.; España, F.; Osa, A.; Almenar, L.; Aznar, J.; Estellés, A. Thrombin-activatable fibrinolysis inhibitor in young patients with myocardial infarction and its relationship with the fibrinolytic function and the protein C system. Br. J. Haematol. 2003, 122, 958–965. [Google Scholar] [CrossRef]
- Lisowski, P.; Małyszko, J.; Hirnle, T.; Lisowska, A.; Jackowski, R.; Małyszko, J.S.; Buzun, L.; Myśliwiec, M. Thrombin activatable fibrinolysis inhibitor (TAFI) in stable angina pectoris patients undergoing coronary artery bypass grafting (CABG). Rocz. Akad. Med. Białymstoku 2005, 50, 166–172. [Google Scholar]
- Malyszko, J.; Malyszko, J.S.; Hryszko, T.; Mysliwiec, M. Thrombin-activatable fibrinolysis inhibitor in kidney transplant recipient with dyslipidemia. Transplant. Proc. 2003, 35, 2219–2221. [Google Scholar] [CrossRef]
- Malyszko, J.; Tymcio, J. Thrombin activatable fibrinolysis inhibitor and other hemostatic parameters in patients with essential arterial hypertension. Pol. Arch. Med. Wewn. 2008, 118, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Antovic, J.P.; Yngen, M.; Östenson, C.G.; Antovic, A.; Wallen, H.N.; Jorneskög, G.; Blombäck, M. Thrombin activatable fibrinolysis inhibitor and hemostatic changes in patients with type I diabetes mellitus with and without microvascular complications. Blood Coagul. Fibrinolysis 2003, 14, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Martí-Fàbregas, J.; Borrell, M.; Cocho, D.; Martínez-Ramírez, S.; Martínez-Corral, M.; Fontcuberta, J.; Martí-Vilalta, J.L. Change in hemostatic markers after recombinant tissue-type plasminogen activator is not associated with the chance of recanalization. Stroke 2008, 39, 234–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meltzer, M.E.; Lisman, T.; De Groot, P.G.; Meijers, J.C.M.; Le Cessie, S.; Doggen, C.J.M.; Rosendaal, F.R. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood 2010, 116, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Meltzer, M.E.; Doggen, C.J.M.; De Groot, P.G.; Meijers, J.C.M.; Rosendaal, F.R.; Lisman, T. Low thrombin activatable fibrinolysis inhibitor activity levels are associated with an increased risk of a first myocardial infarction in men. Haematologica 2009, 94, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Folkeringa, N.; Coppens, M.; Veeger, N.J.G.M.; Bom, V.J.J.; Middeldorp, S.; Hamulyak, K.; Prins, M.H.; Büller, H.R.; Van Der Meer, J. Absolute risk of venous and arterial thromboembolism in thrombophilic families is not increased by high thrombin-activatable fibrinolysis inhibitor (TAFI) levels. Thromb. Haemost. 2008, 100, 38–44. [Google Scholar] [CrossRef]
- Schroeder, V.; Wilmer, M.; Buehler, B.; Kohler, H.P. TAFI activity in coronary artery disease: A contribution to the current discussion on TAFI assays. Thromb. Haemost. 2006, 96, 236–237. [Google Scholar] [CrossRef]
- Schol-gelok, S.; Maat, D.; Biedermann, J.S.; Van Gelder, T.; Frank, W.G.; Lijfering, W.M.; Van Der Meer, F.J.M.; Dingeman, C.; Versmissen, J.; Kruip, J.H.A. Rosuvastatin use increases plasma fibrinolytic potential: A ran- domised clinical trial. Br. J. Haematol. 2020, 1–7. [Google Scholar] [CrossRef]
- Harmanci, A.; Kandemir, N.; Dagdelen, S.; Gonc, N.; Buyukasik, Y.; Alikasifoglu, A.; Kirazli, S.; Ozon, A.; Gurlek, A. Thrombin-activatable fibrinolysis inhibitor activity and global fibrinolytic capacity in Type 1 diabetes: Evidence for normal fibrinolytic state. J. Diabetes Complications 2006, 20, 40–44. [Google Scholar] [CrossRef]
- Skeppholm, M.; Wallén, N.H.; Malmqvist, K.; Kallner, A.; Antovic, J.P. Comparison of two immunochemical assays for measuring thrombin-activatable fibrinolysis inhibitor concentration with a functional assay in patients with acute coronary syndrome. Thromb. Res. 2007, 121, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kochan, J.; Yin, O.; Warren, V.; Zamora, C.; Atiee, G.; Pav, J.; Orihashi, Y.; Vashi, V.; Dishy, V. A first-in-human study of DS-1040, an inhibitor of the activated form of thrombin-activatable fibrinolysis inhibitor, in healthy subjects. J. Thromb. Haemost. 2017, 15, 961–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Limsakun, T.; Yin, O.; Warren, V.; Zamora, C.; Atiee, G.; Kochan, J.; Pav, J.; Kobayashi, F.; Vashi, V.; et al. First-in-Human Study to Assess the Safety, Pharmacokinetics, and Pharmacodynamics of an Oral Formulation of DS-1040, an Inhibitor of the Activated Form of Thrombin-Activatable Fibrinolysis Inhibitor, in Healthy Subjects. J. Clin. Pharmacol. 2019, 59, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Verkleij, C.J.N.; Nieuwdorp, M.; Gerdes, V.E.A.; Mörgelin, M.; Meijers, J.C.M.; Marx, P.F. The effects of hyperglycaemia on thrombin-activatable fibrinolysis inhibitor. Thromb. Haemost. 2009, 102, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Verkleij, C.J.N.; De Bruijn, R.E.; Meesters, E.W.; Gerdes, V.E.A.; Meijers, J.C.M.; Marx, P.F. The hemostatic system in patients with type 2 diabetes with and without cardiovascular disease. Clin. Appl. Thromb. 2011, 17. [Google Scholar] [CrossRef] [Green Version]
- Van Tilburg, N.H.; Rosendaal, F.R.; Bertina, R.M. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood 2000, 95, 2855–2859. [Google Scholar] [CrossRef]
- Meltzer, M.E.; Doggen, C.J.M.; De Groot, P.G.; Rosendaal, F.R.; Lisman, T. Plasma levels of fibrinolytic proteins and the risk of myocardial infarction in men. Blood 2010, 116, 529–536. [Google Scholar] [CrossRef] [Green Version]
- de Bruijne, E.L.E.; Gils, A.; Guimarães, A.H.C.; Dippel, D.W.J.; Deckers, J.W.; van den Meiracker, A.H.; Poldermans, D.; Rijken, D.C.; Declerck, P.J.; de Maat, M.P.M.; et al. The role of thrombin activatable fibrinolysis inhibitor in arterial thrombosis at a young age: The ATTAC study. J. Thromb. Haemost. 2009, 7, 919–927. [Google Scholar] [CrossRef]
- De Bruijne, E.L.E.; Gils, A.; Rijken, D.C.; De Maat, M.P.M.; Guimarães, A.H.C.; Poldermans, D.; Declerck, P.J.; Leebeek, F.W.G. High thrombin activatable fibrinolysis inhibitor levels are associated with an increased risk of premature peripheral arterial disease. Thromb. Res. 2011, 127, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Martini, C.H.; Brandts, A.; De Bruijne, E.L.E.; Van Hylckama Vlieg, A.; Leebeek, F.W.G.; Lisman, T.; Rosendaal, F.R. The effect of genetic variants in the thrombin activatable fibrinolysis inhibitor (TAFI) gene on TAFI-antigen levels, clot lysis time and the risk of venous thrombosis. Br. J. Haematol. 2006, 134, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Jood, K.; Redfors, P.; Gils, A.; Blomstrand, C.; Declerck, P.J.; Jern, C. Convalescent plasma levels of TAFI activation peptide predict death and recurrent vascular events in ischemic stroke survivors. J. Thromb. Haemost. 2012, 10, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Ladenvall, C.; Gils, A.; Jood, K.; Blomstrand, C.; Declerck, P.J.; Jern, C. Thrombin activatable fibrinolysis inhibitor activation peptide shows association with all major subtypes of ischemic stroke and with TAFI gene variation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridge, K.I.; Bollen, L.; Zhong, J.; Hesketh, M.; Macrae, F.L.; Johnson, A.; Philippou, H.; Scott, D.J.; Gils, A.; Ariëns, R.A.S. Thrombin-activatable fibrinolysis inhibitor in human abdominal aortic aneurysm disease. J. Thromb. Haemost. 2017, 15, 2218–2225. [Google Scholar] [CrossRef] [PubMed]
- Rooth, E.; Wallen, H.; Antovic, A.; von Arbin, M.; Kaponides, G.; Wahlgren, N.; Blombäck, M.; Antovic, J. Thrombin activatable fibrinolysis inhibitor and its relationship to fibrinolysis and inflammation during the acute and convalescent phase of ischemic stroke. Blood Coagul. Fibrinolysis 2007, 18, 365–370. [Google Scholar] [CrossRef]
- Brouwers, G.-J.; Leebeek, F.W.G.; Tanck, M.W.T.; Jukema, J.W.; Kluft, C.; de Maat, M.P.M. Association between thrombin-activatable fibrinolysis inhibitor (TAFI) and clinical outcome in patients with unstable angina pectoris. Thromb. Haemost. 2003, 90, 92–100. [Google Scholar] [CrossRef]
- Schroeder, V.; Chatterjee, T.; Mehta, H.; Windecker, S.; Pham, T.; Devantay, N.; Meier, B.; Kohler, H.P. Thrombin activatable fibrinolysis inhibitor (TAFI) levels in patients with coronary artery disease investigated by angiography. Thromb. Haemost. 2002, 88, 1020–1025. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Morange, P.E. Very high TAFI antigen levels are associated with a lower risk of hard coronary events: The PRIME study. J. Thromb. Haemost. 2003, 1, 2243–2244. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Morange, P.E.; Aubert, H.; Henry, M.; Aillaud, M.F.; Alessi, M.C.; Samnegård, A.; Hawe, E.; Yudkin, J.; Margaglione, M.; et al. Plasma thrombin-activatable fibrinolysis inhibitor antigen concentration and genotype in relation to myocardial infarction in the North and South of Europe. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Malyszko, J.; Malyszko, J.S.; Mysliwiec, M. Fluvastin therapy affects TAFI concentration in kidney transplant recipients. Transpl. Int. 2003, 16, 53–57. [Google Scholar] [CrossRef]
- Schroeder, V.; Kucher, N.; Kohler, H.P. Role of thrombin activatable fibrinolysis inhibitor (TAFI) in patients with acute pulmonary embolism. J. Thromb. Haemost. 2003, 1, 492–493. [Google Scholar] [CrossRef]
- Lau, H.K.; Segev, A.; Hegele, R.a.; Sparkes, J.D.; Teitel, J.M.; Chisholm, R.J.; Strauss, B.H. Thrombin-activatable fibrinolysis inhibitor (TAFI): A novel predictor of angiographic coronary restenosis. Thromb. Haemost. 2003, 90, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- Segev, A.; Hegele, R.A.; Lau, H.K.; Sparkes, J.D.; Teitel, J.M.; Chisholm, R.J.; Strauss, B.H. Thr325Ile polymorphism of the TAFI gene is related to TAFI antigen plasma levels and angiographic restenosis after percutaneous coronary interventions. Thromb. Res. 2004, 114, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, G.; Ulusoy, S.; Sönmez, M.; Caner Karahan, S.; Menteşe, A.; Kaynar, K.; Bektaş, O. Thrombin activatable fibrinolysis inhibitor (TAFI) levels in hypertensive patients and a comparison of the effects of amlodipine and ramipril on TAFI levels. Clin. Exp. Hypertens. 2013, 35, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Guven, G.S.; Atalar, E.; Yavuz, B.; Beyazit, Y.; Kekilli, M.; Kilicarslan, A.; Sahiner, L.; Oz, G.; Ozer, N.; Aksoyek, S.; et al. Simvastatin treatment improves endothelial function and increases fibrinolysis in patients with hypercholestrolemia. J. Natl. Med. Assoc. 2006, 98, 627–630. [Google Scholar] [PubMed]
- Kilicarslan, A.; Yavuz, B.; Guven, G.S.; Atalar, E.; Sahiner, L.; Beyazit, Y.; Kekilli, M.; Ozer, N.; Oz, G.; Haznedaroglu, I.C.; et al. Fenofibrate improves endothelial function and decreases thrombin-activatable fibrinolysis inhibitor concentration in metabolic syndrome. Blood Coagul. Fibrinolysis 2008, 19, 310–314. [Google Scholar] [CrossRef]
- Verdú, J.; Marco, P.; Benlloch, S.; Lucas, J. Association between the Thr325Ile and Ala147Thr polymorphisms of the TAFI gene and the risk of venous thromboembolic disease. Clin. Appl. Thromb. 2008, 14, 494–495. [Google Scholar] [CrossRef]
- Eichinger, S.; Schönauer, V.; Weltermann, A.; Minar, E.; Bialonczyk, C.; Hirschl, M.; Schneider, B.; Quehenberger, P.; Kyrle, P.A. Thrombin-activatable fibrinolysis inhibitor and the risk for recurrent venous thromboembolism. Blood 2004, 103, 3773–3776. [Google Scholar] [CrossRef]
- Tàssies, D.; Roqué, M.; Monteagudo, J.; Martorell, T.; Sionis, A.; Arzamendi, D.; Heras, M.; Reverter, J.C. Thrombin-activatable fibrinolysis inhibitor genetic polymorphisms as markers of the type of acute coronary syndrome. Thromb. Res. 2009, 124, 614–618. [Google Scholar] [CrossRef]
- Tregouet, D.A.; Schnabel, R.; Alessi, M.C.; Godefroy, T.; Declerck, P.J.; Nicaud, V.; Munzel, T.; Bickel, C.; Rupprecht, H.J.; Lubos, E.; et al. Activated thrombin activatable fibrinolysis inhibitor levels are associated with the risk of cardiovascular death in patients with coronary artery disease: The Athero Gene study. J. Thromb. Haemost. 2009, 7, 49–57. [Google Scholar] [CrossRef]
- Biswas, A.; Tiwari, A.K.; Ranjan, R.; Meena, A.; Akhter, M.S.; Yadav, B.K.; Behari, M.; Saxena, R. Thrombin activatable fibrinolysis inhibitor gene polymorphisms are associated with antigenic levels in the Asian-Indian population but may not be a risk for stroke. Br. J. Haematol. 2008, 143, 581–588. [Google Scholar] [CrossRef]
- Morange, P.E.; Tregouet, D.A.; Frere, C.; Luc, G.; Arveiler, D.; Ferrieres, J.; Amouyel, P.; Evans, A.; Ducimetiere, P.; Cambien, F.; et al. TAFI gene haplotypes, TAFI plasma levels and future risk of coronary heart disease: The PRIME Study. J. Thromb. Haemost. 2005, 3, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Kitagawa, N.; Gabazza, E.C.; Morioka, K.; Urakawa, H.; Tanaka, T.; Katsuki, A.; Araki-Sasaki, R.; Hori, Y.; Nakatani, K.; et al. Increased plasma thrombin-activatable fibrinolysis inhibitor levels in normotensive type 2 diabetic patients with microalbuminuria. J. Clin. Endocrinol. Metab. 2003, 88, 736–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, Y.; Gabazza, E.C.; Yano, Y.; Katsuki, A.; Suzuki, K.; Adachi, Y.; Sumida, Y. Insulin resistance is associated with increased circulating level of thrombin-activatable fibrinolysis inhibitor in type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2002, 87, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, N.; Yano, Y.; Gabazza, E.C.; Bruno, N.E.; Araki, R.; Matsumoto, K.; Katsuki, A.; Hori, Y.; Nakatani, K.; Taguchi, O.; et al. Different metabolic correlations of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 in non-obese type 2 diabetic patients. Diabetes Res. Clin. Pract. 2006, 73, 150–157. [Google Scholar] [CrossRef]
- Morange, P.E.; Juhan-Vague, I.; Scarabin, P.Y.; Alessi, M.C.; Luc, G.; Arveiler, D.; Ferrieres, J.; Amouyel, P.; Evans, A.; Ducimetiere, P. Association between TAFI antigen and Ala 147Thr polymorphism of the TAFI gene and the angina pectoris incidence: The PRIME Study. Thromb. Haemost. 2003, 89, 554–560. [Google Scholar] [CrossRef]
- Kim, S.H.; Han, S.W.; Kim, E.H.; Kim, D.J.; Lee, K.Y.; Kim, D.I.; Heo, J.H. Plasma fibrinolysis inhibitor levels in acute stroke patients with thrombolysis failure. J. Clin. Neurol. 2005, 1, 142–147. [Google Scholar] [CrossRef]
- Montaner, J.; Ribo, M.; Monasterio, J.; Molina, C.A.; Alvarez-Sabin, J. Thrombin-activable fibrinolysis inhibitor levels in the acute phase of ischemic stroke. Stroke 2003, 34, 1038–1040. [Google Scholar] [CrossRef] [Green Version]
- Shantsila, E.; Montoro-Garcia, S.; Tapp, L.D.; Apostolakis, S.; Wrigley, B.J.; Lip, G.Y.H. Fibrinolytic status in acute coronary syndromes: Evidence of differences in relation to clinical features and pathophysiological pathways. Thromb. Haemost. 2012, 108, 32–40. [Google Scholar] [CrossRef]
- Cellai, A.P.; Antonucci, E.; Liotta, A.A.; Fedi, S.; Marcucci, R.; Falciani, M.; Giglioli, C.; Abbate, R.; Prisco, D. TAFI activity and antigen plasma levels are not increased in acute coronary artery disease patients admitted to a coronary care unit. Thromb. Res. 2006, 118, 495–500. [Google Scholar] [CrossRef]
- Hendriks, D.; Scharpé, S.; van Sande, M.; Lommaert, M.P. Characterisation of a carboxypeptidase in human serum distinct from carboxypeptidase N. J. Clin. Chem. Clin. Biochem. 1989, 27, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.Y.G.; Foley, J.; Hsu, G.; Kim, P.Y.; Nesheim, M.E. An assay for measuring functional activated thrombin-activatable fibrinolysis inhibitor in plasma. Anal. Biochem. 2008, 372, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Heylen, E.; Van Goethem, S.; Augustyns, K.; Hendriks, D. Measurement of carboxypeptidase U (active thrombin-activatable fibrinolysis inhibitor) in plasma: Challenges overcome by a novel selective assay. Anal. Biochem. 2010, 403, 114–116. [Google Scholar] [CrossRef]
- Leenaerts, D.; Bosmans, J.M.; van der Veken, P.; Sim, Y.; Lambeir, A.M.; Hendriks, D. Plasma levels of carboxypeptidase U (CPU, CPB2 or TAFIa) are elevated in patients with acute myocardial infarction. J. Thromb. Haemost. 2015, 13, 2227–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leenaerts, D.; Loyau, S.; Mertens, J.C.; Boisseau, W.; Michel, J.B.; Lambeir, A.M.; Jandrot-Perrus, M.; Hendriks, D. Carboxypeptidase U (CPU, carboxypeptidase B2, activated thrombin-activatable fibrinolysis inhibitor) inhibition stimulates the fibrinolytic rate in different in vitro models. J. Thromb. Haemost. 2018, 16, 2057–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit Dop, F.; Latreille, M.; Guicherd, L.; Mertens, J.; Claesen, K.; Hendriks, D.; Arnaud, E.; Donazzolo, Y. Favourable safety profile of S62798, a potent TAFIa (activated thrombin-activatable fibrinolysis inhibitor) inhibitor, in first-in-man study in healthy subjects. Eur. Heart J. 2020, 41. [Google Scholar] [CrossRef]
- Rigla, M.; Wägner, A.M.; Borrell, M.; Mateo, J.; Foncuberta, J.; de Leiva, A.; Ordóñez-Llanos, J.; Pérez, A. Postprandial thrombin activatable fibrinolysis inhibitor and markers of endothelial dysfunction in type 2 diabetic patients. Metabolism. 2006, 55, 1437–1442. [Google Scholar] [CrossRef]
- Ribo, M.; Montaner, J.; Molina, C.a.; Arenillas, J.F.; Santamarina, E.; Alvarez-Sabín, J. Admission fibrinolytic profile predicts clot lysis resistance in stroke patients treated with tissue plasminogen activator. Thromb. Haemost. 2004, 1146–1151. [Google Scholar] [CrossRef]
- Khalifa, N.M.; Gad, M.Z.; Hataba, A.A.; Mahran, L.G. Changes in ADMA and TAFI levels after stenting in coronary artery disease patients. Mol. Med. Rep. 2012, 6, 855–859. [Google Scholar] [CrossRef]
- Willemse, J.L.; Brouns, R.; Heylen, E.; De Deyn, P.P.; Hendriks, D.F. Carboxypeptidase U (TAFIa) activity is induced in vivo in ischemic stroke patients receiving thrombolytic therapy. J. Thromb. Haemost. 2008, 6, 200–202. [Google Scholar] [CrossRef]
- Mertens, J.C.; Claesen, K.; Leenaerts, D.; Sim, Y.; Lambeir, A.M.; Hendriks, D. Inhibition of the procarboxypeptidase U (proCPU, TAFI, proCPB2) system due to hemolysis. J. Thromb. Haemost. 2019, 17, 878–884. [Google Scholar] [CrossRef]
- Pedersen, A.; Redfors, P.; Lundberg, L.; Gils, A.; Declerck, P.J.; Nilsson, S.; Jood, K.; Jern, C. Haemostatic biomarkers are associated with long-term recurrent vascular events after ischaemic stroke. Thromb. Haemost. 2016, 116, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.P.; An, S.S.A. Detecting activated thrombin activatable fibrinolysis inhibitor (TAFIa) and inactivated TAFIa (TAFIai) in normal and hemophilia a plasmas. Bull. Korean Chem. Soc. 2009, 30, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Lisman, T. Decreased Plasma Fibrinolytic Potential As a Risk for Venous and Arterial Thrombosis. Semin. Thromb. Hemost. 2017, 43, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; Philippou, H.; Undas, A.; de Lange, Z.; Rijken, D.C.; Mutch, N.J. An international study on the feasibility of a standardized combined plasma clot turbidity and lysis assay: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1007–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leenaerts, D.; Aernouts, J.; Van Der Veken, P.; Sim, Y.; Lambeir, A.M.; Hendriks, D. Plasma carboxypeptidase U (CPU, CPB2, TAFia) generation during in vitro clot lysis and its interplay between coagulation and fibrinolysis. Thromb. Haemost. 2017, 117, 1498–1508. [Google Scholar] [CrossRef]
- Leurs, J.; Wissing, B.M.; Nerme, V.; Schatteman, K.; Björquist, P.; Hendriks, D. Different mechanisms contribute to the biphasic pattern of carboxypeptidase U (TAFIa) generation during in vitro clot lysis in human plasma. Thromb. Haemost. 2003, 89, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.B.; Bajzar, L. The intrinsic threshold of the fibrinolytic system is modulated by basic carboxypeptidases, but the magnitude of the antifibrinolytic effect of activated thrombin-activable fibrinolysis inhibitor is masked by its instability. J. Biol. Chem. 2004, 279, 27896–27904. [Google Scholar] [CrossRef] [Green Version]
- Leurs, J.; Nerme, V.; Sim, Y.; Hendriks, D. Carboxypeptidase U (TAFIa) prevents lysis from proceeding into the propagation phase through a threshold-dependent mechanism. J. Thromb. Haemost. 2004, 2, 416–423. [Google Scholar] [CrossRef]
- Akinci, B. Role of Thrombin Activatable Fibrinolysis Inhibitor in Endocrine and Cardiovascular Disorders: An Update. Recent Pat. Endocr. Metab. Immune Drug Discov. 2012, 6, 210–217. [Google Scholar] [CrossRef]
- Meltzer, M.E.; Doggen, C.J.M.; De Groot, P.G.; Rosendaal, F.R.; Lisman, T. Fibrinolysis and the risk of venous and arterial thrombosis. Curr. Opin. Hematol. 2007, 14, 242–248. [Google Scholar] [CrossRef]
- Bajzar, L.; Morser, J.; Nesheim, M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J. Biol. Chem. 1996, 271, 16603–16608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boffa, M.B.; Koschinsky, M.L. Curiouser and curiouser: Recent advances in measurement of thrombin-activatable fibrinolysis inhibitor (TAFI) and in understanding its molecular genetics, gene regulation, and biological roles. Clin. Biochem. 2007, 40, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.S.; Cooper, C.M.; Wood, T.; Shafer, J.A.; Gardell, S.J. Characterization of plasmin-mediated activation of plasma procarboxypeptidase B. Modulation by glycosaminoglycans. J. Biol. Chem. 1999, 274, 35046–35052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meltzer, M.E.; Bol, L.; Rosendaal, F.R.; Lisman, T.; Cannegieter, S.C. Hypofibrinolysis as a risk factor for recurrent venous thrombosis; results of the LETS follow-up study. J. Thromb. Haemost. 2010, 8, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Libourel, E.J.; Bank, I.; Meinardi, J.R.; Baljé-Volkers, C.P.; Hamulyak, K.; Middeldorp, S.; Koopman, M.M.; Van Pampus, E.C.; Prins, M.H.; Büller, H.R.; et al. Co-segregation of thrombophilic disorders in factor V Leiden carriers; the contributions of factor VIII, factor XI, thrombin activatable fibrinolysis inhibitor and lipoprotein(a) to the absolute risk of venous thromboembolism. Haematologica 2002, 87, 1068–1073. [Google Scholar] [PubMed]
- Qian, K.; Xu, J.; Wan, H.; Fu, F.; Lu, J.; Lin, Z.; Liu, Z.; Liu, H. Impact of genetic polymorphisms in thrombin activatable fibrinolysis inhibitor (TAFI) on venous thrombosis disease: A meta-analysis. Gene 2015, 569, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ma, H.; Lu, L.; Sun, G.; Liu, D.; Zhou, Y.; Tong, Y.; Lu, Z. Association between thrombin-activatable fibrinolysis inhibitor gene polymorphisms and venous thrombosis risk: A meta-analysis. Blood Coagul. Fibrinolysis 2016, 27, 419–430. [Google Scholar] [CrossRef]
- Zwingerman, N.; Medina-Rivera, A.; Kassam, I.; Wilson, M.D.; Morange, P.E.; Trégöuet, D.A.; Gagnon, F. Sex-specific effect of CPB2 Ala147Thr but not Thr325Ile variants on the risk of venous thrombosis: A comprehensive meta-Analysis. PLoS ONE 2017, 12, 1–18. [Google Scholar] [CrossRef]
- Arauz, A.; Argüelles, N.; Jara, A.; Guerrero, J.; Barboza, M.A. Thrombin-Activatable Fibrinolysis Inhibitor Polymorphisms and Cerebral Venous Thrombosis in Mexican Mestizo Patients. Clin. Appl. Thromb. 2018, 24, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Orikaza, C.M.; Morelli, V.M.; Matos, M.F.; Lourenço, D.M. Haplotypes of TAFI gene and the risk of cerebral venous thrombosis—A case-control study. Thromb. Res. 2014, 133, 120–124. [Google Scholar] [CrossRef]
- Tokgoz, S.; Zamani, A.G.; Durakbasi-Dursun, H.G.; Yılmaz, O.; Ilhan, N.; Demirel, S.; Tavli, M.; Sinan, A. TAFI gene polymorphisms in patients with cerebral venous thrombosis. Acta Neurol. Belg. 2013, 113, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Verdú, J.; Marco, P.; Benlloch, S.; Sanchez, J.; Lucas, J. Thrombin activatable fibrinolysis inhibitor (TAFI) polymorphisms and plasma TAFI levels measured with an ELISA insensitive to isoforms in patients with venous thromboembolic disease (VTD). Thromb. Haemost. 2006, 95, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Zee, R.Y.L.; Hegener, H.H.; Ridker, P.M. Carboxypeptidase B2 gene polymorphisms and the risk of venous thromboembolism. J. Thromb. Haemost. 2005, 3, 2819–2821. [Google Scholar] [CrossRef] [PubMed]
- Isordia-Salas, I.; Martínez-Marino, M.; Alberti-Minutti, P.; Ricardo-Moreno, M.T.; Castro-Calvo, R.; Santiago-Germán, D.; Alvarado-Moreno, J.A.; Calleja-Carreño, C.; Hernández-Juárez, J.; Leaños-Miranda, A.; et al. Genetic Polymorphisms Associated with Thrombotic Disease Comparison of Two Territories: Myocardial Infarction and Ischemic Stroke. Dis. Markers 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattanawan, C.; Komanasin, N.; Settasatian, N.; Settasatian, C.; Kukongviriyapan, U.; Intharapetch, P.; Senthong, V. Association of TAFI gene polymorphisms with severity of coronary stenosis in stable coronary artery disease. Thromb. Res. 2018, 171, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, A.H.C.; Bertina, R.M.; Rijken, D.C. A new functional assay of thrombin activatable fibrinolysis inhibitor. J. Thromb. Haemost. 2005, 3, 1284–1292. [Google Scholar] [CrossRef]
- Kamal, H.M.; Ahmed, A.S.; Fawzy, M.S.; Mohamed, F.A.; Elbaz, A.A. Plasma thrombin-activatable fibrinolysis inhibitor levels and Thr325ile polymorphism as a risk marker of myocardial infarction in Egyptian patients. Acta Cardiol. 2011, 66, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Kozian, D.H.; Lorenz, M.; März, W.; Cousin, E.; Mace, S.; Deleuze, J.F. Association between the Thr325Ile polymorphism of the thrombin-activatable fibrinolysis inhibitor and stroke in the Ludwigshafen Risk and Cardiovascular Health Study. Thromb. Haemost. 2010, 103, 976–983. [Google Scholar] [CrossRef]
- Fernandez-Cadenas, I.; Alvarez-Sabin, J.; Ribo, M.; Rubiera, M.; Mendioroz, M.; Molina, C.A.; Rosell, A.; Montaner, J. Influence of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 gene polymorphisms on tissue-type plasminogen activator-induced recanalization in ischemic stroke patients. J. Thromb. Haemost. 2007, 5, 1862–1868. [Google Scholar] [CrossRef]
- Leebeek, F.W.G.; Van Goor, M.P.J.; Guimaraes, A.H.C.; Brouwers, G.J.; De Maat, M.P.M.; Dippel, D.W.J.; Rijken, D.C. High functional levels of thrombin-activatable fibrinolysis inhibitor are associated with an increased risk of first ischemic stroke. J. Thromb. Haemost. 2005, 3, 2211–2218. [Google Scholar] [CrossRef]
- Santamaría, A.; Martínez-Rubio, A.; Borrell, M.; Mateo, J.; Ortín, R.; Foncuberta, J. Risk of acute coronary artery disease associated with functional thrombin activatable fibrinolysis inhibitor plasma level. Haematologica 2004, 89, 880–881. [Google Scholar] [PubMed]
- Akatsu, H.; Yamagata, H.; Chen, Y.; Miki, T.; Kamino, K.; Takeda, M.; Campbell, W.; Kondo, I.; Kosaka, K.; Yamamoto, T.; et al. TAFI polymorphisms at amino 147 and 325 are not risk factors for cerebral infarction. Br. J. Haematol. 2004, 127, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Morange, P.E.; Henry, M.; Frere, C.; Juhan-Vague, I. Thr325Ile polymorphism of the TAFIgene does not influence the risk of myocardial infarction. PACE Pacing Clin. Electrophysiol. 2002, 99, 1878–1879. [Google Scholar] [CrossRef]
- Shi, J.; Zhi, P.; Chen, J.; Wu, P.; Tan, S. Genetic variations in the thrombin-activatable fibrinolysis inhibitor gene and risk of cardiovascular disease: A systematic review and meta-analysis. Thromb. Res. 2014, 134, 610–616. [Google Scholar] [CrossRef]
- Wang, S.W.; Zhang, H.H.; Dong, C.Y.; Sun, H.H. Meta-analysis of TAFI polymorphisms and risk of cardiovascular and cerebrovascular diseases. Genet. Mol. Res. 2016, 15, 1–13. [Google Scholar] [CrossRef]
- Agirbasli, M.; Cincin, A.; Baykan, O.A. Short-term effects of angiotensin receptor blockers on blood pressure control, and plasma inflammatory and fibrinolytic parameters in patients taking angiotensin-converting enzyme inhibitors. JRAAS J. Renin-Angiotensin-Aldosterone Syst. 2008, 9, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Santos, I.R.; Fernandes, A.P.; Carvalho, M.G.; Sousa, M.O.; Ferreira, C.N.; Gomes, K.B. Thrombin-activatable fibrinolysis inhibitor (TAFI) levels and its polymorphism rs3742264 are associated with dyslipidemia in a cohort of Brazilian subjects. Clin. Chim. Acta 2014, 433, 76–83. [Google Scholar] [CrossRef]
- Pang, H.; Zhang, C.; Liu, F.; Gong, X.; Jin, X.; Su, C. Reduced thrombin activatable fibrinolysis inhibitor and enhanced proinflammatory cytokines in acute coronary syndrome. Med. Intensiva 2017, 41, 475–482. [Google Scholar] [CrossRef]
- Mattsson, C.; Björkman, J.A.; Abrahamsson, T.; Nerme, V.; Schatteman, K.; Leurs, J.; Scharpé, S.; Hendriks, D. Local proCPU (TAFI) activation during thrombolytic treatment in a dog model of coronary artery thrombosis can be inhibited with a direct, small molecule thrombin inhibitor (melagatran). Thromb. Haemost. 2002, 87, 557–562. [Google Scholar] [CrossRef] [Green Version]
- Mao, S.S.; Colussi, D.; Bailey, C.M.; Bosserman, M.; Burlein, C.; Gardell, S.J.; Carroll, S.S. Electrochemiluminescence assay for basic carboxypeptidases: Inhibition of basic carboxypeptidases and activation of thrombin-activatable fibrinolysis inhibitor. Anal. Biochem. 2003, 319, 159–170. [Google Scholar] [CrossRef]
- Redlitz, A.; Tan, A.K.; Eaton, D.L.; Plow, E.F. Plasma carboxypeptidases as regulators of the plasminogen system. J. Clin. Invest. 1995, 96, 2534–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reverter, D.; Fernández-Catalán, C.; Baumgartner, R.; Pfänder, R.; Huber, R.; Bode, W.; Vendrell, J.; Holak, T.A.; Avilés, F.X. Structure of a novel leech carboxypeptidase inhibitor determined free in solution and in complex with human carboxypeptidase A2. Nat. Struct. Biol. 2000, 7, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Arolas, J.L.; Lorenzo, J.; Rovira, A.; Castellà, J.; Aviles, F.X.; Sommerhoff, C.P. A carboxypeptidase inhibitor from the tick Rhipicephalus bursa: Isolation, cDNA cloning, recombinant expression, and characterization. J. Biol. Chem. 2005, 280, 3441–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemse, J.L.; Heylen, E.; Nesheim, M.E.; Hendriks, D.F. Carboxypeptidase U (TAFIa): A new drug target for fibrinolytic therapy? J. Thromb. Haemost. 2009, 7, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, N.; Sasaki, T.; Sugimoto, K.; Ishii, H.; Yamamoto, K. Design and characterization of a selenium-containing inhibitor of activated thrombin-activatable fibrinolysis inhibitor (TAFIa), a zinc-containing metalloprotease. J. Med. Chem. 2012, 55, 7696–7705. [Google Scholar] [CrossRef]
- Bunnage, M.E.; Owen, D.R. TAFIa inhibitors in the treatment of thrombosis. Curr. Opin. Drug Discov. Devel. 2008, 11, 480–486. [Google Scholar]
- Polla, M.O.; Tottie, L.; Nordén, C.; Linschoten, M.; Müsil, D.; Trumpp-Kallmeyer, S.; Aukrust, I.R.; Ringom, R.; Holm, K.H.; Neset, S.M.; et al. Design and synthesis of potent, orally active, inhibitors of carboxypeptidase U (TAFIa). Bioorganic Med. Chem. 2004, 12, 1151–1175. [Google Scholar] [CrossRef]
- Eriksson, H.; Sandset, P.M.; Jensen, E.; Wall, U.; Andersson, M.; Nerme, V.; Eriksson-Lepkowska, M.; Wahlander, K.; Eriksson, H.; Jensen, E.; et al. CPU inhibition with AZD9684: Profibrinolytic effects in acute pulmonary embolism patients [Abstract]. J. Thromb. Haemost. 2007, 5, P-S-367. [Google Scholar]
- Brink, M.; Dahlén, A.; Olsson, T.; Polla, M.; Svensson, T. Design and synthesis of conformationally restricted inhibitors of TAFIa. Bioorganic Med. Chem. 2014, 22, 2261–2268. [Google Scholar] [CrossRef]
- Suzuki, K.; Muto, Y.; Fushihara, K.; Kanemoto, K.; Iida, H.; Sato, E.; Kikuchi, C.; Matsushima, T.; Kato, E.; Nomoto, M.; et al. Enhancement of Fibrinolysis by EF6265, a Specific Inhibitor of Plasma Carboxypeptidase B. J. Pharmacol. Exp. Ther. 2004, 309, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Muto, Y.; Suzuki, K.; Iida, H.; Sakakibara, S.; Kato, E.; Itoh, F.; Kakui, N.; Ishii, H. EF6265, a novel inhibitor of activated thrombin-activatable fibrinolysis inhibitor, protects against sepsis-induced organ dysfunction in rats. Crit. Care Med. 2009, 37, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Bunnage, M.E.; Blagg, J.; Steele, J.; Owen, D.R.; Allerton, C.; McElroy, A.B.; Miller, D.; Ringer, T.; Butcher, K.; Beaumont, K.; et al. Discovery of potent & selective inhibitors of activated thrombin-activatable fibrinolysis inhibitor for the treatment of thrombosis. J. Med. Chem. 2007, 50, 6095–6103. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.R.; Bull, D.J.; Bunnage, M.E.; Glossop, M.S.; Maguire, R.J.; Strang, R.S. Oxygenated analogues of UK-396082 as inhibitors of activated thrombin activatable fibrinolysis inhibitor. Bioorg. Med. Chem. Lett. 2010, 20, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Barrow, J.C.; Nantermet, P.G.; Stauffer, S.R.; Ngo, P.L.; Steinbeiser, M.A.; Mao, S.; Carroll, S.S.; Bailey, C.; Colussi, D.; Bosserman, M.; et al. Synthesis and evaluation of imidazole acetic acid inhibitors of activated thrombin-activatable fibrinolysis inhibitor as novel antithrombotics. J. Med. Chem. 2003, 46, 5294–5297. [Google Scholar] [CrossRef] [PubMed]
- Nantermet, P.G.; Barrow, J.C.; Lindsley, S.R.; Young, M.; Mao, S.S.; Carroll, S.; Bailey, C.; Bosserman, M.; Colussi, D.; McMasters, D.R.; et al. Imidazole acetic acid TAFIa inhibitors: SAR studies centered around the basic P’1 group. Bioorganic Med. Chem. Lett. 2004, 14, 2141–2145. [Google Scholar] [CrossRef]
- Islam, I.; Bryant, J.; May, K.; Mohan, R.; Yuan, S.; Kent, L.; Morser, J.; Zhao, L.; Vergona, R.; White, K.; et al. 3-Mercaptopropionic acids as efficacious inhibitors of activated thrombin activatable fibrinolysis inhibitor (TAFIa). Bioorganic Med. Chem. Lett. 2007, 17, 1349–1354. [Google Scholar] [CrossRef]
- Wang, Y.-X.; da Cunha, V.; Vincelette, J.; Zhoa, L.; Nagashima, M.; Kawai, K.; Yuan, S.; Emayan, K.; Islam, I.; Hosoya, J.; et al. A novel inhibitor of activated thrombin activatable fibrinolysis inhibitor (TAFIa)—PartII: Enhancement of both exogenous and endogenous fibrinolysis in animal models of thrombosis. Thromb. Haemost. 2007, 97, 54–61. [Google Scholar]
- Wang, Y.; Zhao, L.; Nagashima, M.; Vincelette, J.; Sukovich, D.; Li, W.; Subramanyam, B.; Yuan, S.; Emayan, K.; Islam, I.; et al. A novel inhibitor of activated thrombin-activatable fibrinolysis inhibitor (TAFIa)—Part I: Pharmacological characterization. Thromb. Haemost. 2007, 97, 45–53. [Google Scholar]
- Pogačić Kramp, V. List of drugs in development for neurodegenerative diseases: Update October 2011. Neurodegener. Dis. 2012, 9, 210–283. [Google Scholar] [CrossRef] [Green Version]
- Halland, N.; Brönstrup, M.; Czech, J.; Czechtizky, W.; Evers, A.; Follmann, M.; Kohlmann, M.; Schiell, M.; Kurz, M.; Schreuder, H.A.; et al. Novel Small Molecule Inhibitors of Activated Thrombin Activatable Fibrinolysis Inhibitor (TAFIa) from Natural Product Anabaenopeptin. J. Med. Chem. 2015, 58, 4839–4844. [Google Scholar] [CrossRef]
- Halland, N.; Czech, J.; Czechtizky, W.; Evers, A.; Follmann, M.; Kohlmann, M.; Schreuder, H.A.; Kallus, C. Sulfamide as Zinc Binding Motif in Small Molecule Inhibitors of Activated Thrombin Activatable Fibrinolysis Inhibitor (TAFIa). J. Med. Chem. 2016, 59, 9567–9573. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, H.; Liesum, A.; Lönze, P.; Stump, H.; Hoffmann, H.; Schiell, M.; Kurz, M.; Toti, L.; Bauer, A.; Kallus, C.; et al. Isolation, Co-Crystallization and Structure-Based Characterization of Anabaenopeptins as Highly Potent Inhibitors of Activated Thrombin Activatable Fibrinolysis Inhibitor (TAFIa). Sci. Rep. 2016, 6, 32958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, A.; Chauveau, F.; Cho, T.-H.; Kallus, C.; Wagner, M.; Boutitie, F.; Maucort-Boulch, D.; Berthezène, Y.; Wiart, M.; Nighoghossian, N. Effects of a TAFI-Inhibitor Combined with a Suboptimal Dose of rtPA in a Murine Thromboembolic Model of Stroke. Cerebrovasc. Dis. 2014, 38, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Sansilvestri-Morel, P.; Bertin, F.; Lapret, I.; Neau, B.; Blanc-Guillemaud, V.; Petit-Dop, F.; Tupinon-Mathieu, I.; Delerive, P. S62798, a TAFIa inhibitor, accelerates endogenous thrombolysis in a murine model of pulmonary thromboembolism. Eur. Heart J. 2020, 41. [Google Scholar] [CrossRef]
- Noguchi, K.; Edo, N.; Miyoshi, N.; Isobe, A.; Watanabe, A.; Ito, Y.; Morishima, Y.; Yamaguchi, K. Fibrinolytic potential of DS-1040, a novel orally available inhibitor of activated thrombin-activatable fibrinolysis inhibitor (TAFIa). Thromb. Res. 2018, 168, 96–101. [Google Scholar] [CrossRef]
- Limsakun, T.; Dishy, V.; Mendell, J.; Pizzagalli, F.; Pav, J.; Kochan, J.; Vandell, A.G.; Rambaran, C.; Kobayashi, F.; Orihashi, Y.; et al. Safety and Pharmacokinetics of DS-1040 Drug-Drug Interactions With Aspirin, Clopidogrel, and Enoxaparin. J. Clin. Pharmacol. 2020, 60, 691–701. [Google Scholar] [CrossRef]
- Zhang, X.-S.; Xiang, B.-R. Discontinued drugs in 2007: Cardiovascular drugs. Expert Opin. Investig. Drugs 2008, 17, 1817–1828. [Google Scholar] [CrossRef]
- Davidsson, P.; Nerme, V.K. WO2009082340—Method for Monitoring the Progression of Fibinolysis. International Patent Application No. PCT/SE2008/051487, 2 July 2009. [Google Scholar]
- Mertens, J.C.; Boisseau, W.; Leenaerts, D.; Di Meglio, L.; Loyau, S.; Lambeir, A.; Ducroux, C.; Jandrot-Perrus, M.; Michel, J.; Mazighi, M.; et al. Selective inhibition of carboxypeptidase U may reduce microvascular thrombosis in rat experimental stroke. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Foley, J.H.; Kim, P.Y.; Mutch, N.J.; Gils, A. Insights into thrombin activatable fibrinolysis inhibitor function and regulation. J. Thromb. Haemost. 2013, 11, 306–315. [Google Scholar] [CrossRef]
- Atkinson, J.M.; Pullen, N.; Da Silva-Lodge, M.; Williams, L.; Johnson, T.S. Inhibition of thrombin-activated fibrinolysis inhibitor increases survival in experimental kidney fibrosis. J. Am. Soc. Nephrol. 2015, 26, 1925–1937. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, J.M.; Pullen, N.; Johnson, T.S. An inhibitor of thrombin activated fibrinolysis inhibitor (TAFI) can reduce extracellular matrix accumulation in an in vitro model of glucose induced ECM expansion. Matrix Biol. 2013, 32, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Björquist, P.; Buchanan, M.; Campitelli, M.; Caroll, A.; Hyde, E.; Neve, J.; Polla, M.; Quinn, R. WO2005039617—Use of Cyclic Anabaenopeptin-Type Peptides for the Treatment of a Condition Wherein Inhibition of Carboxypeptidase U is Beneficial, Novel Anabaenopeptin Derivatives and Intermediates Thereof. International Patent Application No. PCT/SE2004/001568, 6 May 2005. [Google Scholar]
- ClinicalTrials.gov. NCT02923115: Study to Assess the Safety, Pharmacokinetics/Dynamics of DS-1040b in Subjects With Acute Submassive Pulmonary Embolism; National Library of Medicine (US); U.S. National Library of Medicine: Bethesda, MD, USA, 2020.
- ClinicalTrials.gov. NCT02586233: Study to Assess the Safety, Pharmacokinetics and Pharmacodynamics of DS-1040b in Subjects With Acute Ischemic Stroke; U.S. National Library of Medicine: Bethesda, MD, USA, 2020. [Google Scholar]
- ClinicalTrials.gov. NCT03198715: Safety of DS-1040b in Acute Ischemic Stroke Patients Treated With Thrombectomy; U.S. National Library of Medicine: Bethesda, MD, USA, 2020.
- Daiichi Sankyo Daiichi Sankyo Group. Clinical Trial Protocols and Study Results; Daiichi Sankyo: Tokyo, Japan, 2020. [Google Scholar]
- Sasaki, T.; Yoshimoto, N.; Sugimoto, K.; Takada, K.; Murayama, N.; Yamazaki, H.; Yamamoto, K.; Ishii, H. Intravenous and oral administrations of DD2 [7-Amino-2-(sulfanylmethyl) heptanoic acid] produce thrombolysis through inhibition of plasma TAFIa in rats with tissue factor-induced microthrombosis. Thromb. Res. 2012, 130, e222–e228. [Google Scholar] [CrossRef] [PubMed]
- Gils, A.; Ceresa, E.; Macovei, A.M.; Marx, P.F.; Peeters, M.; Compernolle, G.; Declerck, P.J. Modulation of TAFI function through different pathways—Implications for the development of TAFI inhibitors. J. Thromb. Haemost. 2005, 3, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Vercauteren, E.; Develter, J.; Bammens, R.; Declerck, P.J.; Gils, A. Identification and characterisation of monoclonal antibodies that impair the activation of human thrombin activatable fibrinolysis inhibitor through different mechanisms. Thromb. Haemost. 2011, 106, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, M.L.V.; De Winter, A.; Buelens, K.; Compernolle, G.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S.; Gils, A.; Declerck, P.J. TAFIa inhibiting nanobodies as profibrinolytic tools and discovery of a new TAFIa conformation. J. Thromb. Haemost. 2011, 9, 2268–2277. [Google Scholar] [CrossRef] [PubMed]
- Buelens, K.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S.; Gils, A.; Declerck, P.J. Generation and characterization of inhibitory nanobodies towards thrombin activatable fibrinolysis inhibitor. J. Thromb. Haemost. 2010, 8, 1302–1312. [Google Scholar] [CrossRef]
- Hendrickx, M.L.V.; Zatloukalova, M.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S.; Gils, A.; Declerck, P.J. Identification of a novel, nanobody-induced, mechanism of TAFI inactivation and its in vivo application. J. Thromb. Haemost. 2014, 12, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, E.; Emmerechts, J.; Peeters, M.; Hoylaerts, M.F.; Declerck, P.J.; Gils, A. Evaluation of the profibrinolytic properties of an anti-TAFI monoclonal antibody in a mouse thromboembolism model. Blood 2011, 117, 4615–4622. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, M.L.V.; Zatloukalova, M.; Hassanzadeh-Ghassabeh, G.; Muyldermans, S.; Gils, A.; Declerck, P.J. In vitro and in vivo characterisation of the profibrinolytic effect of an inhibitory anti-rat TAFI nanobody. Thromb. Haemost. 2014, 111, 824–832. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Gils, A.; Declerck, P.J.; Colucci, M. Monoclonal antibodies targeting the antifibrinolytic activity of activated thrombin-activatable fibrinolysis inhibitor but not the anti-inflammatory activity on osteopontin and C5a. J. Thromb. Haemost. 2013, 11, 2137–2147. [Google Scholar] [CrossRef]
- Denorme, F.; Wyseure, T.; Peeters, M.; Vandeputte, N.; Gils, A.; Deckmyn, H.; Vanhoorelbeke, K.; Declerck, P.J.; De Meyer, S.F. Inhibition of Thrombin-Activatable Fibrinolysis Inhibitor and Plasminogen Activator Inhibitor-1 Reduces Ischemic Brain Damage in Mice. Stroke 2016, 47, 2419–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyseure, T.; Rubio, M.; Denorme, F.; De Lizarrondo, S.M.; Peeters, M.; Gils, A.; De Meyer, S.F.; Vivien, D.; Declerck, P.J. Innovative thrombolytic strategy using a heterodimer diabody against TAFI and PAI-1 in mouse models of thrombosis and stroke. J. Am. Soc. Hematol. 1325, 125, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declerck, P.J.; De Meyer, S.F.; Geukens, N.; Gils, A.; Rubio, M.; Vivien, D.; Wyseure, T.L. WO2015118147—Dual Targeting of TAF and PAI-1. Patent Application No. PCT/EP2015/052624, 13 August 2015. [Google Scholar]
- Schneider, M.; Nesheim, M. Reversible inhibitors of TAFIa can both promote and inhibit fibrinolysis. J. Thromb. Haemost. 2003, 1, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.B.; Hughes, B.; James, I.; Haddock, P.; Kluft, C.; Bajzar, L. Stabilization versus inhibition of TAFIa by competitive inhibitors in vitro. J. Biol. Chem. 2003, 278, 8913–8921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vercauteren, E.; Gils, A. Is there any need for a TAFI(a) inhibitor as thrombolytic drug? Thromb. Res. 2012, 130, 574–575. [Google Scholar] [CrossRef] [PubMed]
- Willemse, J.L.; Hendriks, D.F. A role for carboxypeptidase U (TAFI) in thrombosis. Front. Biosci. 2007, 12, 1973–1987. [Google Scholar] [CrossRef]
- Bird, E.; Tamura, J.; Bostwick, J.S.; Steinbacher, T.E.; Stewart, A.; Liu, Y.; Baumann, J.; Feyen, J.; Tamasi, J.; Schumacher, W.A. Is exogenous tissue plasminogen activator necessary for antithrombotic efficacy of an inhibitor of thrombin activatable fibrinolysis inhibitor (TAFI) in rats? Thromb. Res. 2007, 120, 549–558. [Google Scholar] [CrossRef]
- Kraft, P.; Schwarz, T.; Meijers, J.C.M.; Stoll, G.; Kleinschnitz, C. Thrombin-Activatable Fibrinolysis Inhibitor ( TAFI ) Deficient Mice Are Susceptible to Intracerebral Thrombosis and Ischemic Stroke. PLoS ONE 2010, 5, 10–15. [Google Scholar] [CrossRef]
- Orbe, J.; Alexandru, N.; Roncal, C.; Belzunce, M.; Bibiot, P.; Rodriguez, J.A.; Meijers, J.C.M.; Georgescu, A.; Paramo, J.A. Lack of TAFI increases brain damage and microparticle generation after thrombolytic therapy in ischemic stroke. Thromb. Res. 2015, 136, 445–450. [Google Scholar] [CrossRef]
- Rehill, A.M.; Preston, R.J.S. A new thrombomodulin-related coagulopathy. J. Thromb. Haemost. 2020, 18, 2123–2125. [Google Scholar] [CrossRef]
- Westbury, S.K.; Whyte, C.S.; Stephens, J.; Downes, K.; Turro, E.; Claesen, K.; Mertens, J.C.; Hendriks, D.; Latif, A.L.; Leishman, E.J.; et al. A new pedigree with thrombomodulin-associated coagulopathy in which delayed fibrinolysis is partially attenuated by co-inherited TAFI deficiency. J. Thromb. Haemost. 2020, 18, 2209–2214. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Smith, P.L.; Hsu, M.Y.; Tamasi, J.A.; Bird, E.; Schumacher, W.A. Deficiency in thrombin-activatable fibrinolysis inhibitor (TAFI) protected mice from ferric chloride-induced vena cava thrombosis. J. Thromb. Thrombolysis 2007, 23, 41–49. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Assay Type | Interference or Influence of | Additional Information | Used in | |||||
|---|---|---|---|---|---|---|---|---|
| Thr/Ile325 | CPN | ProCPU | CPU | CPUi | ||||
| ProCPU measurement | ||||||||
| Activity * | Wang et al. 1994 [26] | No | Yes | - | Yes | No | Hip-Arg with detection by RP-HPLC | |
| Mosnier et al. 1998 [11] | No | Yes | - | Yes | No | Hip-Arg with colorimetric detection | [27,28,29] | |
| Schatteman et al. 1999 [30] | No | Yes | - | Yes | No | Hip-Arg with detection by RP-HPLC | [31,32,33] | |
| Schatteman et al. 2001 [34] | No | Yes | - | Yes | No | p-OH-Hip-Arg with colorimetric detection | [35] | |
| Heylen et al. 2010 [9] | No | No | - | Yes | No | Bz-o-cyano-Phe-Arg with detection by RP-HPLC; Not very sensitive to influence of hemolysis | [36] | |
| STA-Stachrom® TAFI (Diagnostica Stago) | No | No | - | Yes | No | Azoformyl–AA2—AA1 (Patent: SERBIO PCT/FR 02/02376) with colorimetric detection | [37] | |
| Actichrome® TAFI (American Diagnostica) | No | No | - | Yes | No | Chromogenic substrate with colorimetric detection; No longer marketed | [38,39,40,41,42,43,44] | |
| Pefakit® TAFI (Pentapharm) | No | No | - | Yes | No | Synthetic substrate with colorimetric detection | [45,46,47,48,49,50,51,52,53] | |
| ELISA | Mosnier et al. 1998 [11] | Unk. | No | - | Unk. | Unk. | Murine monoclonal capture and rabbit polyclonal detection antibody | [54,55] |
| van Tilburg et al. 2000 [56] | No | No | - | Yes | Yes | Electroimmunoassay | [56,57,58,59,60] | |
| Strömqvist et al. 2001 [13] | Unk. | No | - | No | No | No reaction with plasma of other species (guinea pig, rat, dog, pig, hamster) | ||
| Ceresa et al. 2006 [12] | No ** | No | - | No | No | Monoclonal capture and detection antibody | [58,59,61,62,63] | |
| VisuLize® TAFI (Affinity Biologicals) | Yes | No | - | Yes | Yes | Sheep polyclonal capture and detection antibody; Marketed by Milan Analytica | [40,41,42,43,64,65,66,67,68,69,70] | |
| Imuclone® TAFI (American Diagnostica) | Yes | No | - | No | No | [71,72,73,74,75] | ||
| Asserachrom® TAFI-1B1 (Diagnostica Stago) | No | No | - | No | No | Previously marketed by Kordia Laboratory Supplies; No longer marketed | [51,76,77,78,79,80,81,82,83,84] | |
| Zymutest® (Total) TAFI (Hyphen BioMed) | No | No | - | No | No | Previously marketed as Zymutest® proTAFI | [38,85,86,87,88] | |
| Coalize® TAFI (Chromogenix) | Unk. | No | - | Unk. | Unk. | Monoclonal capture and polyclonal detection antibody | [39,89] | |
| Activation peptide | ||||||||
| ELISA | Ceresa et al. 2006 [12] | No *** | No | No | Yes | No | Monoclonal capture and detection antibody; Measures both AP and CPU | [58,59,61,63] |
| Active CPU | ||||||||
| Activity | Hendriks et al. 1989 [90] | No | Yes | No | - | No | p-OH-Hip-Arg with colorimetric detection and Hip-Arg with detection by RP-HPLC | |
| Kim et al. 2008 [91] | No | No | No | - | No | Plasmin-modified fibrin is covalently bound to a quencher molecule and mixed with fluorescein-labeled plasminogen and the plasma sample. The rate of fluorescence increase detected by a plate reader reflects the amount of CPU present in the sample. High sensitive (LOD: 12 pM); Not affected by other hemostatic factors | ||
| Heylen et al. 2010 [92] | No | No | No | - | No | Bz-o-cyano-Phe-Arg with detection by RP-HPLC; High sensitive (LOD: 18 pM); Highly sensitive to influence of hemolysis | [93,94,95] | |
| ELISA | Zymutest® (Activatable) TAFI (Hyphen BioMed) | Yes | Previously marketed as Zymutest® TAFI | [53,96,97] | ||||
| Active & inactive CPU **** | ||||||||
| ELISA | Asserachrom TAFIa/ai (Diagnostica Stago) | No | No | No | - | - | Combined measurement of CPU and CPUi; Not sensitive to influence of hemolysis | [37,63] |
| Imubind® TAFIa/ai (Biomedica Diagnostics) | No | No | No | - | - | No longer marketed | [98] | |
| Venous Thrombosis | ||
| Arauz et al. 2018 [119] | No risk association in CVT cases (N = 113) relative to controls (N = 131) for the +1040C/T polymorphism or in haplotype analysis of the CPB2 gene. | PCR |
| Orikaza et al. 2014 [120] | The +1040C/T polymorphism significantly increased the risk of CVT (N = 72) compared to VTE cases (N = 128) and controls (N = 134). | PCR |
| Tokgoz et al. 2013 [121] | In 59 patients with CVT and 100 healthy control subjects, the association between the +1040C/T polymorphism and CVT was investigated. Frequencies of polymorphic genotype and allele were similar in patients and controls and were not significant for CVT. | PCR |
| Meltzer et al. 2010 [45] | In a study involving 770 patients from the MEGA study (first DVT of the leg or first PE) and 743 controls, high proCPU levels were shown to be an independent risk factor for VTE. | Activity assay (Pentapharm) |
| Meltzer et al. 2010 [114] | The +1040C/T polymorphism was associated with both proCPU levels and increased risk for recurrent VTE in 474 patients diagnosed with first DVT. | PCR + In-house developed activity assay [56] |
| Verdu et al. 2008 [76] | It was found that the Thr/Thr genotype of the Thr/Ile325 polymorphism was associated with an increased risk of VTE. | PCR + ELISA (Diagnostica Stago) |
| Folkeringa et al. 2008 [47] | In a large cohort of thrombophilic families, no correlation was observed between high proCPU levels and the risk of venous or arterial thromboembolism. | Activity assay (Pentapharm) |
| Verdu et al. 2006 [122] | High proCPU levels (>90th percentile of the controls) increased the risk for future DVT 4-fold compared to patients with lower proCPU levels. 60 patients with previous DVT or PE and 62 controls were included in the study. | ELISA (Diagnostica Stago) |
| Martini et al. 2006 [60] | Thr/Ile325 polymorphism is associated with proCPU antigen levels in 471 patients with first DVT, but there was no association with increased risk for DVT. | PCR + In-house developed activity assay [56] |
| Zee et al. 2005 [123] | No evidence was provided for an association between six polymorphisms, including the +1040C/T polymorphism, in the CPB2 gene and the risk for VTE. | PCR |
| Eichinger et al. 2004 [77] | Higher proCPU levels in patients with a previous first spontaneous VTE were associated with an almost 2-fold higher risk for VTE recurrence compared to patients with lower proCPU levels (N = 600 total study population). | ELISA (American Diagnostica) |
| Libourel et al. 2002 [115] | Symptomatic Factor V Leiden carriers (N = 17) had higher proCPU levels compared to asymptomatic carriers (N = 136). High levels of proCPU are a mild risk factor for VTE. | In-house developed activity assay [30] |
| Franco et al. 2001 [29] | A tendency towards protection against DVT was observed for a several CPB2 gene polymorphisms, that paralleled the lower proCPU levels detected in carriers of these polymorphisms (N = 388) compared to controls (N = 388). | PCR + In-house developed ELISA [11] |
| Van Tilburg et al. 2000 [56] | ProCPU levels were similar in patients with a first episode of DVT (N = 474) compared to controls (N = 474). Although, there were more DVT patients than controls with high proCPU levels and high proCPU levels were associated with an increased risk for thrombosis. | In-house developed activity assay [56] |
| Arterial thrombosis and coronary artery disease (CAD) | ||
| Isordia-Salas et al. 2019 [124] | Thr/Ile325 polymorphism in the CPB2 gene was associated with an increased risk for STEMI (N = 244), but not for IIS (N = 250). Genotype and allele distribution were similar in IIS patients and controls (N = 244). | PCR |
| Rattanawan et al. 2018 [125] | The +1040C/T polymorphism was not associated with the severity of coronary artery stenosis. | PCR |
| Khalifa et al. 2012 [98] | ProCPU levels were not correlated with the pre-disposition of in-stent restenosis following coronary stenting in 37 patients with CAD. | CPU + CPUi ELISA (American Diagnostica) |
| Jood et al. 2012 [61] | No association between intact proCPU antigen and survival rate, nor reoccurrence of vascular events (recurrent stroke, transient ischemic attack or coronary event; N = 37) in ischemic stroke survivors (N = 517). Blood was collected 3 months after the index event. Two years after inclusion the survival rates and vascular events were assessed. | In-house developed ELISA [12] |
| De Bruijne et al. 2011 [59] | Increased levels of intact proCPU antigen are associated with an increased risk of premature peripheral arterial disease. Functional proCPU was not significantly higher in patients (N = 47) compared to controls (N = 141). Blood samples were collected 1–3 months after the event. | In-house developed ELISA [12] + In-house developed activity assay [126] |
| Kamal et al. 2011 [127] | Homozygous and heterozygous carriers of the Ile325 were more frequent in patients with AMI (N = 46) than in controls (N = 54). | PCR |
| Kozian et al. 2010 [128] | Homozygosity for the Ile325 allele of the Thr/Ile325 polymorphism was associated with the incidence of stroke and the age at onset of first stroke (N = 3300). | PCR |
| Tassies et al. 2009 [78] | ProCPU polymorphism Thr/Ile325 is related to the type of acute coronary syndrome (N = 248; total cohort). Homozygous 325Ile genotypes are less prevalent in patients with STEMI compared to NSTEMI patients. | ELISA (Diagnostica Stago) + Activity assay (American Diagnostica) |
| De Bruijne et al. 2009 [58] | In young patients with arterial thrombosis (N = 327), the distribution of the Thr/Ile325 SNP was compared to healthy controls (N = 332). In homozygous carriers of the Ile325 allele lower proCPU levels were observed together with a decreased risk of arterial thrombosis compared to homozygous carriers of the Thr325 allele. In the same population total proCPU levels and proCPU activity levels did not differ from controls. Blood samples were collected 1–3 months after the event. | In-house developed ELISA [12] + In-house developed activity assay [126] |
| Tregouet et al. 2009 [79] | In a prospective study on patients with angiographically proven coronary artery disease none of the selected CPB2 gene polymorphisms, including Thr/Ile325, was associated with the occurrence of cardiovascular events (N = 1668). In the same study, the total proCPU antigen was associated with an increased risk of future cardiovascular death. | PCR + ELISA (Diagnostica Stago) |
| Meltzer et al. 2009 [46] | The +1040C/T SNP was not associated with myocardial infarction in men (N = 554) versus controls (N = 643). Also, patients with proCPU levels in the first quartile (lowest levels) display an increased risk of a first myocardial infarction compared to patients with proCPU levels in the fourth quartile. The time between blood sampling after the infarct ranged from 88 days to 5.8 years with a median of 2.6 years. | PCR + Activity assay (Pentapharm) |
| Biswas et al. 2008 [80] | No association was detected between 16 single nucleotide polymorphisms (of which the Thr/Ile325 polymorphism was one) and the risk of cardio-embolic stroke (N = 120). Also, significantly higher proCPU antigen levels were observed in patients with acute onset non-cardioembolic stroke (N = 120) compared to normal individuals (N = 120). Blood samples were collected within 10 days of the stroke and at 3-month follow-up. | ELISA (Diagnostica Stago) |
| Ladenvall et al. 2007 [62] | No association was detected between 11 single nucleotide polymorphisms (including Thr/Ile325 SNP) and overall ischemic stroke risk (N = 600). In addition, increased levels of intact proCPU are found in ischemic stroke patients compared to controls (N = 600). An independent association was found with large vessel disease, cryptogenic stroke and acute-phase small vessel disease subtypes. Increased proCPU levels do not reflect an acute phase response. Blood sampling was conducted within 10 days of the stroke and at 3-months follow-up. | PCR + In-house developed ELISA [12] |
| Fernandez-Cadenas et al. 2007 [129] | Ile/Ile homozygosity for the Thr/Ile325 polymorphism was associated with lower rates of recanalization after rtPA infusion in ischemic stroke patients (N = 139). | PCR |
| Cruden et al. 2006 [38] | Plasma proCPU does not predict reperfusion in patients receiving thrombolytic therapy for acute STEMI (N = 110). Blood was collected prior to administration of thrombolytic therapy. | ELISA (Hyphen Biomed) + Activity assay (American Diagnostica) |
| Schroeder et al. 2006 [48] | Re-evaluation of the study in 2002 which was compromised by genotype-dependent artifacts. Significant associations between proCPU activity and cardiovascular risk factors as well as with coronary artery disease were found. ProCPU activity was higher in coronary artery disease patients (N = 338) than in controls (N = 158). Blood samples were collected during angiography. | Activity assay (Pentapharm) |
| Morange et al. 2005 [81] | No clear relation between coronary heart disease and six proCPU gene polymorphism was found (including Thr/Ile325). A total of 248 cases and 493 controls were used. And no significant association between proCPU levels and angina pectoris or hard coronary events was present after re-analysis of the PRIME data with an ELISA which was shown to be insensitive to proCPU genotype. | PCR + ELISA (Diagnostica Stago) |
| Leebeek et al. 2005 [130] | The proCPU genotype does not seem to predict the risk of ischemic stroke. No difference was found between patients (N = 124) and controls (N = 125) with respect to the distribution of the +1040C/T polymorphism. Also, increased proCPU levels, resulting in decreased fibrinolysis, are associated with an increased risk of first ischemic stroke (ischemic stroke N = 124 vs. controls N = 125). As demonstrated by the persisting elevated proCPU levels three months after the stroke, the increase in functional proCPU levels is not caused by an acute phase reaction. Blood collection between 7 and 14 days after the stroke, second blood collection in a subgroup (N = 36) three months after the stroke. | PCR + In-house developed functional TAFI clot lysis assay [126] |
| Lisowski et al. 2005 [40] | ProCPU levels were significantly higher 7 days after elective CABG in 45 stabile angina pectoris patients with confirmed CAD compared to controls (N = 33). | ELISA (Affinity Biologicals) + Activity assay (American Diagnostica) |
| Kim et al. 2005 [86] | No difference was seen in proCPU levels in acute ischemic stroke patients with (N = 30) or without successful recanalization (N = 13). Blood samples were collected on admission. | ELISA (Hyphen Biomed) |
| Santamaria et al. 2004 [131] | ProCPU levels tended to be higher in patients with acute CAD (N = 174) than in controls (N = 211). Blood samples were collected at least six months after the acute episode. | In-house developed activity assay [11] |
| Segev et al. 2004 [72] | It was shown that the proCPU antigen level is strongly determined by the Thr/Ile325 polymorphism in patients with stable angina pectoris (N = 159). The T/T genotype was the least prevalent and associated with the lowest proCPU levels and the lowest rate of angiographic restenosis in this population. | PCR + ELISA (American Diagnostica) |
| Akatsu et al. 2004 [132] | In 253 patients with confirmed neuropathology that died during hospitalization, no statistical correlation between the Thr/Ile325 polymorphisms and risk for cerebral infarction was found. | PCR |
| Morange et al. 2003 [85] Re-evaluated in 2005 | In France, proCPU levels were significantly higher in men who subsequently developed angina pectoris (N = 81) than in their controls (N = 81), whereas no difference was observed between cases (N = 62) and controls (N = 124) in Northern Ireland. | PCR + ELISA (Hyphen Biomed) |
| Brouwers et al. 2003 [65] | No significant association between the Thr/Ile325 polymorphism was found in non-refractory patients (N = 133) compared to refractory patients (N = 76) with unstable angina pectoris. Higher proCPU levels in patients with non-refractory unstable angina pectoris (N = 133) than in refractory patients (N = 76). Blood samples were obtained on admission. | PCR + ELISA (Affinity Biologicals) |
| Zorio et al. 2003 [39] | No difference according to the Thr/Ile325 polymorphism between young patients with myocardial infarction (N = 127) and controls (N = 99). Patients had higher plasma proCPU activity levels, but lower proCPU antigen in comparison to controls. Blood sample was collected at least 3 months after the myocardial infarction. | PCR + ELISA (Chromogenix) + Activity assay (American Diagnostica) |
| Lau et al. 2003 [71] | Higher preprocedural proCPU plasma levels in patients with restenosis. ProCPU plasma levels correlated with 6-month % diameter stenosis after percutaneous coronary intervention (N = 159). | ELISA (American Diagnostica) |
| Juhan-Vague et al. 2003 [67] | No difference in proCPU plasma concentration in men who subsequently suffered from myocardial infarction or coronary death (N = 159) when compared with their controls (N = 317). Prospective study. | ELISA (Milan Analytica) |
| Santamaria et al. 2003 [28] | Higher proCPU plasma levels in ischemic stroke patients (N = 114) than in healthy controls (N = 150). Blood samples collected at least one month after the acute thrombotic episode | In-house developed activity assay [11] |
| Juhan-Vague et al. 2002 [68] | Patients who suffered from AMI (N = 598) showed lower plasma proCPU antigen values versus controls (N = 653). Blood samples were collected 3–5 months after myocardial infarction. | PCR + ELISA (Milan Analytica) |
| Morange et al. 2002 [133] | The Thr/Ile325 polymorphism does not influence the risk of MI in white male patients younger than 60 years who survived a first MI (N = 533) and male controls of the same age (N = 575). | PCR |
| Schroeder et al. 2002 [66] Re-evaluated in 2006 | Higher proCPU antigen levels in coronary artery disease patients (N = 362) compared to controls with angiographically verified normal coronary vessels (N = 134). The difference was more prominent in intracoronary than in venous blood samples. Blood samples were collected during angiography. | ELISA (Milan Analytica) |
| Silveira et al. 2000 [31] | Higher proCPU plasma concentration in men requiring coronary artery bypass grafting because of stable angina pectoris (N = 110) than in controls (N = 56). Blood samples were collected preoperative. | In-house developed activity assay [30] |
| Arterial Thrombosis and Coronary Artery Disease (CAD) | ||
|---|---|---|
| Alessi et al. 2016 [37] | CPU/CPUi levels were monitored in 109 patients with ischemic stroke, with 41 receiving rtPA. Blood samples were collected post-admission/post-thrombolysis up to day 90 at 8 different time points. AIS patients had higher levels of CPU/CPUi at admission in comparison with the control population. In thrombolysed patients, an increase in CPU/CPUi was observed at the end of thrombolytic therapy, which lasted up to 4 h. In the non-thrombolysed group, CPU/CPUi levels did not differ over time. Both in thrombolysed as in non-thrombolysed patients, higher CPU/CPUi levels were associated with a more severe stroke and unfavorable outcome. No details available on sample collection and the use of inhibitors to prevent ex vivo proCPU activation. | CPU + CPUi ELISA (Diagnostica Stago) |
| Leenaerts et al. 2015 [93] | During the acute phase of myocardial infarction, CPU activity levels are higher in patients with AMI (N = 45) than in controls (N = 42). No association was found between CPU activity and AMI type (NSTEMI vs. STEMI). Intracoronary samples contained higher CPU levels than peripheral samples, indicating increased local CPU generation. Blood samples were collected at the start coronary catheterization. Blood collected in prechilled tubes containing sodium citrate, PPACK and aprotinin. Samples were immediately placed on ice after collection. | In-house developed activity assay [92] |
| Brouns et al. 2009 [33] | In patients with ischemic stroke (N = 12) receiving thrombolytic therapy, the amount of CPU generated is associated with evolution of the neurological deficit as well as with achieved recanalization. Blood samples were taken at 6–9 different time points before, during and after thrombolytic therapy. Blood collected in prechilled tubes containing sodium citrate, PPACK and aprotinin. Samples were immediately placed on ice after collection. | In-house developed activity assay [139] |
| Willemse et al. 2008 [99] | CPU activity is induced during therapeutic thrombolysis of patients with acute ischemic stroke (N = 8). Blood samples were collected at six to nine time points before, during and after thrombolysis. Blood collected in prechilled tubes containing sodium citrate, PPACK and aprotinin. Samples were immediately placed on ice after collection. | In-house developed activity assay [139] |
| Company | Patents | Chemical Structure | Lead Compound | Highest Development Status Reported | Reference |
|---|---|---|---|---|---|
| AstraZeneca | WO/2000/66550 WO/2000/066557 WO/2000/066152 WO/2003/106420 WO/2003/027128 | Phosphor and/or thiol-containing compounds | AZD9684 | Phase II completed; development halted | [36,147,148] |
| WO/2005/039617 | Cyclic anabaenopeptin-type peptides | Unknown | Discovery | ||
| Unknown | Conformationally restricted imidazole derivatives | Compounds 7 and 12 | Preclinical | [149] | |
| Meiji Seika Kaisha/ MediciNova * | WO/2001/19836 | Phosphonic and phosphinic acid derivatives | EF6265/MN-462 * | Preclinical | [150,151] |
| Pfizer | WO/2002/14285 WO/2003/061652 WO/2003/061653 | Imidazole propanoic acid derivatives | UK-396082 | Phase I completed, development halted | [152,153] |
| Merck | WO/2003/013526 | Imidazole acetic acid derivatives | Compound 10j/ Compound (-)-8 | Preclinical | [154,155] |
| Berlex Biosciences (Bayer) | WO/2003/080631 WO/2006/041808 | Aryl guanidinic and mercaptopropionic acid derivatives | BX 528 | Preclinical | [156,157,158] |
| Sanofi | WO/2005/105781 WO/2007/045339 WO/2010/130718 WO/2013/076178 WO/2013/076179 WO/2008/067909 | Imidazole, urea & sulfamide containing compounds | SAR104772 SAR126119 | Phase I completed Phase II initiated in 2013 | [159] |
| Sanofi | WO/2009/146802 WO/2014/198620 | Macrocyclic urea derivatives | Unknown | Preclinical | [160,161,162] |
| Unknown | FFC.HTZ4.059 | Preclinical | [163] | ||
| Servier | WO/2010/149888 | 2-mercapto-cyclopentane carboxylic acid compounds | Unknown | Discovery | |
| WO/2017/121969 | Phosphinane & azaphosphinane derivatives | S62798 | Phase I completed | [95,164] | |
| Taisho Pharmaceutical | WO/2011/034215 | Dihydroimidazo-quinoline compounds | Unknown | Discovery | |
| Daiichi Sankyo | WO/2011/115064 WO/2011/115065 WO/2013/039202 | Cycloalkyl-substituted imidazole, cyclopropanecarboxylic acid, and acrylic acid derivatives | DS-1040 | Phase Ib/II completed; development halted | [52,53,165,166] |
| WO/2016/204239 WO/2016/104678 | Combinations of a CPU inhibitor with other compounds | DS-1040 + rtPA DS-1040 + Edoxaban | |||
| WO/2017/170460 | CPU inhibitor application in inflammatory bowel disease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claesen, K.; Mertens, J.C.; Leenaerts, D.; Hendriks, D. Carboxypeptidase U (CPU, TAFIa, CPB2) in Thromboembolic Disease: What Do We Know Three Decades after Its Discovery? Int. J. Mol. Sci. 2021, 22, 883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020883
Claesen K, Mertens JC, Leenaerts D, Hendriks D. Carboxypeptidase U (CPU, TAFIa, CPB2) in Thromboembolic Disease: What Do We Know Three Decades after Its Discovery? International Journal of Molecular Sciences. 2021; 22(2):883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020883
Chicago/Turabian StyleClaesen, Karen, Joachim C. Mertens, Dorien Leenaerts, and Dirk Hendriks. 2021. "Carboxypeptidase U (CPU, TAFIa, CPB2) in Thromboembolic Disease: What Do We Know Three Decades after Its Discovery?" International Journal of Molecular Sciences 22, no. 2: 883. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020883