
Synthesis, In Silico Prediction and In Vitro Evaluation of Antimicrobial Activity, DFT Calculation and Theoretical Investigation of Novel Xanthines and Uracil Containing Imidazolone Derivatives


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Activity
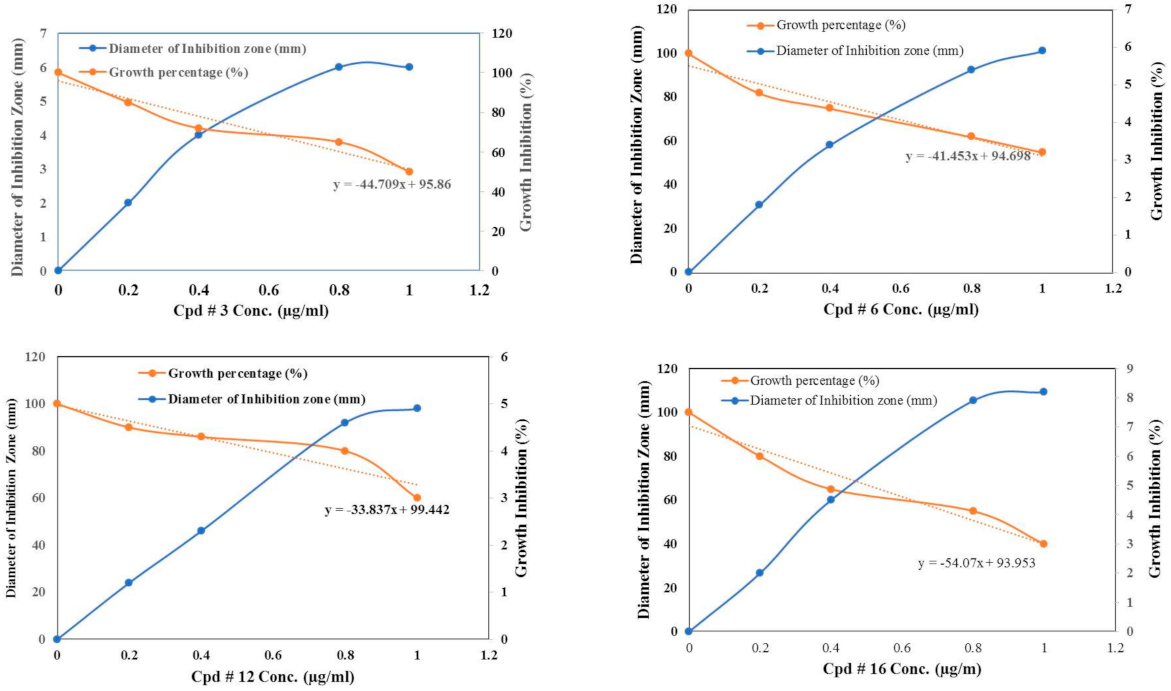
2.2.1. Antimicrobial Activity
2.2.2. TEM Analysis of Microorganisms in Response to the Tested Compounds
2.2.3. Cytotoxicity Analysis
Cytotoxicity Assay
2.3. Molecular Docking Study
2.4. Pharmacokinetics and ADME Activity
2.5. Computational Details
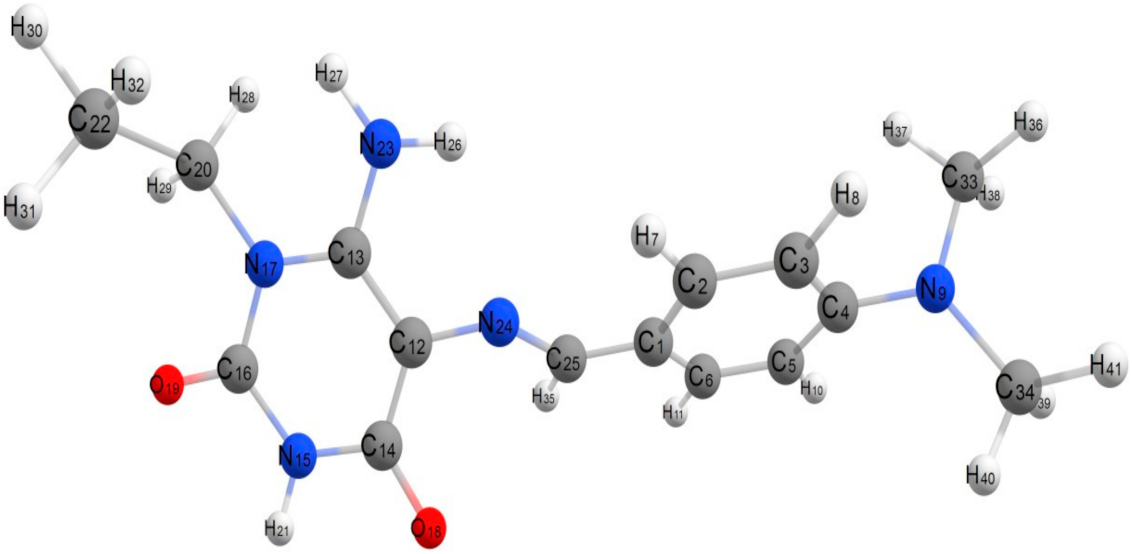
2.5.1. Structural Parameters and Models
6-Amino-5-(4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)-1-ethylpyrimidine-2,4(1H,3H)-dione, compound 12
6-Amino-5-(4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)-1-methyl-2-thioxo-2,3-dihydropyrimidin-4(1H)-one, compound 13
6-Amino-5-((4-(dimethylamino)benzylidene)amino)-1-ethylpyrimidine-2,4(1H,3H)-dione, compound (16)
2.5.2. Charge Distribution Analysis
2.5.3. Molecular Orbitals and Frontier
2.5.4. Excited State
3. Material and Methods
3.1. Chemistry
3.1.1. Materials and Instruments
3.1.2. Synthetic Procedures
3.1.3. N-Substituted-8-purinyl-2-(arylvinyl)acetamides 3-5
3.1.4. N-(1-(3-Benzyl-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)-2-(4-chlorophenyl)vinyl) acetamide (3)
3.1.5. N-(2-(4-Chlorophenyl)-1-(3-methyl-6-oxo-2-thioxo-2,3,6,7-tetrahydro-1H-purin-8-yl) vinyl) acetamide (4)
3.1.6. N-(1-(3-(2-Chlorobenzyl)-2,6-dioxo-2,3,6,7-tetrahydro-1H-purin-8-yl)-2-(4-chlorophenyl) vinyl)acetamide (5)
3.1.7. 1-Substituted-6-amino-5-(4-arylidene-2-methyl)imidazolylpyrimidinediones 6-13
3.1.8. 6-Amino-5-(4-(4-chlorobenzylidene)-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)-1,3-dimethylpyrimidine-2,4-(1H,3H)-dione (6)
3.1.9. 6-Amino-1-(2-chlorobenzyl)-5-(2-methyl-4-(3-nitrobenzylidene)-5-oxo-4,5-dihydro-1H-imidazol-1-yl)pyrimidine-2,4(1H,3H)-dione (7)
3.1.10. 6-Amino-1-benzyl-5-(2-methyl-4-(3-nitrobenzylidene)-5-oxo-4,5-dihydro-1H-imidazol-1-yl)pyrimidine-2,4(1H,3H)-dione (8)
3.1.11. 6-Amino-1-ethyl-5-(2-methyl-4-(3-nitrobenzylidene)-5-oxo-4,5-dihydro-1H-imidazol-1-yl) pyrimidine-2,4(1H,3H)-dione (9)
3.1.12. 6-Amino-1-benzyl-5-(4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)pyrimidine-2,4(1H,3H)-dione (10)
3.1.13. 6-Amino-5-(4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)-1-(2-chlorobenzyl)pyrimidine-2,4(1H,3H)-dione (11)
3.1.14. 6-Amino-5-(4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)-1-ethylpyrimidine-2,4(1H,3H)-dione (12)
3.1.15. 6-Amino-5-(4-benzylidene-2-methyl-5-oxo-4,5-dihydro-1H-imidazol-1-yl)-1-methyl-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (13)
3.1.16. 1-Substituted-6-amino-5-((4-(dimethylamino)benzylidene)aminopyrimidindiones 14-16
3.1.17. 6-Amino-5-((4-(dimethylamino)benzylidene)amino)-1-methyl-2-thioxo-2,3-dihydropyrimidin-4(1H)-one [44] (14)
3.1.18. 6-Amino-1-benzyl-5-((4-(dimethylamino)benzylidene)amino)pyrimidine-2,4(1H,3H)-dione (15)
3.1.19. 6-Amino-5-((4-(dimethylamino)benzylidene)amino)-1-ethylpyrimidine-2,4(1H,3H)-dione (16)
3.2. Biological Activity
3.2.1. Antibacterial and Antifungal Activity
3.2.2. Transmission Electron Microscopic Analysis
3.2.3. Cytotoxicity Assay
3.3. Molecular Docking Study
3.4. Pharmacokinetics and ADME Activity
3.5. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alghamdi, S.S.; Suliman, R.S.; Almutairi, K.; Kahtani, K.; Aljatli, D. Imidazole as a Promising Medicinal Scaffold: Current Status and Future Direction. Drug Des. Dev. Ther. 2021, ume 15, 3289–3312. [Google Scholar] [CrossRef]
- Sherer, C.; Snape, T.J. Heterocyclic scaffolds as promising anticancer agents against tumours of the central nervous system: Exploring the scope of indole and carbazole derivatives. Eur. J. Med. Chem. 2015, 97, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Report of the WHO-China Joint Mission on Coronavirus Disease 2019 (COVID-19). Available online: https://www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report.pdf (accessed on 24 February 2020).
- Desai, N.C.; Wadekar, K.R.; Mehta, H.K.; Pandit, U.P. Design, Synthesis, and Antimicrobial Activity of Novel Fluorine-Containing Imidazolones. Russ. J. Org. Chem. 2021, 57, 976–985. [Google Scholar] [CrossRef]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A popula-tion-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia JLiu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observa-tional study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Lescure, F.-X.; Bouadma, L.; Nguyen, D.; Parisey, M.; Wicky, P.-H.; Behillil, S.; Gaymard, A.; Bouscambert-Duchamp, M.; Donati, F.; Le Hingrat, Q.; et al. Clinical and virological data of the first cases of COVID-19 in Europe: A case series. Lancet Infect. Dis. 2020, 20, 697–706. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, C.R.; Chughtai, A.A.; Barnes, M.; Ridda, I.; Seale, H.; Toms, R.; Heywood, A. The role of pneumonia and secondary bacterial infection in fatal and serious outcomes of pandemic influenza a(H1N1)pdm09. BMC Infect. Dis. 2018, 18, 637. [Google Scholar] [CrossRef] [Green Version]
- Molina, P.; Tárraga, A.; Otón, F. Imidazole derivatives: A comprehensive survey of their recognition properties. Org. Biomol. Chem. 2012, 9, 1711–1724. [Google Scholar] [CrossRef]
- Sanad, S.M.H.; Mekky, A.E.M. Efficient synthesis and characterization of novel bis-heterocyclic derivatives and benzo-fused macrocycles containing oxazolone or imidazolone subunits. J. Heterocycl. Chem. 2020, 57, 3930–3942. [Google Scholar] [CrossRef]
- Mikhaylov, A.A.; Solyev, P.N.; Kuleshov, A.V.; Kublitskii, V.S.; Korlyukov, A.A.; Lushpa, V.A.; Baranov, M.S. Im-idazolone-activated donor-acceptor cyclopropanes with a peripheral stereocenter. A study on stereoselectivity of cycloaddition with aldehydes. Chem. Heterocycl. Compd. 2020, 56, 1092–1096. [Google Scholar] [CrossRef]
- Kumar, S.; Aghara, J.C.; Manoj, A.; Alex, A.T.; Mathew, A.J.; Joesph, A. Novel Quinolone Substituted Imidaz-ol-5(4H)-ones as Anti-inflammatory, Anticancer Agents: Synthesis, Biological Screening and Molecular Dock-ing Studies. Indian J. Pharm. Educ. Res. 2020, 54, 771–780. [Google Scholar] [CrossRef]
- Zaitseva, E.R.; Smirnov, A.Y.; Ivanov, I.A.; Mineev, K.S.; Baranov, M.S. Synthesis of 5-(aminomethylidene) imid-azol -4-ones by using N,N-dialkylformamide acetals. Chem. Heterocycl. Compd. 2020, 56, 1097–1099. [Google Scholar] [CrossRef]
- Snieckus, V.; Richardson, P. Synthesis of C2-Substituted Imidazolones. Synfacts 2020, 16, 1149. [Google Scholar] [CrossRef]
- Desai, N.; Joshi, V.; Rajpara, K.; Makwana, A.H. A new synthetic approach and in vitro antimicrobial evaluation of novel imidazole incorporated 4-thiazolidinone motifs. Arab. J. Chem. 2017, 10, S589–S599. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Huang, C.H.; Shao, B.; Qin, L.; Xu, D.; Li, F.; Qu, N.; Xie, L.N.; Kalyanaraman, B.; Zhu, B.Z. Potent Oxidation of DNA by Haloquinoid Disinfection Byproducts to the More Mutagenic Imidazolone dIz via an Unprecedent-ed Haloquinone-Enoxy Radical-Mediated Mechanism. Environ. Sci. Technol. 2020, 54, 6244. [Google Scholar] [CrossRef]
- Abdelhameid, M.K.; Zaki, I.; Mohammed, M.R.; Mohamed, K.O. Design, synthesis, and cytotoxic screening of novel azole derivatives on hepatocellular carcinoma (HepG2 Cells). Bioorganic Chem. 2020, 101, 103995. [Google Scholar] [CrossRef]
- Hebert, S.P.; Schlegel, H.B. Computational Investigation into the Oxidation of Guanine to Form Imidazolone (Iz) and Related Degradation Products. Chem. Res. Toxicol. 2020, 33, 1010–1027. [Google Scholar] [CrossRef]
- Zaitseva, E.R.; Smirnov, A.Y.; Myasnyanko, I.; Sokolov, A.I.; Baranov, M.S. Synthesis of 2-arylideneimidazo [1,2-a]pyrazine- 3,6,8(2H,5H,7H)-triones as a result of oxidation of 4-arylidene-2-methyl-1H-imidazol- 5(4H)-ones with selenium dioxide. Chem. Heterocycl. Compd. 2020, 56, 116–119. [Google Scholar] [CrossRef]
- Xu, B.; Lee, E.M.; Medina, A.; Sun, X.; Wang, D.; Tang, H.; Zhou, G.-C. Inhibition of zika virus infection by fused tricyclic derivatives of 1,2,4,5-tetrahydroimidazo[1,5-a]quinolin-3(3aH)-one. Bioorganic Chem. 2020, 104, 104205. [Google Scholar] [CrossRef]
- Desai, N.C.; Vaghani, H.V.; Rajpara, K.M.; Joshi, V.V.; Satodiya, H.M. Novel approach for synthesis of potent an-timicrobial hybrid molecules containing pyrimidine-based imidazole scaffolds. Med. Chem. Res. 2014, 23, 4395. [Google Scholar] [CrossRef]
- Desai, N.C.; Vaghani, H.V.; Karkar, T.J.; Patel, B.Y.; Jadeja, K.A. Synthesis and antimicrobial studies of 1, 2, 3, 4-tetrahydropyrimidine bearing imidazole analogues. Indian J. Chem. Sect. B 2017, 56, 438. [Google Scholar]
- Desai, N.C.; Vaja, D.V.; Jadeja, K.A.; Joshi, S.B.; Khedkar, V.M. Synthesis, Biological Evaluation and Molecular Docking Study of Pyrazole, Pyrazoline Clubbed Pyridine as Potential Antimicrobial Agents. Anti-Infect. Agents 2020, 18, 306–314. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.A.; Fayed, E.A.; Abdel-Razek, A.S. One pot synthesis, antimicrobial and antioxidant activities of fused uracils: Pyrimidodiazepines, lumazines, triazolouracil and xanthines. Chem. Cent. J. 2017, 11, 1–13. [Google Scholar] [CrossRef]
- Singh, G.; Sharma, G.; Kalra, S.P.; Satija, P.; Singh, P.B.; Wreidt, M. Click-Derived Uracil-Appended Organosi-latranyl Scaffolds: Synthesis, Antibacterial Characteristics, Pb2+ Binding and Fabrication of Hybrid Silica Na-noparticles. Chem. Select. 2020, 5, 284–292. [Google Scholar]
- Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 2006, 71, 941–948. [Google Scholar] [CrossRef]
- Deshmukh, M.B.; Salunkhe, S.M.; Patil, D.R.; Anbhule, P.V. A novel and efficient one step synthesis of 2-amino-5-cyano- 6-hydroxy-4-aryl pyrimidines and their anti-bacterial activity. Eur. J. Med. Chem. 2009, 44, 2651–2654. [Google Scholar] [CrossRef]
- Buron, F.; Merour, J.Y.; Akssira, M.; Guillaumet, G.; Routier, S. ChemInform Abstract: Recent Advances in the Chemistry and Biology of Pyridopyrimidines. Angew. Chem. 2015, 46, 9576–9595. [Google Scholar] [CrossRef]
- Lu, X.; Chen, Y.; Guo, Y.; Liu, Z.; Shi, Y.; Xu, Y.; Wang, X.; Zhang, Z.; Liu, J. The design and synthesis of N-1-alkylated-5- aminoaryalkylsubstituted-6-methyluracils as potential non-nucleoside HIV-1 RT inhibitors. Bioorganic Med. Chem. 2007, 15, 7399–7407. [Google Scholar] [CrossRef]
- Ding, Y.; Girardet, J.-L.; Smith, K.L.; Larson, G.; Prigaro, B.; Wu, J.Z.; Yao, N. Parallel synthesis of 5-cyano-6-aryl-2-thiouracil derivatives as inhibitors for hepatitis C viral NS5B RNA-dependent RNA polymerase. Bioorganic Chem. 2006, 34, 26–38. [Google Scholar] [CrossRef]
- El-Brollosy, N.R.; Al-Deeb, O.A.; El-Emam, A.A.; Pedersen, E.B.; La Colla, P.; Collu, G.; Sanna, G.; Loddo, R. Synthesis of Novel Uracil Non-Nucleoside Derivatives as Potential Reverse Transcriptase Inhibitors of HIV-1. Arch. Pharm. 2009, 342, 663–670. [Google Scholar] [CrossRef]
- El-Brollosy, N.R.; Al-Omar, M.A.; Al-Deeb, O.A.; El-Emam, A.A.; Nielsen, C. Synthesis of novel uracil non-nucleosides analogues of 3,4-dihydro2-alkylthio-6-benzyl-4-oxo-pyrimidines and 6-benzyl-1- ethoxyme-thyl-5-isopropyluracil. J. Chem. Res. 2007, 5, 263–267. [Google Scholar] [CrossRef]
- Nuñez, M.; Gavilan, M.D.; Conejo-García, A.; Cruz-Lopez, O.; Gallo, M.; Espinosa, A.; Campos, J. Design, Synthesis and Anticancer Activity Against the MCF-7 Cell Line of Benzo-Fused 1,4-Dihetero Seven- and Six-Membered Tethered Pyrimidines and Purines. Curr. Med. Chem. 2008, 15, 2614–2631. [Google Scholar] [CrossRef]
- Boisdron-Celle, M.; Remaud, G.; Traore, S.; Poirier, A.L.; Gamelin, L.; Morel, A.; Gamelin, E. 5-Fluorouracil-related severe toxicity: A comparison of diferent methods for the pretherapeutic detection of dihydropyrimidine dehy-drogenase deficiency. Cancer Lett. 2007, 249, 271–282. [Google Scholar] [CrossRef]
- Semenov, V.E.; Voloshina, A.D.; Toroptzova, E.M.; Kulik, N.V.; Zobov, V.V.; Giniyatullin, R.K.; Mikhailov, A.S.; Nikolaev, A.E.; Akamsin, V.D.; Reznik, V.S. Antibacterial and antifungal activity of acyclic and macrocyclic uracil de-rivatives with quaternized nitrogen atoms in spacers. Eur. J. Med. Chem. 2006, 41, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Antonello, M.; Dante, R.; Silvio, M.; Gerald, B.; Giovanna, S.; Claudio, P.; Anna, T.P. Discovery of uracil-based histone deacetylase inhibitors able to reduce acquired an-tifungal resistance and trailing growth in Candida albicans. Bioorg. Med. Chem. Lett. 2007, 17, 1221–1225. [Google Scholar]
- El-Kalyoubi, S.; Agili, F. Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido[2,3-d]pyrimidine, Xanthine and Lumazine Derivatives. Molecules 2020, 25, 5205. [Google Scholar] [CrossRef] [PubMed]
- El-Kalyoubi, S.; Fayed, E. Synthesis and evaluation of antitumor activities of novel fused tri- and tetracyclicura-cil derivatives. J. Chem. Res. 2016, 40, 771–777. [Google Scholar] [CrossRef]
- El-Sayed, A.S.A.; Ali, D.M.I.; Yassin, M.A.; Zayed, R.A.; Ali, G.S. Sterol inhibitor “Fluconazole” enhance the Taxol yield and molecular expression of its encoding genes cluster from Aspergillus flavipes. Process Biochem. 2019, 76, 55–67. [Google Scholar] [CrossRef]
- Phalke, P.L.; Kale, R.B.; Modhave, M.B.; Sonawane, P.S.; Khaire, S.R.; Jadhav, S.L. One pot synthesis, antimicrobial and antioxidant activities of fused uracils: Pyrimidodiazepines, lumazines, triazolouracil and xanthines. J. Drug Deliv. Therap. 2019, 9, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Isabella, F.S.; Marra, P.; Pedro De Castro, G.W. Amarante, Recent Advances in Azlactone Transformations. Eur. J. Org. Chem. 2019, 2019, 5829. [Google Scholar] [CrossRef]
- Muthuboopathi, G.; Shanmugarajan, T.S. Synthesis, characterization, and biological evaluation of oxazolone an-alogs. Asian J. Pharm. Clin. Res. 2018, 11, 159–162. [Google Scholar]
- Yoneda, F.; Nagamatsu, T.; Yamasaki, H. Facile and General Synthesis of 8-Substituted 2-Methylthiopurin-6-ones. Heterocycles 1992, 33, 775. [Google Scholar] [CrossRef]
- Wang, B.; Guo, F.; Huang, C.; Zhao, H. Unraveling the iterative type I polyketide synthases hidden in Streptomyces. Proc. Natl. Acad. Sci. USA 2020, 117, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, M.; Unniraman, S.; Mahadevan, S.; Nagaraja, V. Effect of different classes of inhibitors on DNA gyrase from Mycobacterium smegmatis. J. Antimicrob. Chemother. 2001, 48, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sayed, A.S.A.; Mohamed, N.Z.; Safan, S.; Yassin, M.A.; Shaban, L.; Shindia, A.A.; Ali, G.S.; Sitohy, M.Z. Restoring the taxol biosynthetic machinery of Aspergillus terreus by Podocarpus gracilior pilger microbiome, with retrieving the ribosome biogenesis proteins of WD40 superfamily. Sci. Rep. 2019, 9, 11534. [Google Scholar] [CrossRef] [Green Version]
- Turel, I.; Golič, L.; Bukovec, P.; Gubina, M. Antibacterial tests of Bismuth(III)–Quinolone (Ciprofloxacin, cf) compounds against Helicobacter pylori and some other bacteria. Crystal structure of (cfH2)2[Bi2Cl10]·4H2O. J. Inorg. Biochem. 1998, 71, 53–60. [Google Scholar] [CrossRef]
- Turel, I.; Bukovec, P.; Quirós, M. Crystal structure of ciprofloxacin hexahydrate and its characterization. Int. J. Pharm. 1997, 152, 59–65. [Google Scholar] [CrossRef]
- Yue, Y.-F.; Sun, W.; Gao, E.-Q.; Fang, C.-J.; Xu, S.; Yan, C.-H. Syntheses and crystal structures of three Mn(II) complexes with 2-hydroxynicotinate. Inorganica Chim. Acta 2007, 360, 1466–1473. [Google Scholar] [CrossRef]
- Top, S.; Tang, J.; Vessieres, A.; Carrez, C.; Provot, C.; Jaouen, G. Ferrocenyl hydroxytamoxifen: A prototype for a new range of oestradiol receptor site-directed cytotoxics. Chem. Commun. 1996, 8, 955–956. [Google Scholar] [CrossRef]
- Pfab, W.; Fischer, E.O. Zur Kristallstruktur der Di-cyclopentadienyl-verbindungen des zweiwertigen Eisens, Kobalts und Nickels. Z. Anorg. Allg. Chem. 1953, 274, 316–322. [Google Scholar] [CrossRef]
- Becka, M.; Vilkova, M.; Soral, M.; Potocnak, I.; Breza, M.; Beres, T.; Imrich, J. Synthesis and isomerisation of acridin substituted 1, 3-thiazolidin-4-ones and 4-oxo-1,3- thiazolidin-5-ylidene acetates. An experimental and computational study. J. Mol. Struc. 2018, 1154, 152–164. [Google Scholar] [CrossRef]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; Wiley: London, UK, 2011. [Google Scholar]
- Kurtaran, R.; Odabasoglu, S.; Azizoglu, A.; Kara, H.; Atako, O.L. Experimental and computational study on [2, 6-bis (3, 5-dimethyl-N-pyrazolyl) pyridine]-(dithiocyanato) mercury (II). Polyhedron 2007, 26, 5069–5074. [Google Scholar] [CrossRef]
- El-Sayed, A.S.A.; Ali, G.S. Aspergillus flavipes is a novel efficient biocontrol agent of Phytophthora parasitica. Biol. Control. 2020, 140, 104072. [Google Scholar] [CrossRef]
- Ciofini, I.; Lainé, P.P.; Bedioui, F.; Adamo, C. Photoinduced Intramolecular Electron Transfer in Ruthenium and Osmium Polyads: Insights from Theory. J. Am. Chem. Soc. 2004, 126, 10763–10777. [Google Scholar] [CrossRef] [PubMed]
- Ciofini, I.; Daul, C.; Adam, J. Phototriggered Linkage Isomerization in Ruthenium− Dimethylsulfoxyde Complexes: Insights from Theory. J. Phys. Chem. A 2003, 107, 11182–11190. [Google Scholar] [CrossRef]
- El Baz, A.F.; Shetaia, Y.M.; Elkhouli, R.R. Xylitol production by Candida tropicalis under different statistically optimized growth conditions. Afr. J. Biotechnol. 2011, 10, 16617–16625. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- El-Sayed, A.S.A.; Shindia, A.A.; Zeid, A.A.A.; Yassin, A.M.; Sitohy, M.Z.; Sitohy, B. Aspergillus nidulans thermostable arginine deiminase-Dextran conjugates with enhanced molecular stability, proteolytic resistance, pharmacokinetic properties and anticancer activity. Enzym. Microb. Technol. 2019, 131, 109432. [Google Scholar] [CrossRef]
- Bergey, D.H.; Bergey’s, J.G.H. Manual of Determinative Bacteriology, 9th ed.; Williams & Wilkins: Baltimore, MD, USA, 1994. [Google Scholar]
- Hassan, A.; Sorour, N.M.; El-Baz, A.; Shetaia, Y. Simple synthesis of bacterial cellulose/magnetite nanoparticles composite for the removal of antimony from aqueous solution. Int. J. Environ. Sci. Technol. 2019, 16, 1433–1448. [Google Scholar] [CrossRef]
- El-Sayed, A.S.A.; Abdel-Azeim, S.; Ibrahim, H.M.; Yassin, M.A.; Abdel-Ghany, S.E.; Esener, S.; Ali, G.S. Biochemical stability and molecular dynamic characterization of Aspergillus fumigatus cystathionine γ-lyase in response to various reaction effectors. Enzym. Microb. Technol. 2015, 81, 31–46. [Google Scholar] [CrossRef] [Green Version]
- El-Sayed, A.S.A.; Akbar, A.; Iqrar, I.; Ali, R.; Norman, D.; Brennan, M.; Ali, G.S. A glucanolytic Pseudomonas sp. associated with Smilax bona-nox L. displays strong activity against Phytophthora parasitica. Microbiol. Res. 2018, 207, 140–152. [Google Scholar] [CrossRef]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA – An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Kattan, S.W.; Nafie, M.S.; Elmgeed, G.A.; Alelwani, W.; Badar, M.; Tantawy, M.A. Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: Implication of PI3K/AKT/mTOR pathway. J. Steroid Biochem. Mol. Biol. 2020, 198, 105604. [Google Scholar] [CrossRef] [PubMed]
- Tantawy, M.A.; Sroor, F.M.; Mohamed, M.F.; El-Naggar, M.E.; Saleh, F.M.; Hassaneen, H.M.; Abdelhamid, I.A. Molecular Docking Study, Cytotoxicity, Cell Cycle Arrest and Apoptotic Induction of Novel Chalcones Incorporating Thiadiazolyl Isoquinoline in Cervical Cancer. Anti-Cancer Agents Med. Chem. 2020, 20, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Nafie, M.S.; Tantawy, M.A.; Elmgeed, G.A. Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents. Steroids 2019, 152, 108485. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Motaal, M.; Almohawes, K.; Tantawy, M.A. Antimicrobial evaluation and docking study of some new substituted benzimidazole-2yl derivatives. Bioorganic Chem. 2020, 101, 103972. [Google Scholar] [CrossRef]
- Nagy, M.; Darwish, K.; Kishk, S.; Tantawy, M.; Nasr, A.; Qushawy, M.; Swidan, S.; Mostafa, S.; Salama, I. Design, Synthesis, Anticancer Activity, and Solid Lipid Nanoparticle Formulation of Indole- and Benzimidazole-Based Compounds as Pro-Apoptotic Agents Targeting Bcl-2 Protein. Pharmaceuticals 2021, 14, 113. [Google Scholar] [CrossRef]
- Alsayed, M.; El-Kady, D.S.; ATantawy, M.; Abdelhalim, M.M.; Elazabawy, S.R.; Abdallah, A.E.M.; AElmegeed, G. Novel Melatonin Derivatives: Synthesis, Anticancer Evaluations and Molecular-Docking Study. Egypt. J. Chem. 2020, 64, 1517–1533. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 98, Revision A.9; Gaussian INc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- El-Baz, A.F.; Sorour, N.M.; Shetaia, Y.M. Trichosporon jirovecii-mediated synthesis of cadmium sulfide nanoparticles. J. Basic Microbiol. 2016, 56, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Flurry, R.L., Jr. Molecular Orbital Theory of Bonding in Organic Molecules; Marcel Dekker: New York, NY, USA, 1968. [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diameter of Inhibition Zone (mm) | |||||||
|---|---|---|---|---|---|---|---|
| Fungi | Gram Negative Bacteria | Gram Positive Bacteria | |||||
| Cpd | C. albicans | A. flavus | E. coli | Salmonella sp. | Pseudomonas sp. | S. aureus | Bacillus sp. |
| 10% DMSO | - | - | - | - | - | - | - |
| 3 | 10 ± 0.8 | 2 | 4 ± 0.2 | 0.2 ± 0.01 | 0 | 2 ± 0.01 | 0.7 ± 0.01 |
| 4 | 2 | 0 | 2 | 0.1 | 0 | 1 ± 0.01 | 0.1 |
| 5 | 2 | 0 | 2 | 0 | 0 | 0.8 ± 0.01 | 0.8 ± 0.04 |
| 6 | 10 ± 0.6 | 2 | 4 ± 0.1 | 0 | 0 | 1 ± 0.01 | 0.9 ± 0.01 |
| 7 | 2 | 0 | 2 | 0 | 0 | 0 | 0 |
| 8 | 2 | 3 | 2 | 0 | 1.8 ± 0.01 | 0.2 | 0 |
| 9 | 5 ± 0.2 | 2 | 3± 0.3 | 0 | 0.9 ± 0.05 | 0 | 0.8 ± 0.03 |
| 10 | 4 ± 0.2 | 0 | 4 ± 0.23 | 0 | 0 | 0 | 0 |
| 11 | 6 ± 0.3 | 3 | 3 ± 0.3 | 0 | 0 | 0 | 0 |
| 12 | 8 ± 0.4 | 4 | 5 ± 0.22 | 0 | 1.5 ± 0.01 | 0.3 | 0 |
| 13 | 6 ± 0.2 | 3 | 4 ± 0.23 | 0 | 0 | 0.4 | 0.9 ± 0.01 |
| 14 | 2 | 2 | 2 | 0 | 1.9 ± 0.03 | 0 | 0 |
| 15 | 7 ± 0.5 | 3 | 3 ± 0.1 | 0.1 ± 0.01 | 1.8 ± 0.03 | 0 | 0.8 ± 0.03 |
| 16 | 12 ±0.3 | 10 | 7 ± 0.2 | 0.2 ± 0.01 | 1.9 ± 0.04 | 0.7 | 1.2 ± 0.04 |
| Fluconazole | 2.1 ± 0.1 | 2.9 ± 0.1 | 0 | 0 | 0 | 0 | 0 |
| Bacimethrin | 0.9 ± 0.2 | 0.6 ± 0.2 | 0.8 ± 0.2 | 0.9 ± 0.2 | 0.7 ± 0.2 | ||
| Primsol | 0.6 ± 0.09 | 0.5 ± 0.02 | 0.4 ± 0.02 | 0.3 ± 0.01 | 0.4 ± 0.3 | ||
| IC50 Values of the Synthesized Compounds | |||||||
|---|---|---|---|---|---|---|---|
| Fungi | Gram Negative Bacteria | Gram Positive Bacteria | |||||
| Cpd | C. albicans | A. flavus | E. coli | Salmonella sp. | Pseudomonas sp. | S. aureus | Bacillus sp. |
| 3 | 1.07 | 4.9 | 1.8 | 3.9 | 5.2 | 2.8 | 2.4 |
| 4 | 5.0 | 6.0 | 4.8 | 2.1 | 5.7 | 2.1 | 3.2 |
| 5 | 6.0 | 7.5 | 4.5 | 5.0 | 6.1 | 2.9 | 2.8 |
| 6 | 1.02 | 4.2 | 1.9 | 4.9 | 5.1 | 2.9 | 2.2 |
| 7 | 5.3 | 8.0 | 5.1 | 6.2 | 7.1 | 5.2 | 5.2 |
| 8 | 5.1 | 7.5 | 4.9 | 7.9 | 1.9 | 5.4 | 5.5 |
| 9 | 4.9 | 3.8 | 4.1 | 8.0 | 4.2 | 6.0 | 3.8 |
| 10 | 4.2 | 8.0 | 2.1 | 7.9 | 2.9 | 6.2 | 4.9 |
| 11 | 3.2 | 3.2 | 2.2 | 7.0 | 3.9 | 6.3 | 5.5 |
| 12 | 1.46 | 2.9 | 2.11 | 7.2 | 2.2 | 3.1 | 6.9 |
| 13 | 3.3 | 3.9 | 1.9 | 2.3 | 6.9 | 3.2 | 2.8 |
| 14 | 4.2 | 3.9 | 5.1 | 8.1 | 2.8 | 4.0 | 4.9 |
| 15 | 4.1 | 4.2 | 4.9 | 7.2 | 2.9 | 5.1 | 5.3 |
| 16 | 0.82 | 1.2 | 1.98 | 1.5 | 2.1 | 2.67 | 2.1 |
| Fluconazole | 0.6 ± 0.1 | 6.0 ± 0.1 | 0 | 0 | 0 | 0 | 0 |
| Bacimethrin | 0 | 0 | 3.9 ± 0.6 | 4.8 ± 0.1 | 5.1 ± 0.1 | 4.2 ± 0.1 | 3.9 ± 0.1 |
| Primsol | 0 | 0 | 4.3 ± 0.4 | 4.8 ± 0.1 | 4.6 ± 0.1 | 5.0 ± 0.1 | 4.3 ± 0.1 |
| Compounds | APC Protein (PDB: 2KR5) | |||
|---|---|---|---|---|
| Free Binding of Energy (Kcal/mol) | H-Bond | |||
| No of H-Bond | Amino Acid Residues | Length Å | ||
| 3 | −5.1 | 1 | GLU65 | 2.261 |
| 12 | −5.1 | 1 | GLU65 | 2.348 |
| 13 | −5.2 | 1 | GLU65 | 2.358 |
| 16 | −4.5 | 1 | GLU65 | 1.984 |
| PNS | −4.5 | 2 | GLU65 | 2.265 |
| ASP71 | 2.559 | |||
| Compounds | DNAg Protein (PDB: 1KZN) | |||
| Free Binding of Energy (Kcal/mol) | H-Bond | |||
| No of H-Bond | Amino Acid Residues | Length Å | ||
| 3 | −8.2 | 1 | THR165 | 3.074 |
| 12 | −7.2 | 2 | ASP49 | 2.576 |
| ASN46 | 3.395 | |||
| 13 | −7.4 | 2 | ASP49 | 2.573 |
| ASN46 | 3.368 | |||
| 16 | −7.1 | 2 | ASP73 | 2.276 |
| ASN46 | 2.392 | |||
| CBN | −9.3 | 4 | ASN46 | 3.142 |
| ARG136 | 2.930 | |||
| ARG136 | 2.923 | |||
| ARG136 | 3.244 | |||
| Compounds | PBP Protein (PDB: 3UDI) | |||
| Free Binding of Energy (Kcal/mol) | H-Bond | |||
| No of H-Bond | Amino Acid Residues | Length Å | ||
| 3 | −8.7 | 1 | ASN674 | 2.006 |
| 12 | −8.2 | 2 | SER470 | 2.118 |
| THR672 | 2.797 | |||
| 13 | −7.6 | 2 | SER470 | 2.458 |
| THR672 | 3.040 | |||
| 16 | −7.3 | 3 | SER470 | 2.697 |
| SER487 | 3.278 | |||
| THR672 | 2.916 | |||
| PNS | −7.2 | 5 | TYR670 | 3.006 |
| TYR670 | 3.035 | |||
| SER434 | 3.078 | |||
| THR672 | 3.128 | |||
| THR672 | 2.310 | |||
| Properties | Compound 12 |
|---|---|
| Formula | C17H17N5O3 |
| Molecular weight | 339.35 |
| Num. of heavy atoms | 25 |
| Num. of aromatic heavy atoms | 12 |
| Num. of rotatable bonds | 3 |
| Num. H-bond donors (HBD) | 2 |
| Num. H-bond acceptor (HBA) | 4 |
| Molar reactivity | 103.26 |
| Topological Polar Surface area (TPSA) | 113.55 |
| Lipophilicity (Log P) | 2.04 |
| Water solubility (Log S) | −2.39 |
| Bond Length (Å) | |||||
| C1-C2 C2-C3 C3-C4 C4-C6 C5-C6 C1-C5 C1-Cl7 C4-C8 C8-C9 C9-C10 C9-N12 | 1.346 1.345 1.348 1.354 1.343 1.341 1.726 1.351 1.353 1.346 1.354 | C13-N12 C13-O14 C13-C15 C10-N11 N11-C16 C16-C18 N17-C18 C10-N17 C16-C19 C19-N20 C19-O23 | 1.384 1.209 1.521 1.339 1.334 1.338 1.348 1.269 1.349 1.377 1.210 | N20-C21 C21-N22 C21-O24 C18-N22 N22-C25 C25-C26 C26-C27 C27-C28 C28-C29 C29-C31 C30-C31 C26-C30 | 1.389 1.395 1.208 1.351 1.453 1.556 1.346 1.342 1.341 1.342 1.342 1.346 |
| Bond Angle (°) | |||||
| C3C4C8 C6C4C8 C4C8C9 C8C9N12 C8C9C10 | 124.63 118.83 131.89 113.55 126.94 | C9N12C13 N12C13C15 N12C13O14 O14C13C15 C9C10N17 | 124.18 122.51 118.39 119.04 125.31 | C9C10N11 C18N22C25 C25N22C21 C26C25N22 C25C26C27 C25C26C30 | 123.47 120.23 120.71 113.98 119.35 122.79 |
| Dihedral Angles (°) | |||||
| C3C4C8C9 C6C4C8C9 C4C8C9N12 C4C8C9C10 C8C9N12C13 O23C19N20C21 | 18.74 −165.54 −179.95 20.23 −105.95 178.85 | C10C9N12C13 C9N12C13C15 N12C9C10N11 N12C9C10N17 C10N17C18N22 O24C21N20C19 | 82.58 −19.77 23.75 −154.23 178.05 179.53 | C26C25N22C18 C26C25N22C21 N22C25C26C20 N22C25C26C27 C8C9N10N17 | −84.71 87.88 −4.19 175.85 102.31 |
| Mulliken charges | |||||
| C1 Cl7 N12 C13 C9 O14 | 0.237 −0.295 −0.284 0.424 0.114 −0.488 | C10 N11 N17 C16 C18 N22 | 0.217 −0.107 −0.334 −0.111 0.246 −0.198 | C21 N20 O24 C19 O23 | 0.501 −0.324 −0.432 0.425 −0.446 |
| Total energy/k cal/mol Heat of formation k cal/mol Total dipole moment/D | −172,059.722 −10,607.049 3.078 | ||||
| Parameters | Compound 3 | Compound 12 | Compound 13 | Compound 16 |
|---|---|---|---|---|
| HOMO, H | −0.319 | −0.321 | −0.315 | −0.309 |
| LUMO, L | −0.237 | −0.245 | −0.254 | −0.237 |
| I = −H | 0.319 | 0.321 | 0.315 | 0.309 |
| A = −L | 0.237 | 0.245 | 0.254 | 0.237 |
| ∆E = L − H | 0.082 | 0.067 | 0.061 | 0.072 |
| η = (I − A)/2 | 0.041 | 0.034 | 0.031 | 0.036 |
| χ = −(H − L/2) | 0.278 | 0.283 | 0.285 | 0.237 |
| σ = 1/η | 24.390 | 29.412 | 32.258 | 27.778 |
| S = 1/2 η | 12.195 | 14.706 | 16.129 | 13.889 |
| Pi = −χ | −0.278 | −0.283 | −0.285 | −0.237 |
| ω = (Pi)2/2 η | 0.942 | 1.178 | 1.310 | 0.780 |
| ∆Nmax = χ/η | 6.780 | 8.324 | 9.194 | 6.583 |
| Compound 3 | Compound 12 | ||||
|---|---|---|---|---|---|
| H | L | H | L | ||
| 2,6-dioxopurine ring C10, N11, C16-O24 | 55.5% | 53.7% | 2,4-dioxo-6-aminopyrimidine ring C1-O8, N12 | 25.6% | 0.0% |
| Chlorophenyl ring C1-Cl7 | 15.4% | 21.8% | 2-methyl-5-oxoimidazol ring N13-C19 | 61.2% | 51.9% |
| Vinyl group C8-C9 | 7.9% | 15.9% | Benzylidene ring C20-C26 | 13.2% | 48.1% |
| acetamide group N12-C15 | 20.2% | 8.6% | Ethyl group C9, C11 | 0.0% | 0.0% |
| Benzyl group C25-C31 | 1% | 0.0% | |||
| Compound 13 | Compound 16 | ||||
| H | L | H | L | ||
| 2-thio-4-oxo-6-aminopyrimidine ring C1-S8, N11 | 56.3% | 0.0% | 2,4-dioxo-6-aminopyrimidine ring C13-O19, N23 | 41.3% | 19.5% |
| 2-methyl-5-oxoimidazol ring N12-C18 | 41.9% | 52.7% | dimethylaminobenzylidene ring C1-N9, C33-C34 | 33.6% | 58.7% |
| Benzylidene ring C19-C25 | 1.8% | 31.4% | Amino group N24-C25 | 25.1% | 21.8% |
| Methyl group C9 | 0.0% | 15.9% | Ethyl group C20, C22 | 0.0% | 0.0% |
| Compound | eV | nm | Major Contributions | Assignment | λmax, nm |
|---|---|---|---|---|---|
| 3 | 4.9546 | 250.24 | H-11 → L(4.5%), H-11 → L + 6(2.9%), H → L(4.9%), H-7 → L(3.2%), H-5→L(34.5%), H-3 → L(3.7%), H-1 → L(14.7%), H → L(4.4%) | π-π * | 265 |
| 4.7874 | 258.98 | H-11 → L (7.8%), H-10 → L (2.5%), H-5 → L (2.8%), H-1→L (78.4%), H → L(4,6%), H → L + 1(3.6%) | π-π * | 275 | |
| 4.4144 | 280.86 | H-11 → L + 7 (4.9%), H-10 → L + 6 (14.1%), H-10 → L + 7 (41.8%), H-7 → L + 7 (6.7%), H-6 → L + 6 (9.6%), H-6 → L + 7 (26.7%) | n-π * | 290 | |
| 4.0996 | 302.43 | H-9 → L (5.2%), H-9 → L + 5 (61.6%), H-9 → L + 6 (21.3%), H-9 → L + 7 (2.7%), H-8 → L + 5(6.3%), H-8 → L + 6 (2.8%) | π-π * | 305 | |
| 3.4797 | 356.3 | H-11 → L + 2 (5.3%), H-10 → L (6.3%), H-10 → L + 1 (10.4%), H-6 → L (22.9%), H-6 → L + 1 (38.3%), H-6 → L + 7 (8.8%), H-6 → L + 10 (7.8%) | n-π * | 368 | |
| 3.2847 | 377.47 | H-1 → L (4.5%), H → L(93.4%) | π-π * | 383 | |
| 12 | 4.7935 | 258.65 | H-8 → L + 1 (2.4%), H-1 → L + 1 (54.4%), H → L + 1 (43%) | π-π * | 269 |
| 4.4183 | 280.61 | H-6 → L (5.8%), H-4 → L (9.8%), H-3 → L (8.6%), H-2 → L(2.7%), H-1 → L(72.8%) | π-π * | 278 | |
| 4.3485 | 285.12 | H-9 → L + 3 (20.5%), H-9 → L + 4 (12.4%), H-8 → L + 3 (7.6%), H-8 → L + 4 (4%), H-7 → L + 3 (27.3%), H-3 → L + 4 (16.6%), H-5 → L + 3 (7%), H-5 → L + 4 (4.2%) | n-π * | 300 | |
| 4.0172 | 308.64 | H-10 → L (5%), H-8 → L (35.2%), H-8 → L + 3 (3.3%), H-8 → L + 4 (7.3%), H-3 → L (15.4%), H-3 → L + 3 (8.5%), H-3 → L + 4 (8.2%), H-3 → L + 5 (7%) | n-π * | 315 | |
| 3.8482 | 322.23 | H-6 → L + 1 (6.1%), H-5 → L + 1 (35.8%), H-5 → L + 3 (13.5%), H-5 → L + 6 (13.6%), H-4 → L + 1 (18.3%), H-4 → L + 3 (6.3%), H-4 → L + 6 (6%) | n-π * | 322 | |
| 3.7319 | 332.23 | H → L (100%) | π-π * | 338 | |
| 3.6207 | 342.43 | H-8 → L (14.5%), H-8 → L + 5 (8.9%), H-6 → L (3.3%), H-5 → L(11.7%), H-5 → L + 5 (3%), H-4 → L (5.7%), H-3 → L (36.7%), H-3 → L + 5 (3.5%) | n-π * | 350 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Kalyoubi, S.; Agili, F.; Zordok, W.A.; El-Sayed, A.S.A. Synthesis, In Silico Prediction and In Vitro Evaluation of Antimicrobial Activity, DFT Calculation and Theoretical Investigation of Novel Xanthines and Uracil Containing Imidazolone Derivatives. Int. J. Mol. Sci. 2021, 22, 10979. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010979
El-Kalyoubi S, Agili F, Zordok WA, El-Sayed ASA. Synthesis, In Silico Prediction and In Vitro Evaluation of Antimicrobial Activity, DFT Calculation and Theoretical Investigation of Novel Xanthines and Uracil Containing Imidazolone Derivatives. International Journal of Molecular Sciences. 2021; 22(20):10979. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010979
Chicago/Turabian StyleEl-Kalyoubi, Samar, Fatimah Agili, Wael A. Zordok, and Ashraf S. A. El-Sayed. 2021. "Synthesis, In Silico Prediction and In Vitro Evaluation of Antimicrobial Activity, DFT Calculation and Theoretical Investigation of Novel Xanthines and Uracil Containing Imidazolone Derivatives" International Journal of Molecular Sciences 22, no. 20: 10979. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222010979