
Full Length Transcriptome Highlights the Coordination of Plastid Transcript Processing

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. A Protocol to Sequence the Full Length Plastid Transcriptome
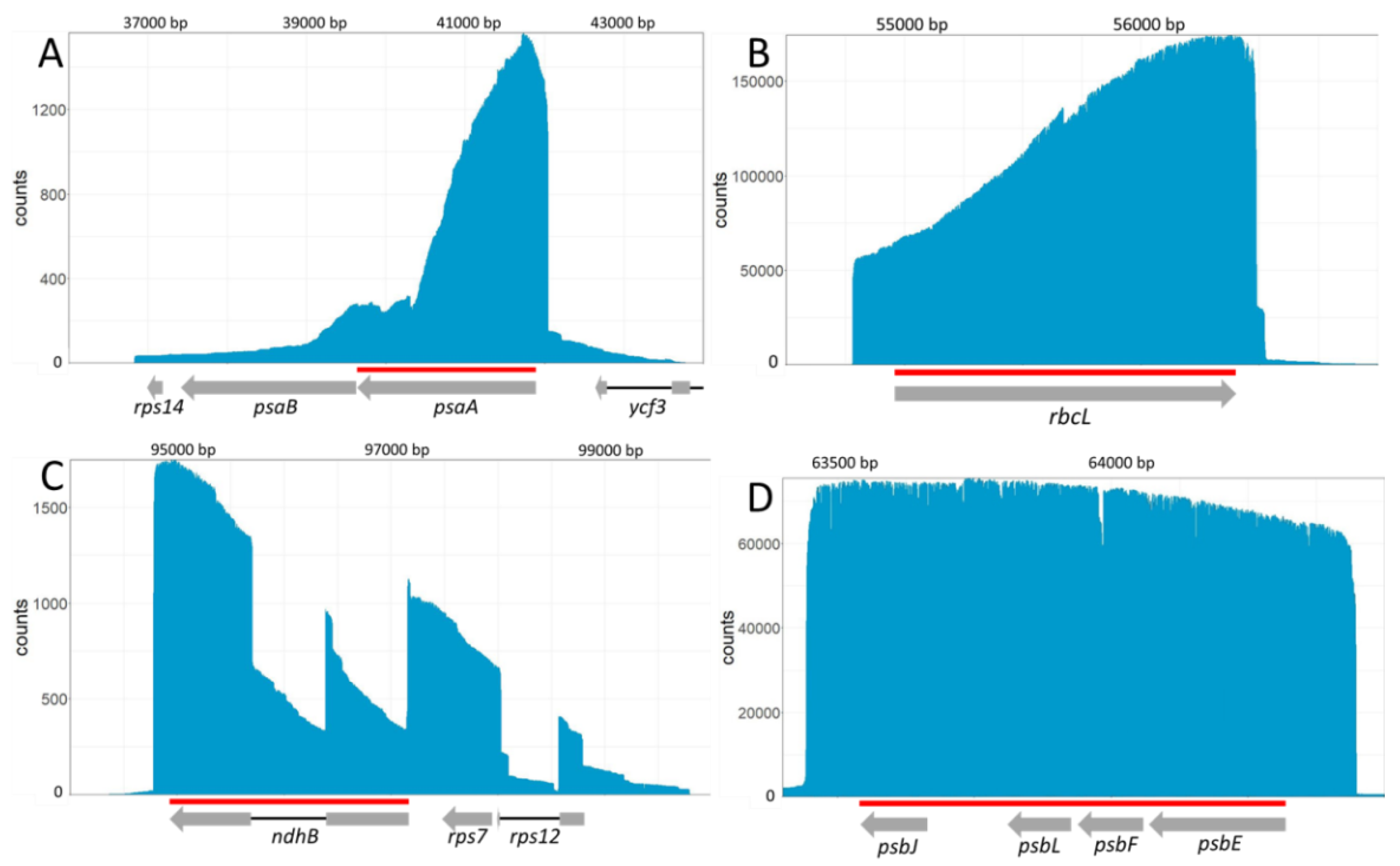
2.2. A Representative Picture of the Plastid Transcriptome


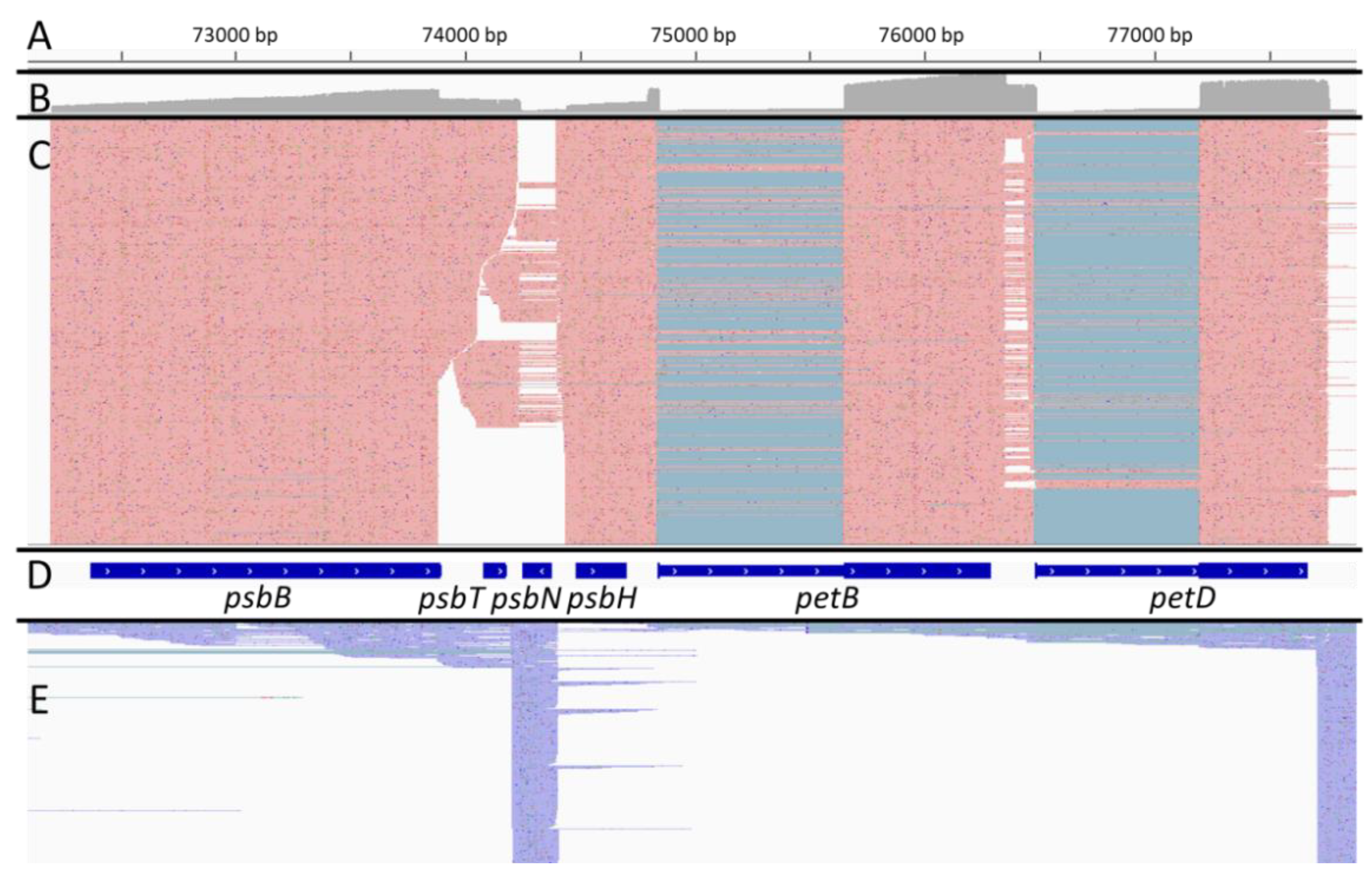
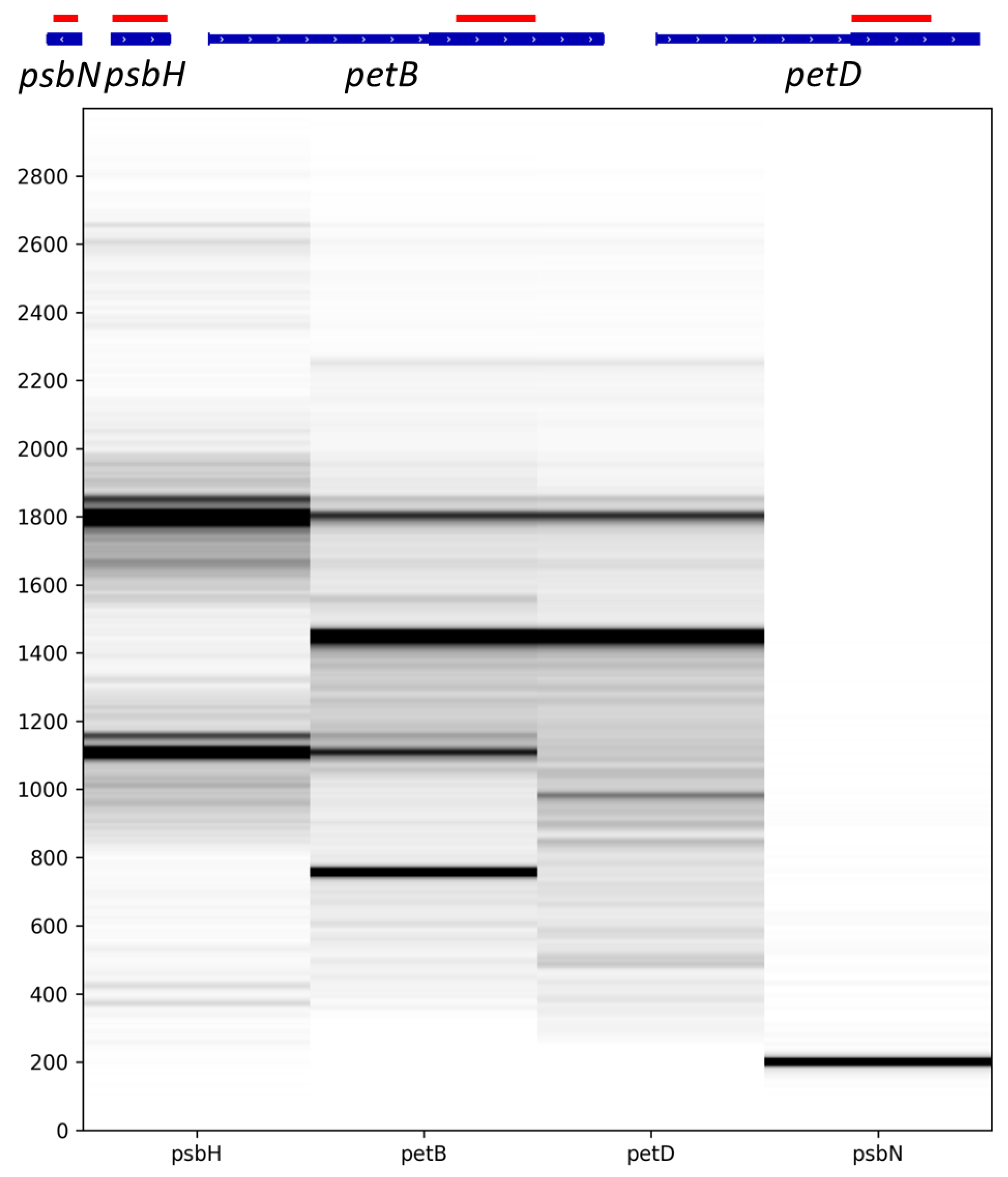
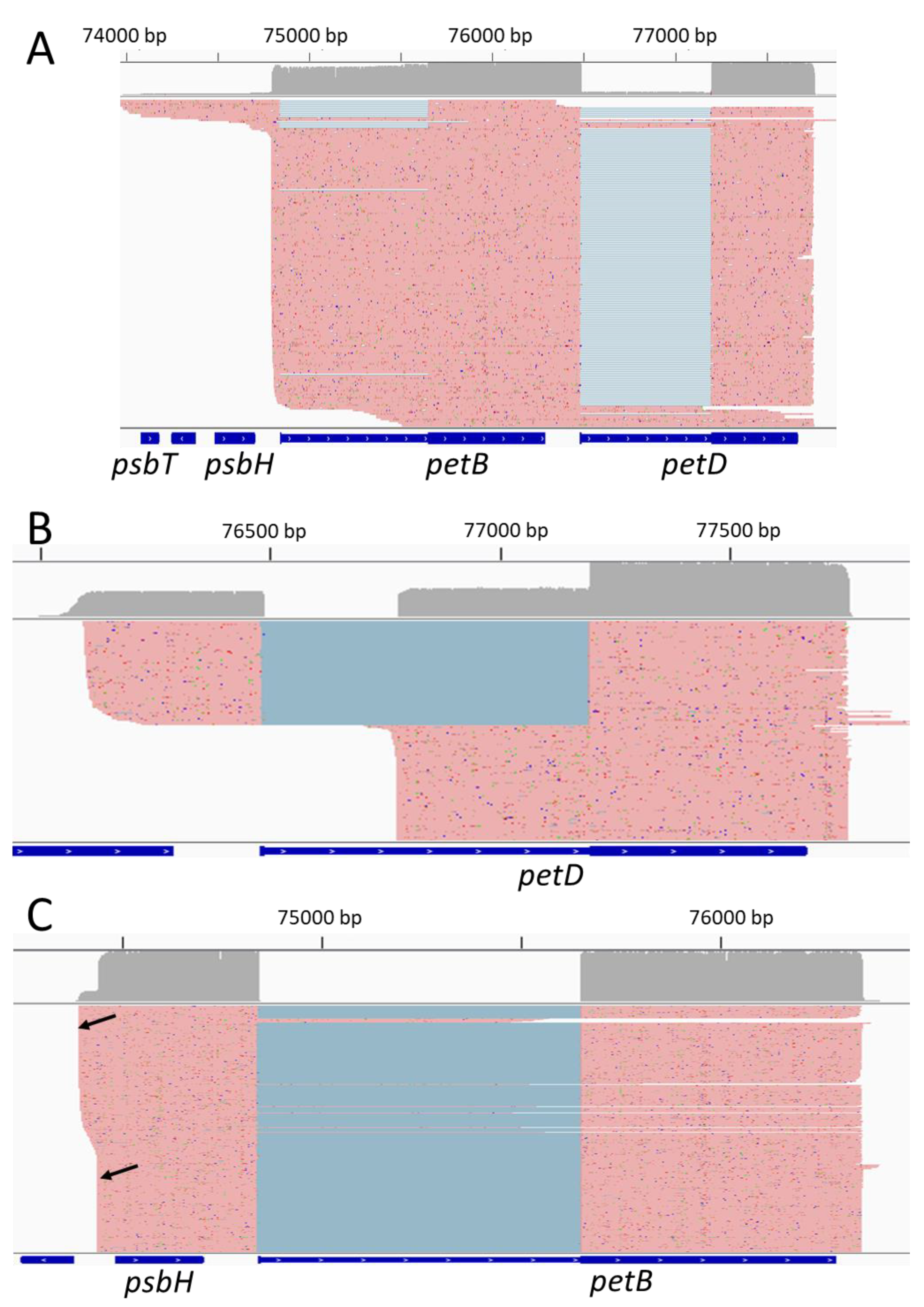
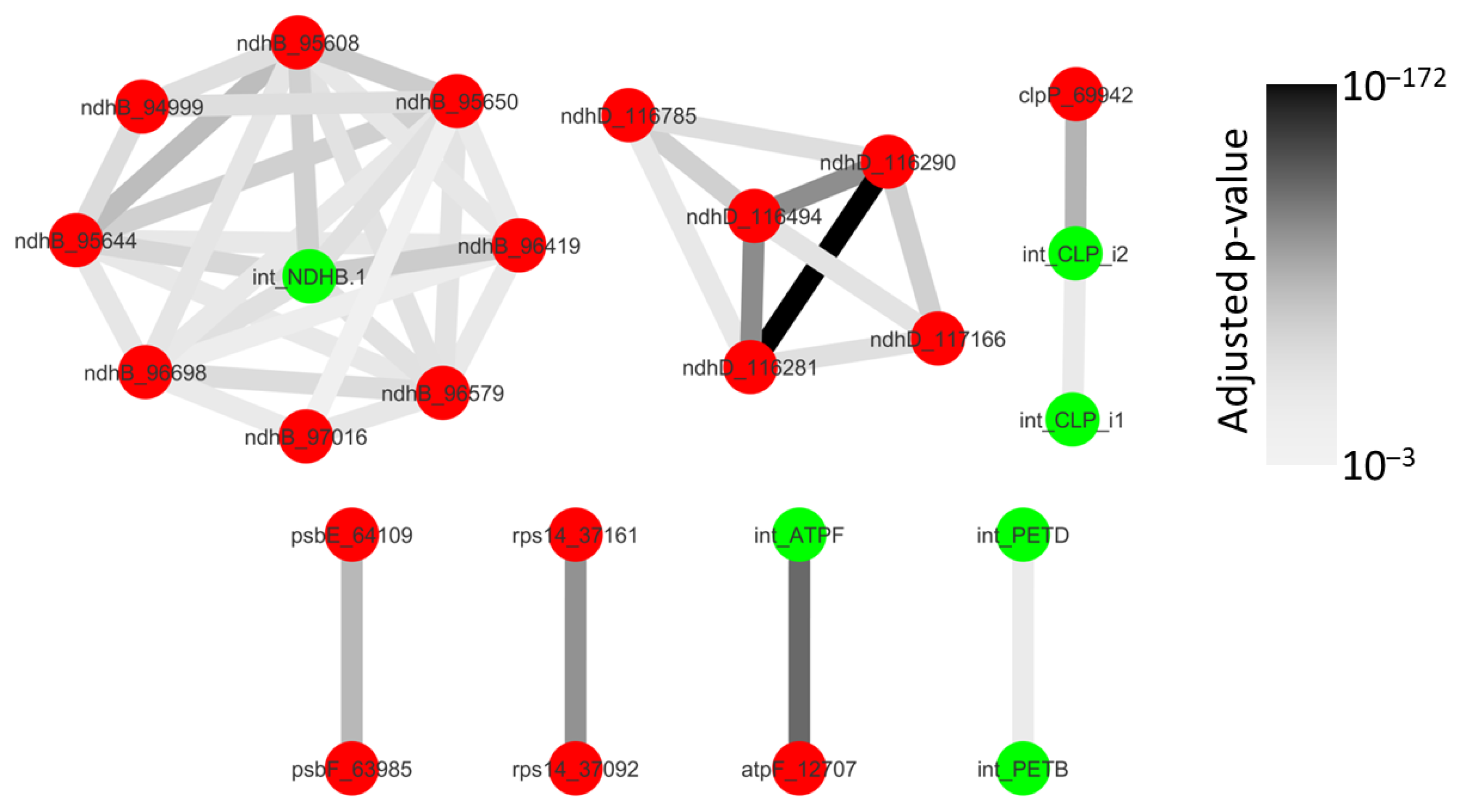
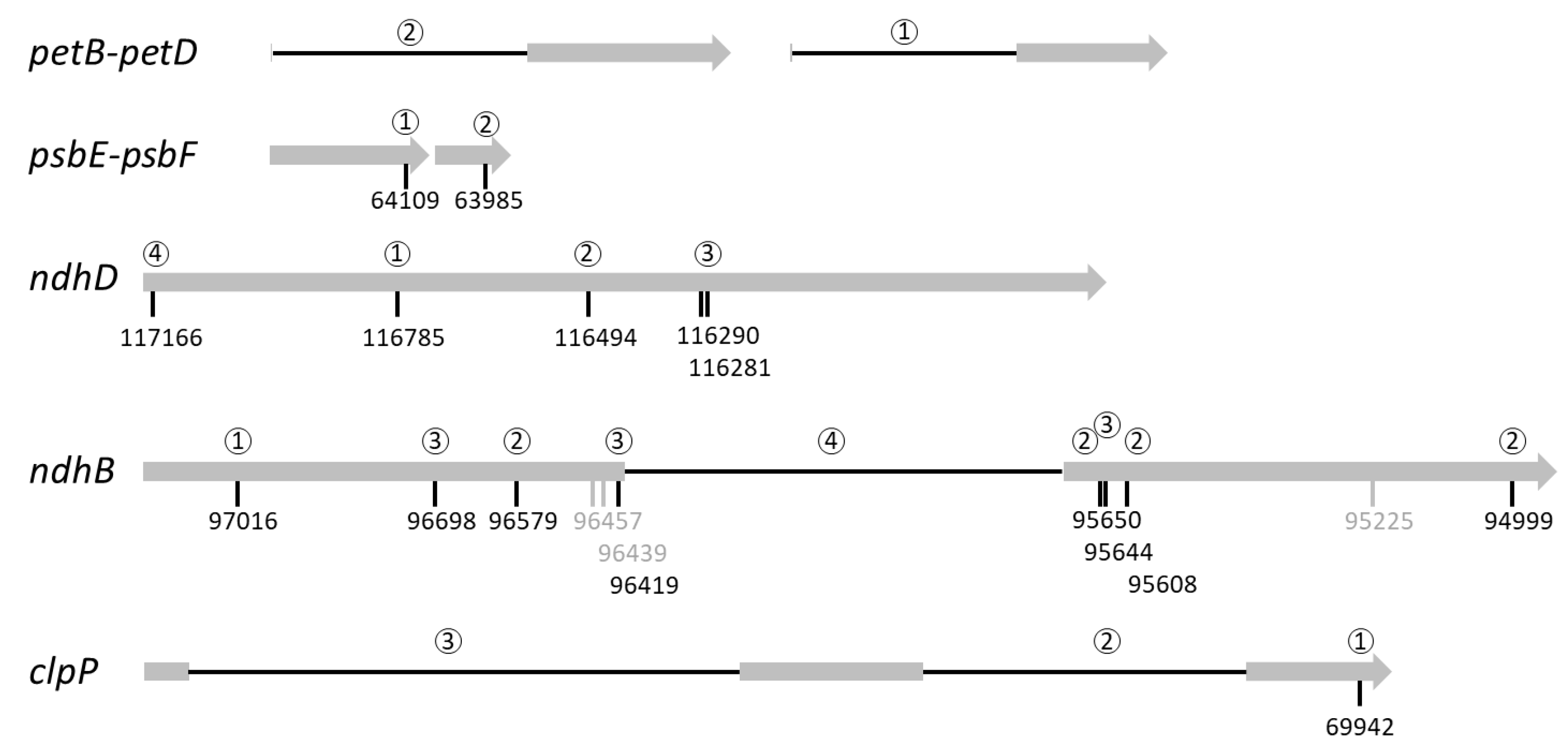
2.3. Some Post-Transcriptional Events Are Coordinated and Ordered
3. Discussion
4. Materials and Methods
4.1. Plant Growth and RNA Extraction
4.2. Nanopore Sequencing
4.3. Bioinformatics and Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Green, B.R. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Maier, U.G.; Bozarth, A.; Funk, H.T.; Zauner, S.; Rensing, S.A.; Schmitz-Linneweber, C.; Börner, T.; Tillich, M. Complex chloroplast RNA metabolism: Just debugging the genetic programme? BMC Biol. 2008, 6, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, D.B.; Goldschmidt-Clermont, M.; Hanson, M.R. Chloroplast RNA Metabolism. Annu. Rev. Plant Biol. 2010, 61, 125–155. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A. Expression of Plastid Genes: Organelle-Specific Elaborations on a Prokaryotic Scaffold. Plant Physiol. 2011, 155, 1520–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Longevialle, A.F.; Small, I.D.; Lurin, C. Nuclearly-encoded splicing factors implicated in RNA splicing in higher plant organelles. Mol. Plant 2010, 3, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Bentolila, S.; Hanson, M.R. The Unexpected Diversity of Plant Organelle RNA Editosomes. Trends Plant Sci. 2016, 21, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Germain, A.; Hotto, A.M.; Barkan, A.; Stern, D.B. RNA processing and decay in plastids. Wiley Interdiscip. Rev. RNA 2013, 4, 295–316. [Google Scholar] [CrossRef]
- MacIntosh, G.C.; Castandet, B. Organellar and Secretory Ribonucleases: Major Players in Plant RNA Homeostasis. Plant Physiol. 2020, 183, 1438. [Google Scholar] [CrossRef]
- Majeran, W.; Friso, G.; Asakura, Y.; Qu, X.; Huang, M.; Ponnala, L.; Watkins, K.P.; Barkan, A.; van Wijk, K.J. Nucleoid-enriched proteomes in developing plastids and chloroplasts from maize leaves: A new conceptual framework for nucleoid functions. Plant Physiol. 2012, 158, 156–189. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Linneweber, C.; Tillich, M.; Herrmann, R.G.; Maier, R.M. Heterologous, splicing-dependent RNA editing in chloroplasts: Allotetraploidy provides trans-factors. EMBO J. 2001, 20, 4874–4883. [Google Scholar] [CrossRef] [Green Version]
- Yap, A.; Kindgren, P.; Colas Des Francs-Small, C.; Kazama, T.; Tanz, S.K.; Toriyama, K.; Small, I. AEF1/MPR25 is implicated in RNA editing of plastid atpF and mitochondrial nad5, and also promotes atpF splicing in Arabidopsis and rice. Plant J. 2015, 81, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Ruwe, H.; Castandet, B.; Schmitz-Linneweber, C.; Stern, D.B. Arabidopsis chloroplast quantitative editotype. FEBS Lett. 2013, 587, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Marechal-Drouard, L.; Cosset, A.; Remacle, C.; Ramamonjisoa, D.; Dietrich, A. A single editing event is a prerequisite for efficient processing of potato mitochondrial phenylalanine tRNA. Mol. Cell. Biol. 1996, 16, 3504–3510. [Google Scholar] [CrossRef] [Green Version]
- Malbert, B.; Burger, M.; Lopez-Obando, M.; Baudry, K.; Launay-Avon, A.; Härtel, B.; Verbitskiy, D.; Jörg, A.; Berthomé, R.; Lurin, C.; et al. The Analysis of the Editing Defects in the dyw2 Mutant Provides New Clues for the Prediction of RNA Targets of Arabidopsis E+-Class PPR Proteins. Plants 2020, 9, 280. [Google Scholar] [CrossRef] [Green Version]
- Ichinose, M.; Sugita, C.; Yagi, Y.; Nakamura, T.; Sugita, M. Two DYW subclass PPR proteins are involved in RNA editing of ccmFc and atp9 transcripts in the moss Physcomitrella patens: First complete set of PPR editing factors in plant mitochondria. Plant Cell Physiol. 2013, 54, 1907–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schallenberg-Rüdinger, M.; Kindgren, P.; Zehrmann, A.; Small, I.; Knoop, V. A DYW-protein knockout in Physcomitrella affects two closely spaced mitochondrial editing sites and causes a severe developmental phenotype. Plant J. 2013, 76, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castandet, B.; Hotto, A.M.; Strickler, S.R.; Stern, D.B. ChloroSeq, an Optimized Chloroplast RNA-Seq Bioinformatic Pipeline, Reveals Remodeling of the Organellar Transcriptome Under Heat Stress. G3 Genes Genomes Genet. 2016, 6, 2817–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, E.J.S.S.; Hotto, A.M.; Strickler, S.R.; Stern, D.B.; Castandet, B. A Guide to the Chloroplast Transcriptome Analysis Using RNA-Seq. Methods Mol. Biol. 2018, 1829, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Malbert, B.; Rigaill, G.; Brunaud, V.; Lurin, C.; Delannoy, E. Bioinformatic Analysis of Chloroplast Gene Expression and RNA Posttranscriptional Maturations Using RNA Sequencing. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1829, pp. 279–294. [Google Scholar]
- Castandet, B.; Germain, A.; Hotto, A.M.; Stern, D.B. Systematic sequencing of chloroplast transcript termini from Arabidopsis thaliana reveals >200 transcription initiation sites and the extensive imprints of RNA-binding proteins and secondary structures. Nucleic Acids Res. 2019, 47, 11889–11905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chotewutmontri, P.; Barkan, A. Dynamics of Chloroplast Translation during Chloroplast Differentiation in Maize. PLoS Genet. 2016, 12, e1006106. [Google Scholar] [CrossRef] [Green Version]
- Ruwe, H.; Wang, G.; Gusewski, S.; Schmitz-Linneweber, C. Systematic analysis of plant mitochondrial and chloroplast small RNAs suggests organelle-specific mRNA stabilization mechanisms. Nucleic Acids Res. 2016, 44, 7406–7417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhelyazkova, P.; Sharma, C.M.; Forstner, K.U.; Liere, K.; Vogel, J.; Borner, T. The Primary Transcriptome of Barley Chloroplasts: Numerous Noncoding RNAs and the Dominating Role of the Plastid-Encoded RNA Polymerase. Plant Cell 2012, 24, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shen, N.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 2020, 16, 85. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Jia, J.; Mo, W.; Jin, X.; Zhai, J. FLEP-seq: Simultaneous detection of RNA polymerase II position, splicing status, polyadenylation site and poly(A) tail length at genome-wide scale by single-molecule nascent RNA sequencing. Nat. Protoc. 2021, 16, 4355–4381. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Long, Y.; Zhang, H.; Li, Z.; Liu, Z.; Zhao, Y.; Lu, D.; Jin, X.; Deng, X.; Xia, R.; et al. Post-transcriptional splicing of nascent RNA contributes to widespread intron retention in plants. Nat. Plants 2020, 6, 780–788. [Google Scholar] [CrossRef]
- Zhu, Y.; Machleder, E.; Chenchik, A.; Li, R.; Siebert, P. Reverse transcriptase template switching: A SMART approach for full-length cDNA library construction. Biotechniques 2001, 30, 892–897. [Google Scholar] [CrossRef] [Green Version]
- Schuster, G.; Stern, D. RNA Polyadenylation and Decay in Mitochondria and Chloroplasts. Prog. Mol. Biol. Transl. Sci. 2009, 85, 393–422. [Google Scholar] [CrossRef]
- Hotto, A.M.; Castandet, B.; Gilet, L.; Higdon, A.; Condon, C.; Stern, D.B. Arabidopsis Chloroplast Mini-Ribonuclease III Participates in rRNA Maturation and Intron Recycling. Plant Cell 2015, 27, 724–740. [Google Scholar] [CrossRef]
- Felder, S.; Meierhoff, K.; Sane, A.P.; Meurer, J.; Driemel, C.; Plücken, H.; Klaff, P.; Stein, B.; Bechtold, N.; Westhoff, P. The Nucleus-Encoded HCF107 Gene of Arabidopsis Provides a Link between Intercistronic RNA Processing and the Accumulation of Translation-Competent psbH Transcripts in Chloroplasts. Plant Cell 2001, 13, 2127. [Google Scholar] [CrossRef] [Green Version]
- Stoppel, R.; Meurer, J. Complex RNA metabolism in the chloroplast: An update on the psbB operon. Planta 2013, 237, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Lezhneva, L.; Meurer, J. The nuclear factor HCF145 affects chloroplast psaA-psaB-rps14 transcript abundance in Arabidopsis thaliana. Plant J. 2004, 38, 740–753. [Google Scholar] [CrossRef]
- Guillaumot, D.; Lopez-Obando, M.; Baudry, K.; Avon, A.; Rigaill, G.; Falcon De Longevialle, A.; Broche, B.; Takenaka, M.; Berthomé, R.; De Jaeger, G.; et al. Two interacting PPR proteins are major Arabidopsis editing factors in plastid and mitochondria. Proc. Natl. Acad. Sci. USA 2017, 114, 8877–8882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüdinger, M.; Funk, H.T.; Rensing, S.A.; Maier, U.G.; Knoop, V. RNA editing: Only eleven sites are present in the Physcomitrella patens mitochondrial transcriptome and a universal nomenclature proposal. Mol. Genet. Genomics 2009, 281, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, F.; Fuchs, R.T.; Sun, Z.; Zheng, Y.; Robb, G.B. Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Res. 2012, 40, e54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyer, R.; Hoch, B.; Neckermann, K.; Maier, R.M.; Kössel, H. RNA editing in maize chloroplasts is a processing step independent of splicing and cleavage to monocistronic mRNAs. Plant J. 1993, 4, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Zeltz, P.; Kössel, H. Complete RNA editing of unspliced and dicistronic transcripts of the intron-containing reading frame IRF170 from maize chloroplasts. Proc. Natl. Acad. Sci. USA 1994, 91, 2295–2299. [Google Scholar] [CrossRef] [Green Version]
- Maréchal-Drouard, L.; Kumar, R.; Remacle, C.; Small, I. RNA editing of larch mitochondrial tRNA His precursors is a prerequisite for processing. Nucleic Acids Res. 1996, 24, 3229–3234. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Hardel, S.L.; Kupsch, C.; Armbruster, U.; Delannoy, E.; Gualberto, J.M.; Lehwark, P.; Leister, D.; Small, I.D.; Schmitz-Linneweber, C. Chloroplast ribonucleoprotein CP31A is required for editing and stability of specific chloroplast mRNAs. Proc. Natl. Acad. Sci. USA 2009, 106, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Karcher, D.; Bock, R. Site-selective inhibition of plastid RNA editing by heat shock and antibiotics: A role for plastid translation in RNA editing. Nucleic Acids Res. 1998, 26, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, M.; Neuwirt, J.; Brennicke, A. Complex cis-elements determine an RNA editing site in pea mitochondria. Nucleic Acids Res. 2004, 32, 4137–4144. [Google Scholar] [CrossRef] [Green Version]
- Castandet, B.; Choury, D.; Bégu, D.; Jordana, X.; Araya, A. Intron RNA editing is essential for splicing in plant mitochondria. Nucleic Acids Res. 2010, 38, 7112–7121. [Google Scholar] [CrossRef] [Green Version]
- Farré, J.-C.; Aknin, C.; Araya, A.; Castandet, B. RNA Editing in Mitochondrial Trans-Introns Is Required for Splicing. PLoS ONE 2012, 7, e52644. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.; Börner, T. Lariat formation and a hydrolytic pathway in plant chloroplast group II intron splicing. EMBO J. 2002, 21, 3794–3803. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.; Schöttler, M.A.; Karcher, D.; Thiele, W.; Bock, R. Elimination of a group II intron from a plastid gene causes a mutant phenotype. Nucleic Acids Res. 2011, 39, 5181–5192. [Google Scholar] [CrossRef]
- Verbitskiy, D.; Takenaka, M.; Neuwirt, J.; Van Der Merwe, J.A.; Brennicke, A. Partially edited RNAs are intermediates of RNA editing in plant mitochondria. Plant J. 2006, 47. [Google Scholar] [CrossRef]
- Castandet, B.; Araya, A. The RNA editing pattern of cox2 mRNA is affected by point mutations in plant mitochondria. PLoS ONE 2011, 6, e20867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudinger, M.; Bolle, N.; Kempken, F. Mitochondrial electroporation and in organello RNA editing of chimeric atp6 transcripts. Mol. Genet. Genom. 2005, 273, 130–136. [Google Scholar] [CrossRef]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 2019, 101, 1040–1056. [Google Scholar] [CrossRef] [PubMed]
- Planchard, N.; Bertin, P.; Quadrado, M.; Dargel-Graffin, C.; Hatin, I.; Namy, O.; Mireau, H. The translational landscape of Arabidopsis mitochondria. Nucleic Acids Res. 2018, 46, 6218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkan, A. Proteins encoded by a complex chloroplast transcription unit are each translated from both monocistronic and polycistronic mRNAs. EMBO J. 1988, 7, 2637–2644. [Google Scholar] [CrossRef]
- Li, M.; Xia, L.; Zhang, Y.; Niu, G.; Li, M.; Wang, P.; Zhang, Y.; Sang, J.; Zou, D.; Hu, S.; et al. Plant editosome database: A curated database of RNA editosome in plants. Nucleic Acids Res. 2019, 47, D170. [Google Scholar] [CrossRef] [Green Version]
- Anreiter, I.; Mir, Q.; Simpson, J.T.; Janga, S.C.; Soller, M. New Twists in Detecting mRNA Modification Dynamics. Trends Biotechnol. 2021, 39, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Manavski, N.; Vicente, A.; Chi, W.; Meurer, J. The chloroplast epitranscriptome: Factors, sites, regulation, and detection methods. Genes 2021, 39, 72–89. [Google Scholar]
- Kadumuri, R.V.; Janga, S.C. Epitranscriptomic Code and Its Alterations in Human Disease. Trends Mol. Med. 2018, 24, 886–903. [Google Scholar] [CrossRef] [PubMed]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Wang, L.; Nie, J.; Sicotte, H.; Li, Y.; Eckel-Passow, J.E.; Dasari, S.; Vedell, P.T.; Barman, P.; Wang, L.; Weinshiboum, R.; et al. Measure transcript integrity using RNA-seq data. BMC Bioinform. 2016, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling The False Discovery Rate—A Practical And Powerful Approach To Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup, 1000 Genome Project Data Processing The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Maturation Rate | Maturation Rate (Guillaumot et al., 2017) | Maturation Rate (Ruwe et al., 2013) |
|---|---|---|---|---|
| int_RPS16 | splicing | 4% | 4% | NA |
| int_ATPF | splicing | 89% | 82% | NA |
| int_RPOC1 | splicing | 64% | 19% | NA |
| int_YCF3_i2 | splicing | 79% | 42% | NA |
| int_YCF3_i1 | splicing | 63% | 45% | NA |
| int_CLP_i2 | splicing | 60% | 71% | NA |
| int_CLP_i1 | splicing | 69% | 62% | NA |
| int_PETB | splicing | 91% | 58% | NA |
| int_PETD | splicing | 97% | 62% | NA |
| int_RPL16 | splicing | 69% | 12% | NA |
| int_RPL2.1 | splicing | 66% | 52% | NA |
| int_NDHB.1 | splicing | 68% | 55% | NA |
| int_RPS12C | splicing | 92% | 81% | NA |
| int_NDHA | splicing | 68% | 27% | NA |
| matK_2931 | editing | 53% | 79% | 93% |
| atpF_12707 | editing | 89% | 91% | 95% |
| atpH_UTR_13210 | editing | 5% | 3% | 4% |
| rpoC1_21806 | editing | 33% | 21% | 15% |
| rpoB_23898 | editing | 87% | 82% | 85% |
| rpoB_25779 | editing | 64% | 83% | 86% |
| rpoB_25992 | editing | 69% | 76% | 94% |
| psbZ_35800 | editing | 93% | 90% | 95% |
| rps14_37092 | editing | 89% | 93% | 94% |
| rps14_37161 | editing | 92% | 97% | 96% |
| ycf3_i2_43350 | editing | 16% | 10% | 12% |
| rps4_UTR_45095 | editing | 6% | 3% | 10% |
| ndhK_ndhJ_49209 | editing | 4% | 4% | 6% |
| accD_57868 | editing | 90% | 95% | 99% |
| accD_58642 | editing | 76% | 75% | 83% |
| psbF_63985 | editing | 90% | 98% | 98% |
| psbE_64109 | editing | 95% | 100% | 100% |
| petL_65716 | editing | 79% | 91% | 86% |
| rps18_UTR_68453 | editing | 3% | 4% | NA |
| rps12_69553 | editing | 21% | 26% | 27% |
| clpP_69942 | editing | 82% | 72% | 81% |
| rpoA_78691 | editing | 78% | 76% | 91% |
| rpl23_86055 | editing | 34% | 74% | 75% |
| ycf2_as_91535 | editing | 3% | 4% | NA |
| ndhB_UTR_94622 | editing | 8% | 0% | NA |
| ndhB_94999 | editing | 88% | 93% | 94% |
| ndhB_95225 | editing | 95% | 98% | 99% |
| ndhB_95608 | editing | 87% | 84% | 80% |
| ndhB_95644 | editing | 78% | 87% | 81% |
| ndhB_95650 | editing | 88% | 91% | 84% |
| ndhB_96419 | editing | 75% | 94% | 92% |
| ndhB_96439 | editing | 6% | 4% | 6% |
| ndhB_96457 | editing | 6% | 3% | 5% |
| ndhB_96579 | editing | 90% | 89% | 90% |
| ndhB_96698 | editing | 81% | 88% | 82% |
| ndhB_97016 | editing | 94% | 94% | 95% |
| ndhF_112349 | editing | 85% | 93% | 96% |
| ndhD_116281 | editing | 76% | 83% | 92% |
| ndhD_116290 | editing | 77% | 84% | 90% |
| ndhD_116494 | editing | 88% | 90% | 93% |
| ndhD_116785 | editing | 94% | 97% | 98% |
| ndhD_117166 | editing | 35% | 33% | 45% |
| ndhG_118858 | editing | 69% | 78% | 85% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guilcher, M.; Liehrmann, A.; Seyman, C.; Blein, T.; Rigaill, G.; Castandet, B.; Delannoy, E. Full Length Transcriptome Highlights the Coordination of Plastid Transcript Processing. Int. J. Mol. Sci. 2021, 22, 11297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011297
Guilcher M, Liehrmann A, Seyman C, Blein T, Rigaill G, Castandet B, Delannoy E. Full Length Transcriptome Highlights the Coordination of Plastid Transcript Processing. International Journal of Molecular Sciences. 2021; 22(20):11297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011297
Chicago/Turabian StyleGuilcher, Marine, Arnaud Liehrmann, Chloé Seyman, Thomas Blein, Guillem Rigaill, Benoit Castandet, and Etienne Delannoy. 2021. "Full Length Transcriptome Highlights the Coordination of Plastid Transcript Processing" International Journal of Molecular Sciences 22, no. 20: 11297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011297