
CC16 Regulates Inflammation, ROS Generation and Apoptosis in Bronchial Epithelial Cells during Klebsiella pneumoniae Infection

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
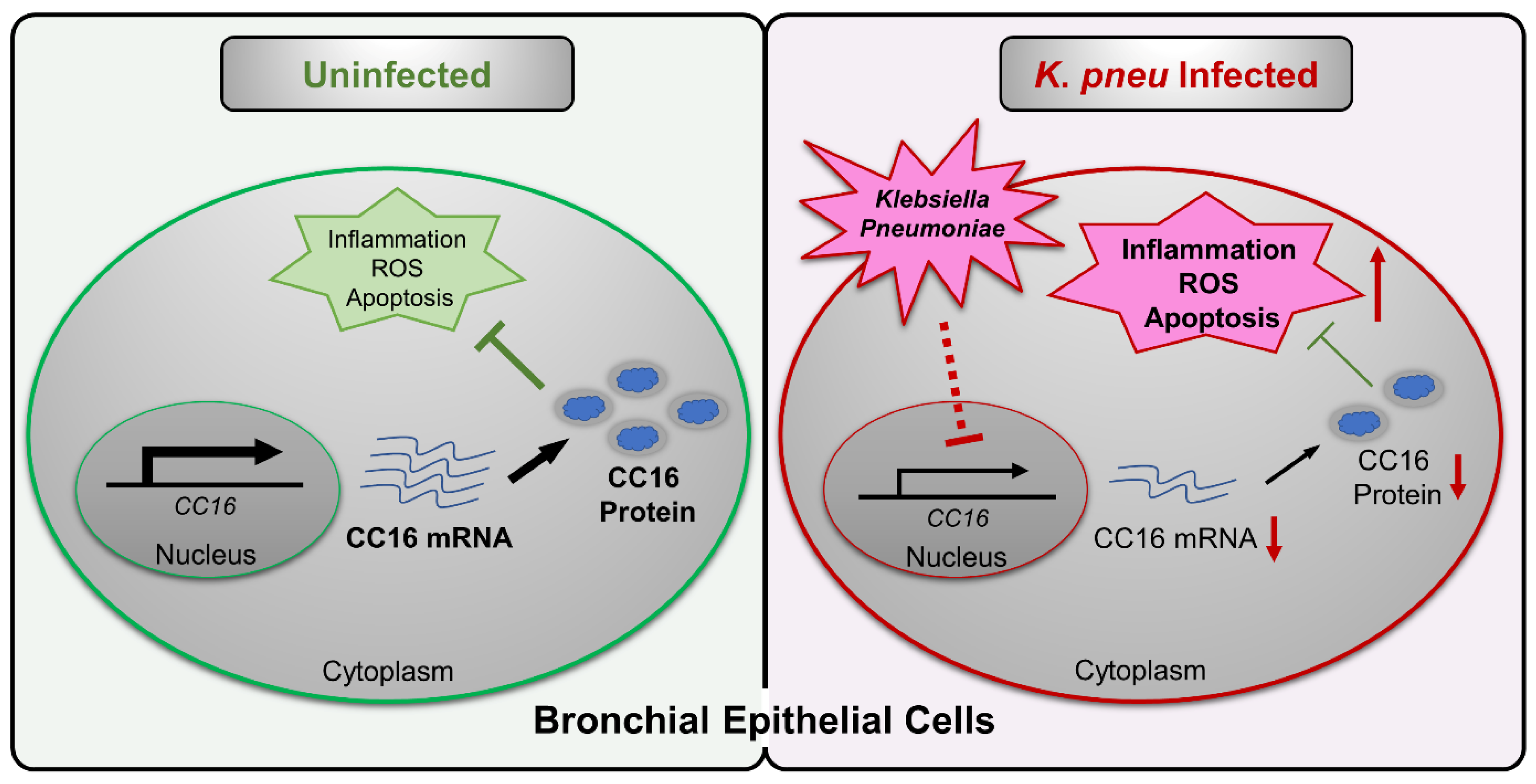
Abstract
:1. Introduction
2. Results
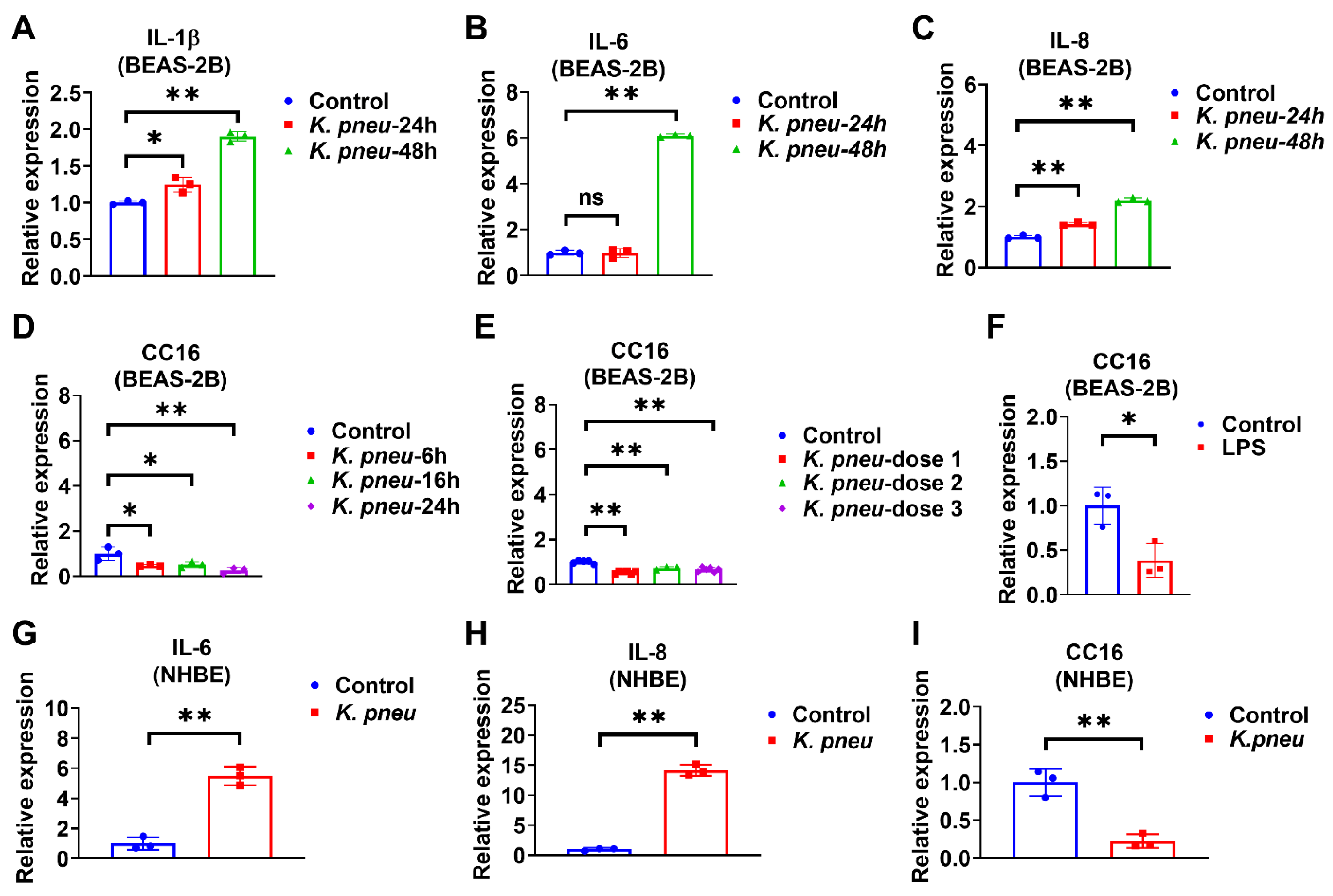
2.1. K. pneu Infection Decreases CC16 Expression in Bronchial Epithelial Cells and Increases Proinflammatory Cytokines
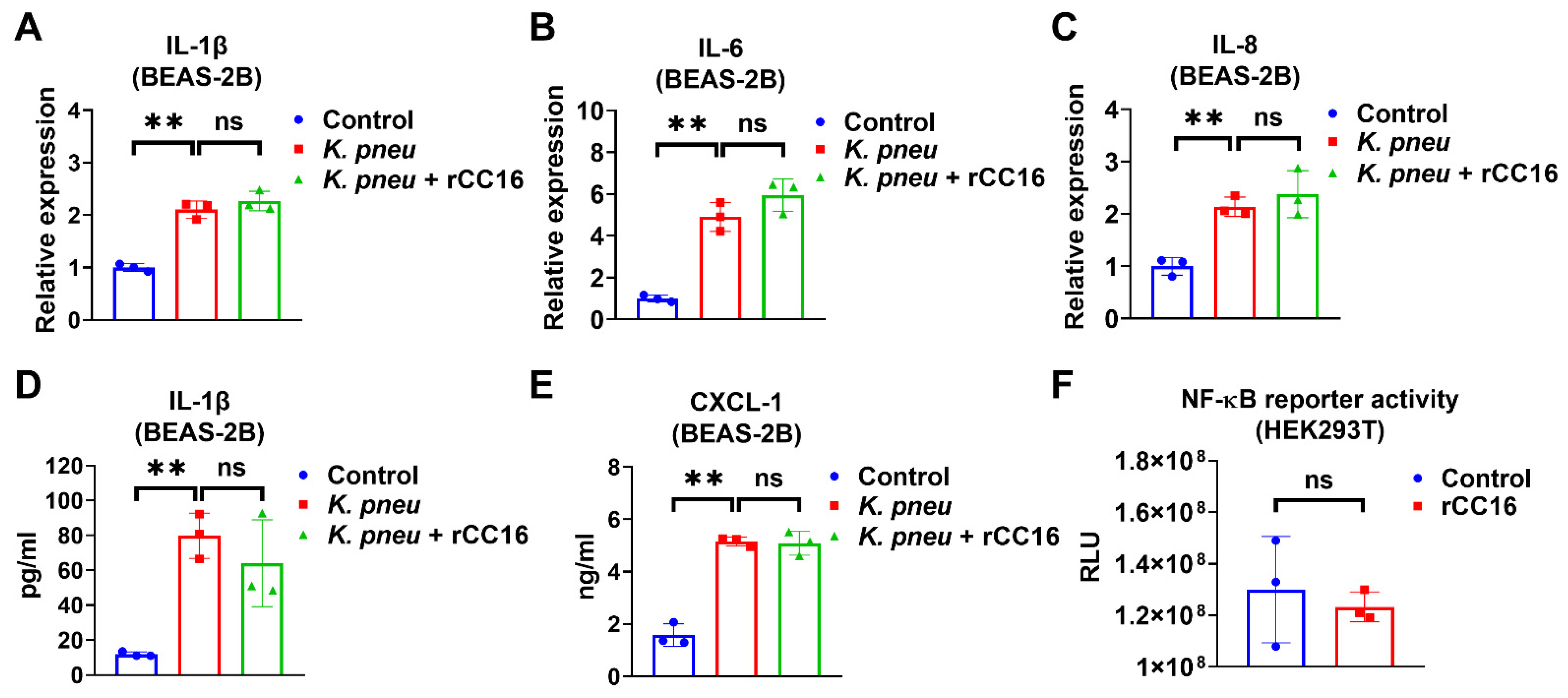
2.2. rCC16 Protein Does Not Reduce K. pneu-Induced Inflammation
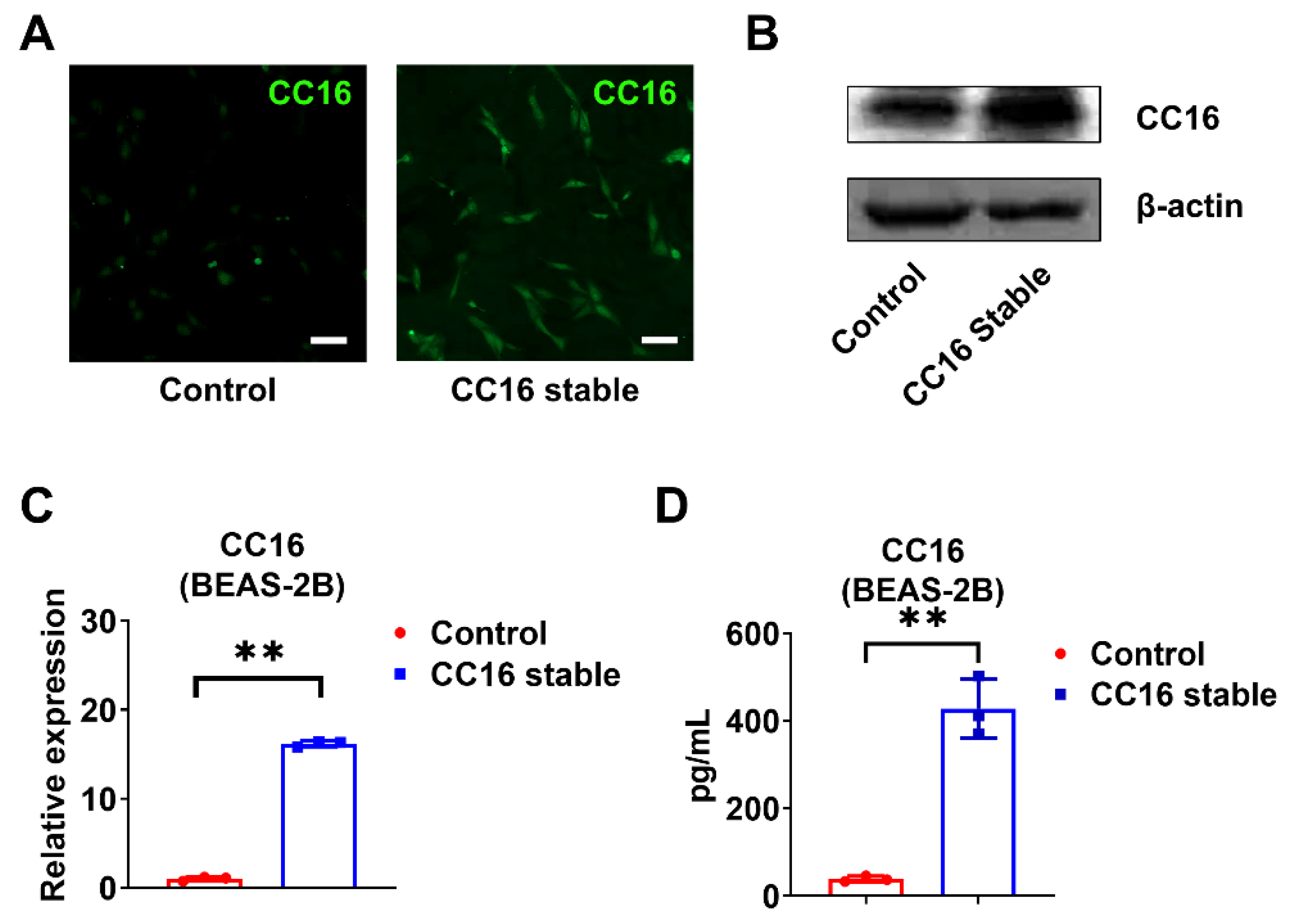
2.3. Development of Stable BEAS-2B Cells Overexpressing CC16
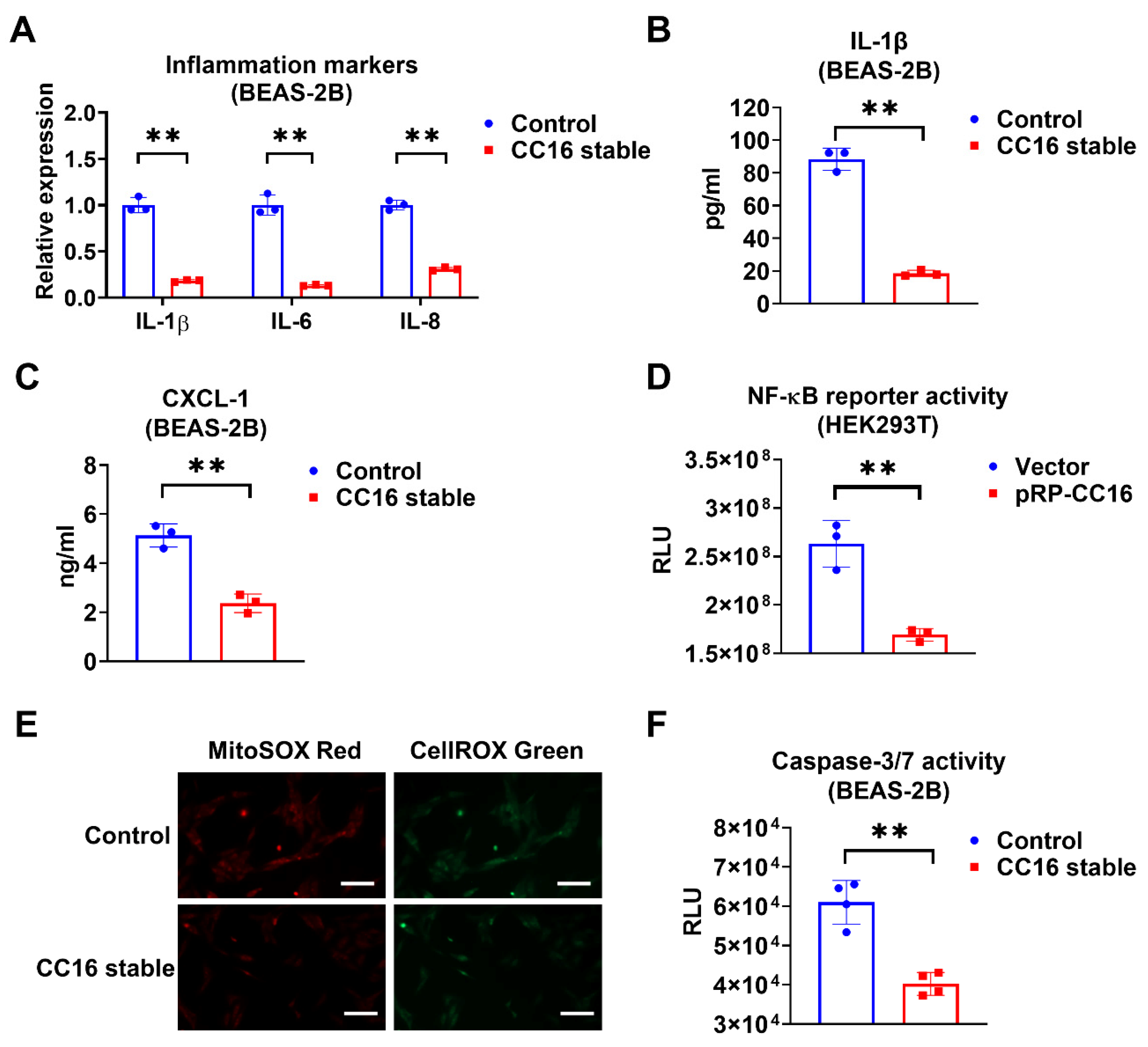
2.4. Overexpression of CC16 Decreases Inflammation, ROS, and Apoptosis during K. pneu Infection
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Isolation and Reverse Transcriptase-Polymerase Chain Reaction
4.3. Western Blot and ELISA
4.4. Bacterial Count and In Vitro Bacterial Infection
4.5. NF-κB Reporter Assay
4.6. Stable Cell Generation
4.7. Immunofluorescence Staining
4.8. Measurement of Cellular and Mitochondrial ROS Production
4.9. Determination of Caspase-3/7 Activities
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sattar, S.B.A.; Sharma, S. Bacterial Pneumonia. In StatPearls; Treasure Island (FL): Las Vegas, NV, USA, 2021. [Google Scholar]
- Olasupo, O.; Xiao, H.; Brown, J.D. Relative Clinical and Cost Burden of Community-Acquired Pneumonia Hospitalizations in Older Adults in the United States—A Cross-Sectional Analysis. Vaccines 2018, 6, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassetti, M.; Welte, T.; Wunderink, R. Treatment of Gram-negative pneumonia in the critical care setting: Is the beta-lactam antibiotic backbone broken beyond repair? Crit. Care 2016, 20, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.; Reygaert, W.C. Gram Negative Bacteria. In StatPearls; Treasure Island (FL): Las Vegas, NV, USA, 2021. [Google Scholar]
- Prasad, K.N.; Mishra, A.M.; Gupta, D.; Husain, N.; Husain, M.; Gupta, R.K. Analysis of microbial etiology and mortality in patients with brain abscess. J. Infect. 2006, 53, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Broad, J.; Le Doare, K.; Heath, P.T.; Hallchurch, P.; Whelan, I.; Boyd, H.; Carruthers, E.; Sharland, M.; Ladhani, S. The current state of immunization against Gram-negative bacteria in children: A review of the literature. Curr. Opin. Infect. Dis. 2020, 33, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Bachman, M.A. Colonization, Infection, and the Accessory Genome of Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.N. Microbial Etiologies of Hospital-Acquired Bacterial Pneumonia and Ventilator-Associated Bacterial Pneumonia. Clin. Infect. Dis. 2010, 51 (Suppl. 1), S81–S87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navon-Venezia, S.; Kondratyeva, K.; Carattoli, A. Klebsiella pneumoniae: A major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol. Rev. 2017, 41, 252–275. [Google Scholar] [CrossRef] [PubMed]
- Klug, J.; Beier, H.M.; Bernard, A.; Chilton, B.S.; Fleming, T.P.; Lehrer, R.I.; Miele, L.; Pattabiraman, N.; Singh, G. Uteroglobin/Clara cell 10-kDa family of proteins: Nomenclature committee report. Ann. N. Y. Acad. Sci. 2000, 923, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Broeckaert, F.; Bernard, A. Clara cell secretory protein (CC16): Characteristics and perspectives as lung peripheral biomarker. Clin. Exp. Allergy 2000, 30, 469–475. [Google Scholar] [CrossRef] [Green Version]
- Park, H.Y.; Churg, A.; Wright, J.L.; Li, Y.; Tam, S.; Man, S.F.P.; Tashkin, D.; Wise, R.; Connett, J.E.; Sin, D.D. Club Cell Protein 16 and Disease Progression in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2013, 188, 1413–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, S.; Halonen, M.; Vasquez, M.M.; Spangenberg, A.; Stern, D.A.; Morgan, W.J.; Wright, A.L.; Lavi, I.; Tarès, L.; Carsin, A.-E.; et al. Relation between circulating CC16 concentrations, lung function, and development of chronic obstructive pulmonary disease across the lifespan: A prospective study. Lancet Respir. Med. 2015, 3, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Lomas, D.A.; Silverman, E.K.; Edwards, L.D.; Miller, B.E.; Coxson, H.O.; Tal-Singer, R. On behalf of the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) investigators Evaluation of serum CC-16 as a biomarker for COPD in the ECLIPSE cohort. Thorax 2008, 63, 1058–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laing, I.A.; Hermans, C.; Bernard, A.; Burton, P.R.; Goldblatt, J.; Le Souëf, P.N. Association between Plasma CC16 Levels, the A38G Polymorphism, and Asthma. Am. J. Respir. Crit. Care Med. 2000, 161, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Lee, Y.S.; Min, K.H.; Hur, G.Y.; Lee, S.Y.; Kang, K.H.; Rhee, C.K.; Park, S.J.; Shim, J.J. Decreased serum club cell secretory protein in asthma and chronic obstructive pulmonary disease overlap: A pilot study. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3411–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almuntashiri, S.; Zhu, Y.; Han, Y.; Wang, X.; Somanath, P.R.; Zhang, D. Club Cell Secreted Protein CC16: Potential Applications in Prognosis and Therapy for Pulmonary Diseases. J. Clin. Med. 2020, 9, 4039. [Google Scholar] [CrossRef] [PubMed]
- Lesur, O.; Critical Care Research Group of the Québec Respiratory Health Network; Langevin, S.; Berthiaume, Y.; Légaré, M.; Skrobik, Y.; Bellemare, J.-F.; Lévy, B.; Fortier, Y.; Lauzier, F.; et al. Outcome value of Clara cell protein in serum of patients with acute respiratory distress syndrome. Intensiv. Care Med. 2006, 32, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, W.; Wang, L.; Tian, F. Diagnostic and prognostic values of Club cell protein 16 (CC16) in critical care patients with acute respiratory distress syndrome. J. Clin. Lab. Anal. 2018, 32, e22262. [Google Scholar] [CrossRef] [Green Version]
- Buendía-Roldán, I.; Ruiz, V.; Sierra, P.; Montes, E.; Ramírez, R.; Vega, A.; Salgado, A.; Vargas, M.H.; Mejía, M.; Pardo, A.; et al. Increased Expression of CC16 in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0168552. [Google Scholar] [CrossRef] [PubMed]
- Kokuho, N.; Ishii, T.; Kamio, K.; Hayashi, H.; Kurahara, M.; Hattori, K.; Motegi, T.; Azuma, A.; Gemma, A.; Kida, K. Diagnostic Values For Club Cell Secretory Protein (CC16) in Serum of Patients of Combined Pulmonary Fibrosis and Emphysema. COPD 2015, 12, 347–354. [Google Scholar] [CrossRef]
- Hermans, C.; Petrek, M.; Kolek, V.; Weynand, B.; Pieters, T.; Lambert, M.; Bernard, A. Serum Clara cell protein (CC16), a marker of the integrity of the air-blood barrier in sarcoidosis. Eur. Respir. J. 2001, 18, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, S.; Kristjansson, S.; Bjarnarson, S.P.; Wennergren, G.; Rudin, A. Clara cell protein 16 (CC16) serum levels in infants during respiratory syncytial virus infection. Acta Paediatr. 2009, 98, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Liu, H.; Li, T.; Wang, D.; Hu, X.; Zhang, X.; Yu, B.; Guo, R.; Wang, H. Recombinant club cell protein 16 (CC16) ameliorates cigarette smoke-induced lung inflammation in a murine disease model of COPD. Mol. Med. Rep. 2018, 18, 2198–2206. [Google Scholar] [CrossRef] [PubMed]
- Gamez, A.S.; Gras, D.; Petit, A.; Knabe, L.; Molinari, N.; Vachier, I.; Chanez, P.; Bourdin, A. Supplementing Defect in Club Cell Secretory Protein Attenuates Airway Inflammation in COPD. Chest 2015, 147, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Zhang, Z.; Mukherjee, A.B. Mice lacking uteroglobin are highly susceptible to developing pulmonary fibrosis. FEBS Lett. 2006, 580, 4515–4520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, C.R.; Gewolb, I.H.; Allen, K.; Welch, R.W.; Melby, J.M.; Pollack, S.; Shaffer, T.H.; Pilon, A.L.; Davis, J.M. Safety, Pharmacokinetics, and Anti-inflammatory Effects of Intratracheal Recombinant Human Clara Cell Protein in Premature Infants with Respiratory Distress Syndrome. Pediatr. Res. 2005, 58, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, C.M.; Roan, E.; Navajas, D. Mechanobiology in Lung Epithelial Cells: Measurements, Perturbations, and Responses. Compr. Physiol. 2012, 2, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadnyk, A.W. Cytokine production by epithelial cells. FASEB J. 1994, 8, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xu, R.; Zhang, H.; Peng, Z.; Feng, M.; Yu, B.; Wang, Y.; Shi, T.; Zhou, Y.; Liu, Y. Exogenous Clara cell protein 16 attenuates silica particles-induced inflammation in THP-1 macrophages by down-regulating NF-κB and caspase-1 activation. J. Toxicol. Sci. 2020, 45, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Yuan, Y.; Wang, D.; Li, T.; Wang, D.; Shi, X.; Guo, M.; Wang, C.; Zhang, X.; Zheng, G.; et al. Recombinant CC16 protein inhibits the production of pro-inflammatory cytokines via NF-κB and p38 MAPK pathways in LPS-activated RAW264.7 macrophages. Acta Biochim. Biophys. Sin. 2017, 49, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta–Bioenerg. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Athanazio, R. Airway disease: Similarities and differences between asthma, COPD and bronchiectasis. Clinics 2012, 67, 1335–1343. [Google Scholar] [CrossRef]
- Rokicki, W.; Rokicki, M.; Wojtacha, J.; Dżeljijli, A. The role and importance of club cells (Clara cells) in the pathogenesis of some respiratory diseases. Pol. J. Cardio-Thorac. Surg. 2016, 1, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, T.; De Andrade, J.; Zhou, Y.; Luckhardt, T.; Thannickal, V.J. Alveolar epithelial disintegrity in pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L185–L191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughman, R.P.; Culver, D.A.; Judson, M. A Concise Review of Pulmonary Sarcoidosis. Am. J. Respir. Crit. Care Med. 2011, 183, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, J.N.; Lucas, R.; Verin, A.D. The Acute Respiratory Distress Syndrome: Mechanisms and Perspective Therapeutic Approaches. Austin J. Vasc. Med. 2015, 2, 2. [Google Scholar]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, Y.; Takemura, T.; Ferrans, V.J. Evolution of metaplastic squamous cells of alveolar walls in pulmonary fibrosis produced by paraquat. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1989, 58, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, J.; Soundararajan, R.; Leung, J.; Cox, R.; Mahendrasah, S.; Muthavarapu, N.; Herrin, T.; Czachor, A.; Tan, L.C.; Hosseinian, N.; et al. The role of club cell phenoconversion and migration in idiopathic pulmonary fibrosis. Aging 2016, 8, 3091–3109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellum, J.A.; Kong, L.; Fink, M.P.; Weissfeld, L.A.; Yealy, D.M.; Pinsky, M.R.; Fine, J.; Krichevsky, A.; Delude, R.L.; Angus, D.C. Understanding the Inflammatory Cytokine Response in Pneumonia and Sepsis: Results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch. Intern. Med. 2007, 167, 1655–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puren, A.J.; Feldman, C.; Savage, N.; Becker, P.J.; Smith, C. Patterns of Cytokine Expression in Community-Acquired Pneumonia. Chest 1995, 107, 1342–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calbo, E.; Alsina, M.; Rodríguez-Carballeira, M.; Lite, J.; Garau, J. Systemic Expression of Cytokine Production in Patients with Severe Pneumococcal Pneumonia: Effects of Treatment with a β-Lactam versus a Fluoroquinolone. Antimicrob. Agents Chemother. 2008, 52, 2395–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glynn, P.; Coakley, R.; Kilgallen, I.; Murphy, N.; O’Neill, S. Circulating interleukin 6 and interleukin 10 in community acquired pneumonia. Thorax 1999, 54, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordon, J.; Aliberti, S.; Fernandez-Botran, R.; Uriarte, S.M.; Rane, M.J.; Duvvuri, P.; Peyrani, P.; Morlacchi, L.C.; Blasi, F.; Ramirez, J. Understanding the roles of cytokines and neutrophil activity and neutrophil apoptosis in the protective versus deleterious inflammatory response in pneumonia. Int. J. Infect. Dis. 2013, 17, e76–e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.-A.; Kim, D.-H. Klebsiella pneumoniae increases the risk of inflammation and colitis in a murine model of intestinal bowel disease. Scand. J. Gastroenterol. 2011, 46, 684–693. [Google Scholar] [CrossRef]
- Shashikant, B.N.; Miller, T.L.; Welch, R.W.; Pilon, A.L.; Shaffer, T.H.; Wolfson, M.R. Dose response to rhCC10-augmented surfactant therapy in a lamb model of infant respiratory distress syndrome: Physiological, inflammatory, and kinetic profiles. J. Appl. Physiol. 2005, 99, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, J.; Shu, M.; Wu, W.; Zhang, W.; Dou, Q.; Wu, J.; Zeng, X. The rCC16 Protein Protects Against LPS-Induced Cell Apoptosis and Inflammatory Responses in Human Lung Pneumocytes. Front. Pharmacol. 2020, 11, 1060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lee, H.; Wang, X.; Groot, M.; Sharma, L.; Cruz, C.S.D.; Jin, Y. A potential role of microvesicle-containing miR-223/142 in lung inflammation. Thorax 2019, 74, 865–874. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almuntashiri, S.; Han, Y.; Zhu, Y.; Dutta, S.; Niazi, S.; Wang, X.; Siddiqui, B.; Zhang, D. CC16 Regulates Inflammation, ROS Generation and Apoptosis in Bronchial Epithelial Cells during Klebsiella pneumoniae Infection. Int. J. Mol. Sci. 2021, 22, 11459. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111459
Almuntashiri S, Han Y, Zhu Y, Dutta S, Niazi S, Wang X, Siddiqui B, Zhang D. CC16 Regulates Inflammation, ROS Generation and Apoptosis in Bronchial Epithelial Cells during Klebsiella pneumoniae Infection. International Journal of Molecular Sciences. 2021; 22(21):11459. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111459
Chicago/Turabian StyleAlmuntashiri, Sultan, Yohan Han, Yin Zhu, Saugata Dutta, Sara Niazi, Xiaoyun Wang, Budder Siddiqui, and Duo Zhang. 2021. "CC16 Regulates Inflammation, ROS Generation and Apoptosis in Bronchial Epithelial Cells during Klebsiella pneumoniae Infection" International Journal of Molecular Sciences 22, no. 21: 11459. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111459