
IL-15 Prevents Renal Fibrosis by Inhibiting Collagen Synthesis: A New Pathway in Chronic Kidney Disease?

 and
and 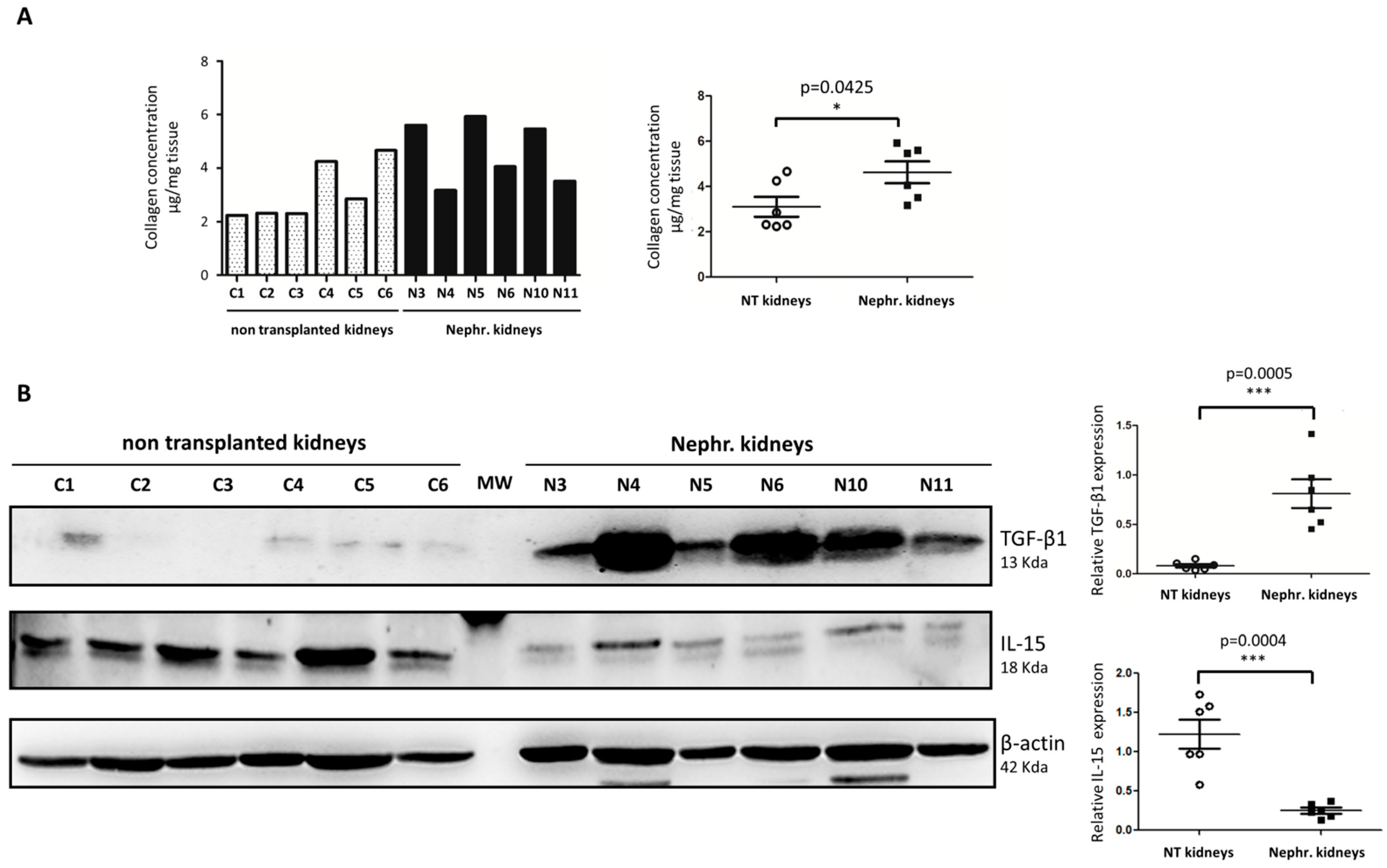{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. IL-15 Expression Is Reduced in Detransplanted Human Renal Grafts
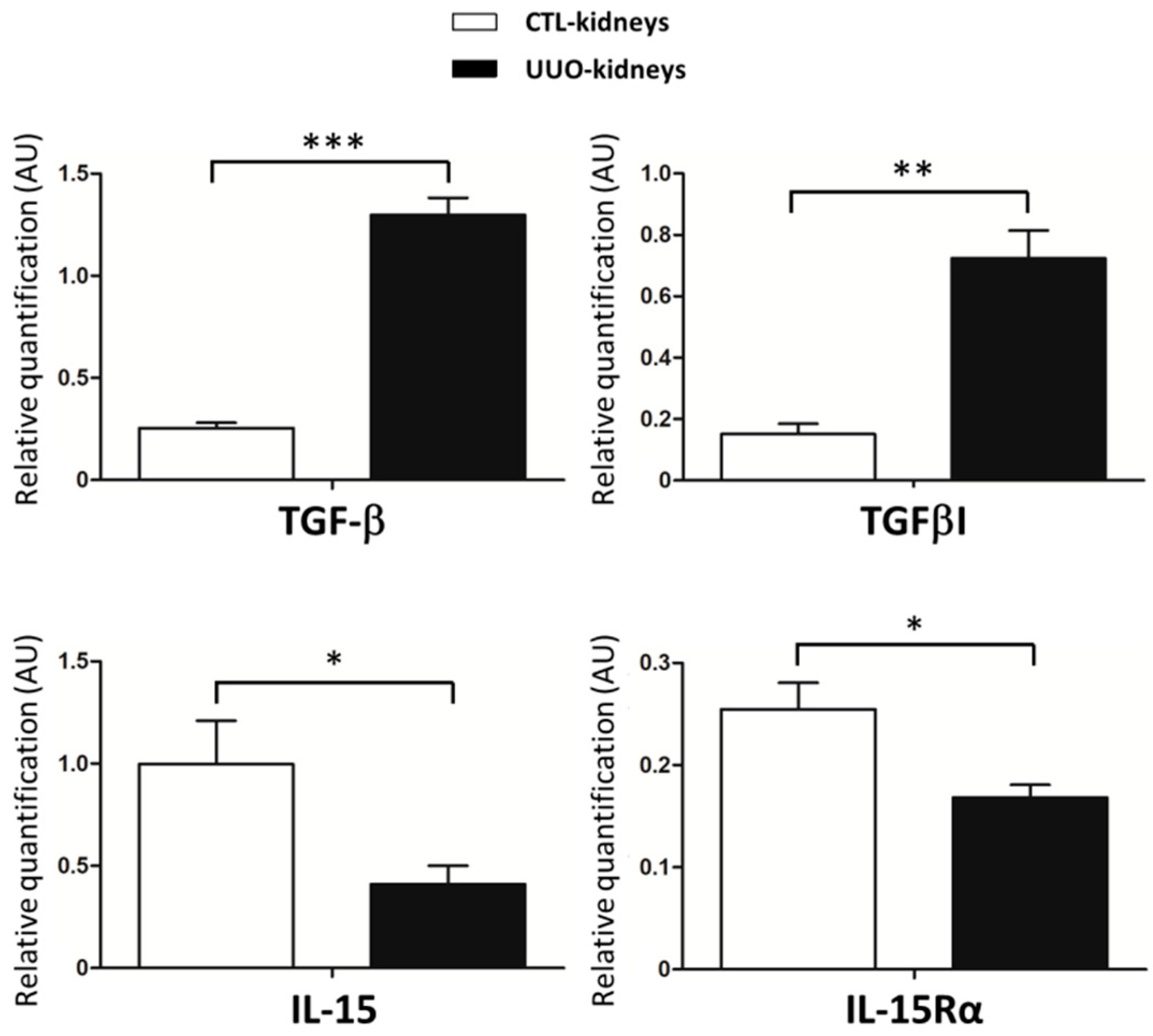
2.2. IL-15/IL-15Rα Expression Decreases in the Obstructed Kidneys in UUO Model
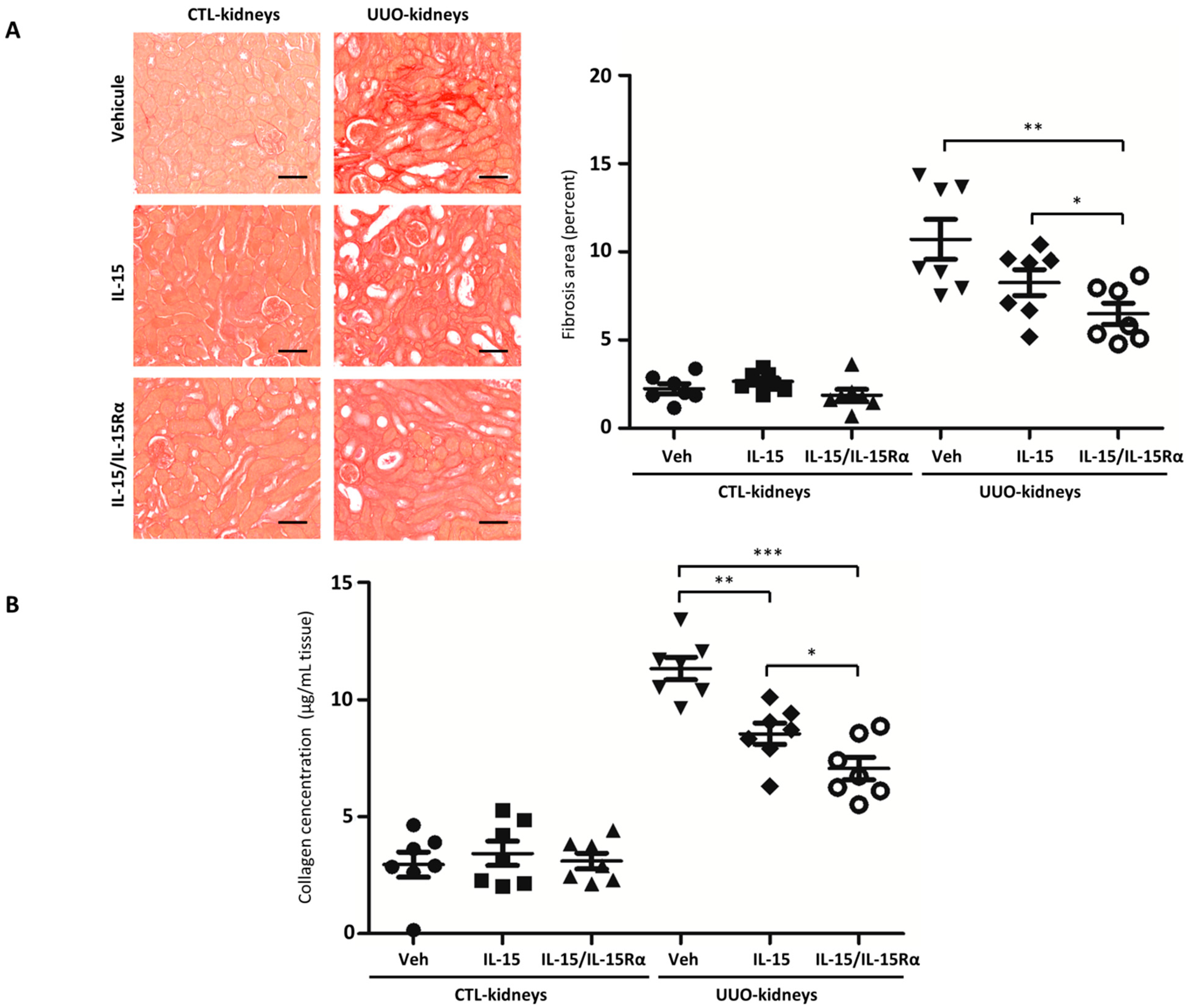
2.3. IL-15/IL-15Rα Treatment Decreases Renal Fibrosis in the UUO Model
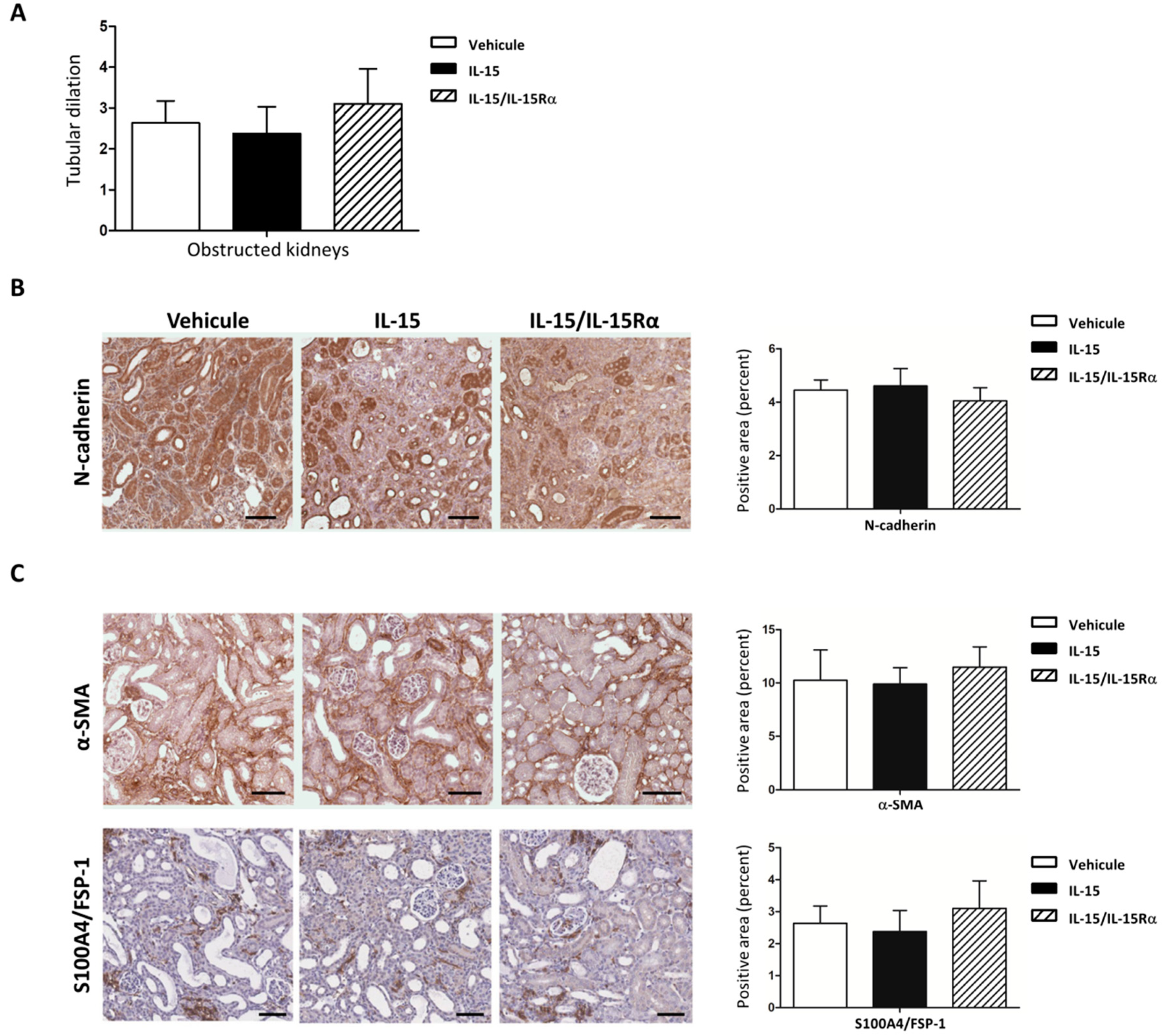
2.4. The IL-15/IL-15Rα Antifibrotic Effect in UUO Does Not Impact the Number of Myofibroblasts Accumulation
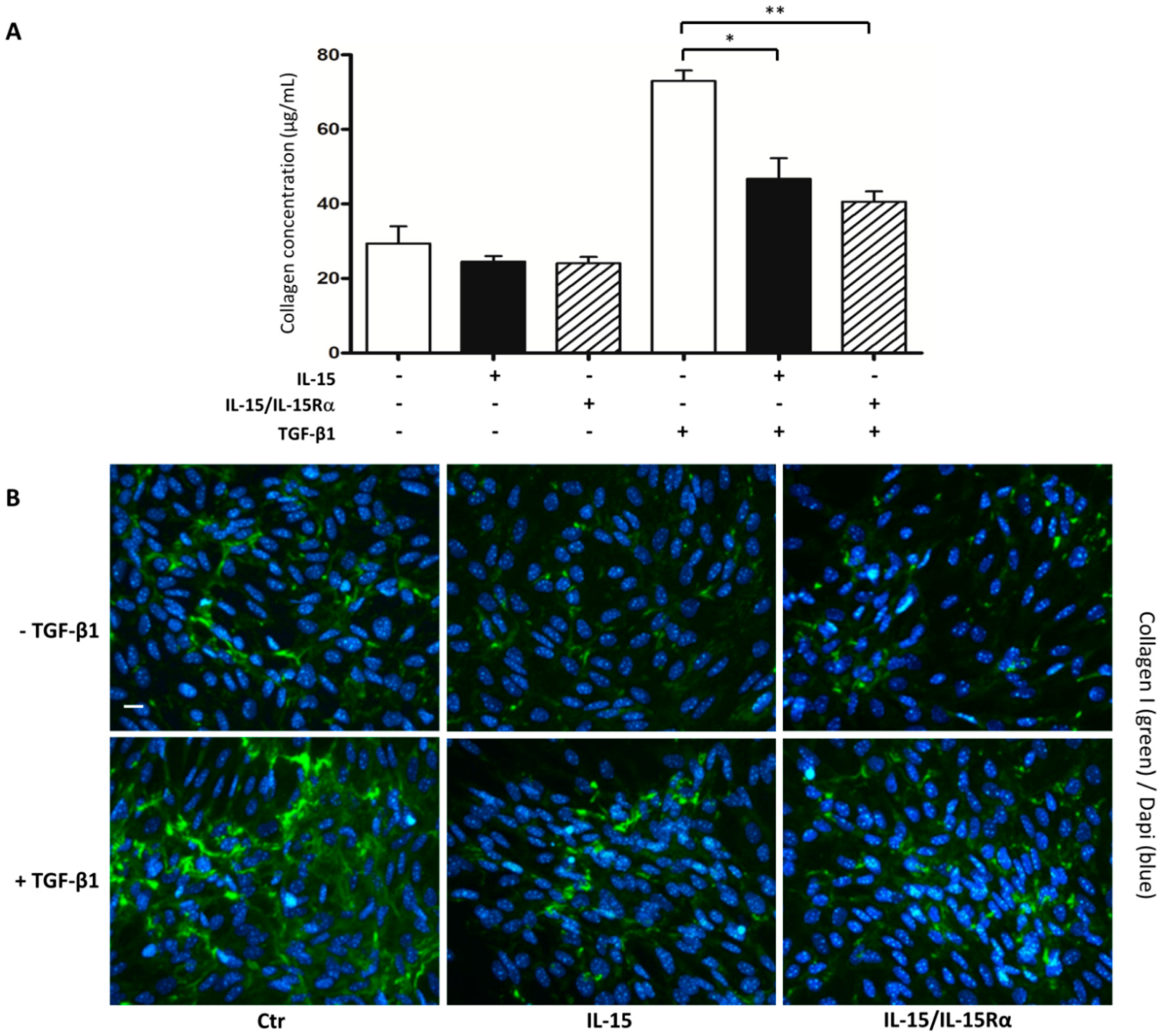
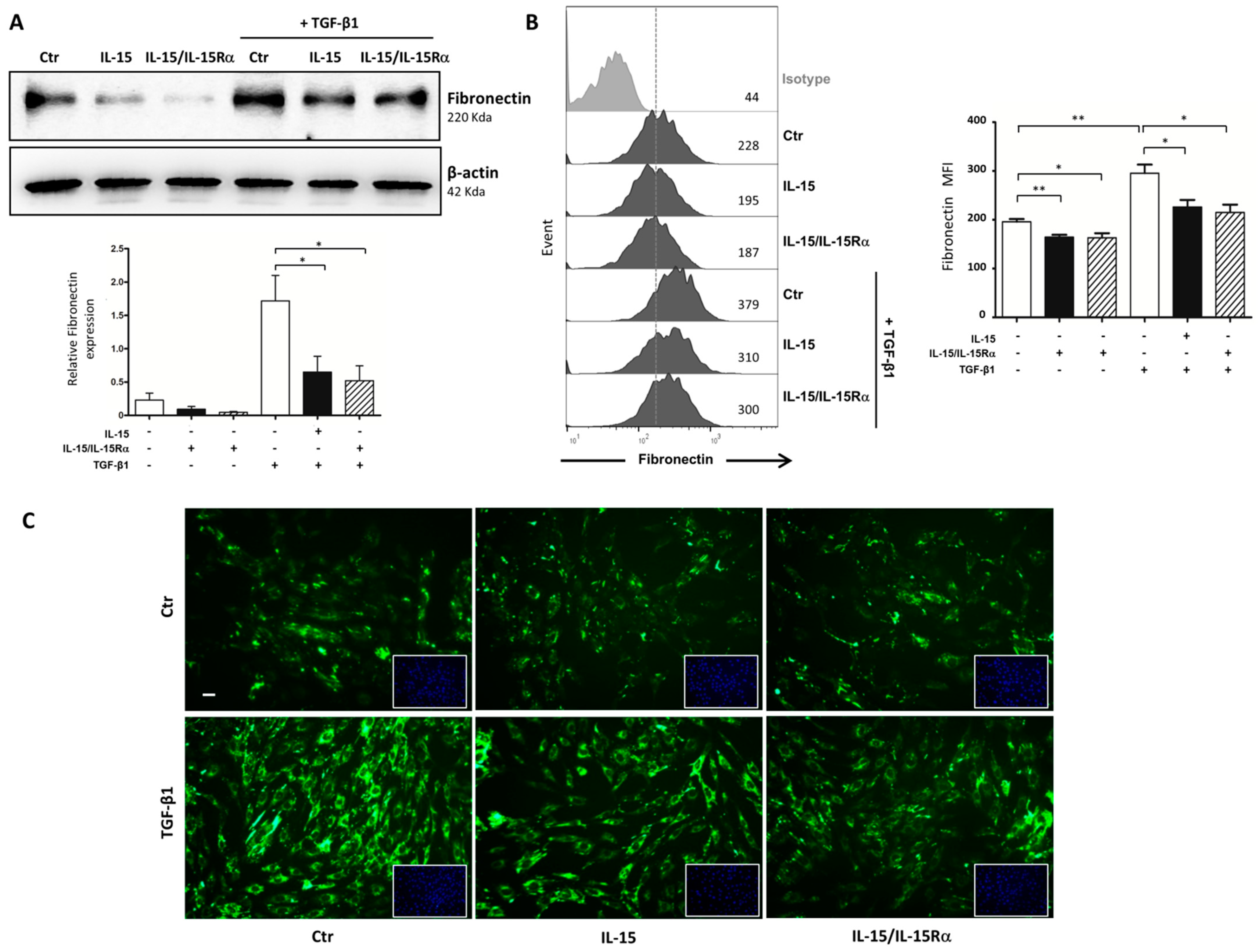
2.5. The Role of IL-15 in Reducing Renal Fibrosis Involves a Direct Action on Renal Myofibroblasts
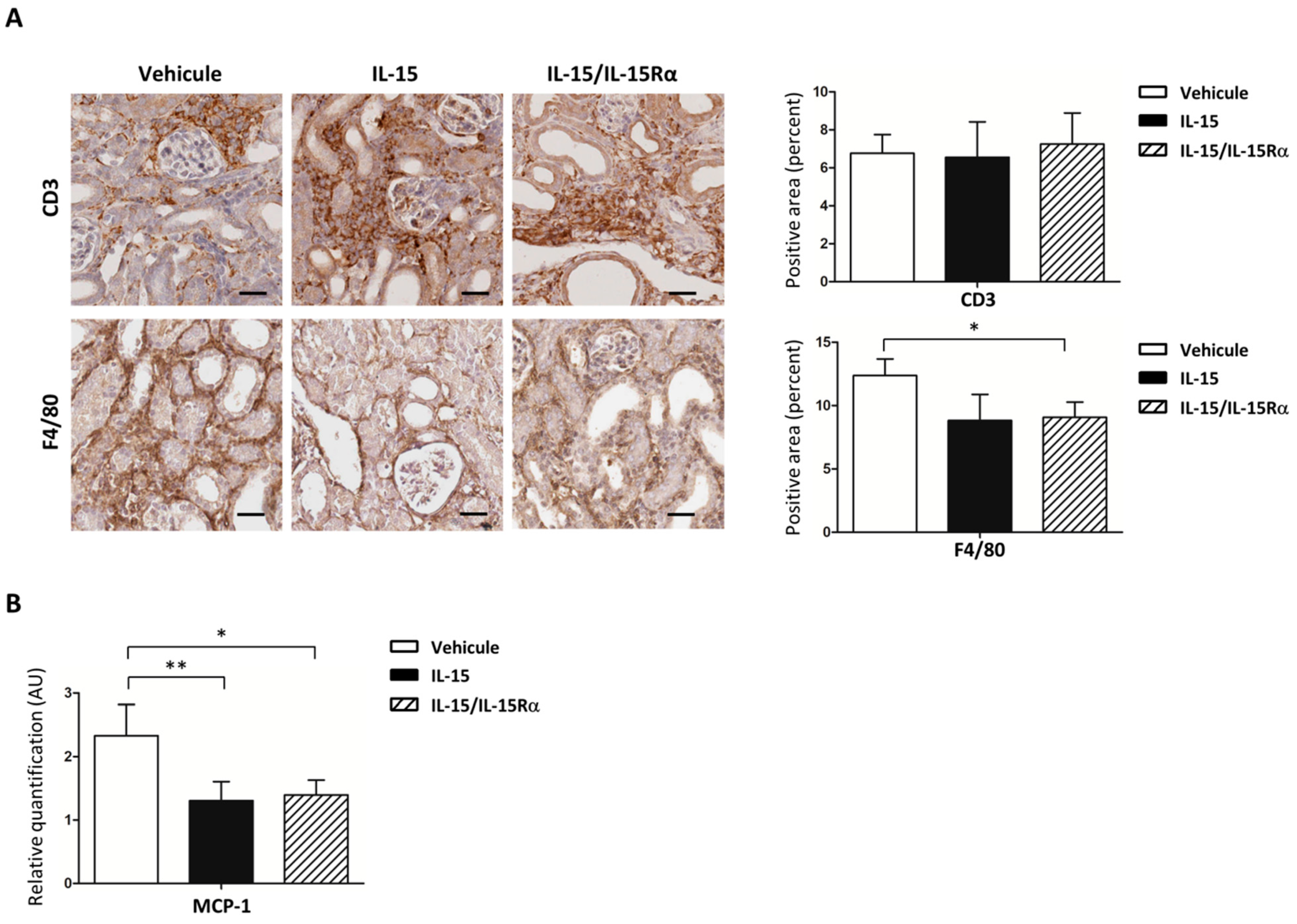
2.6. IL-15 Decreases Both Macrophage Infiltration and MCP-1 Expression in Obstructed Kidneys
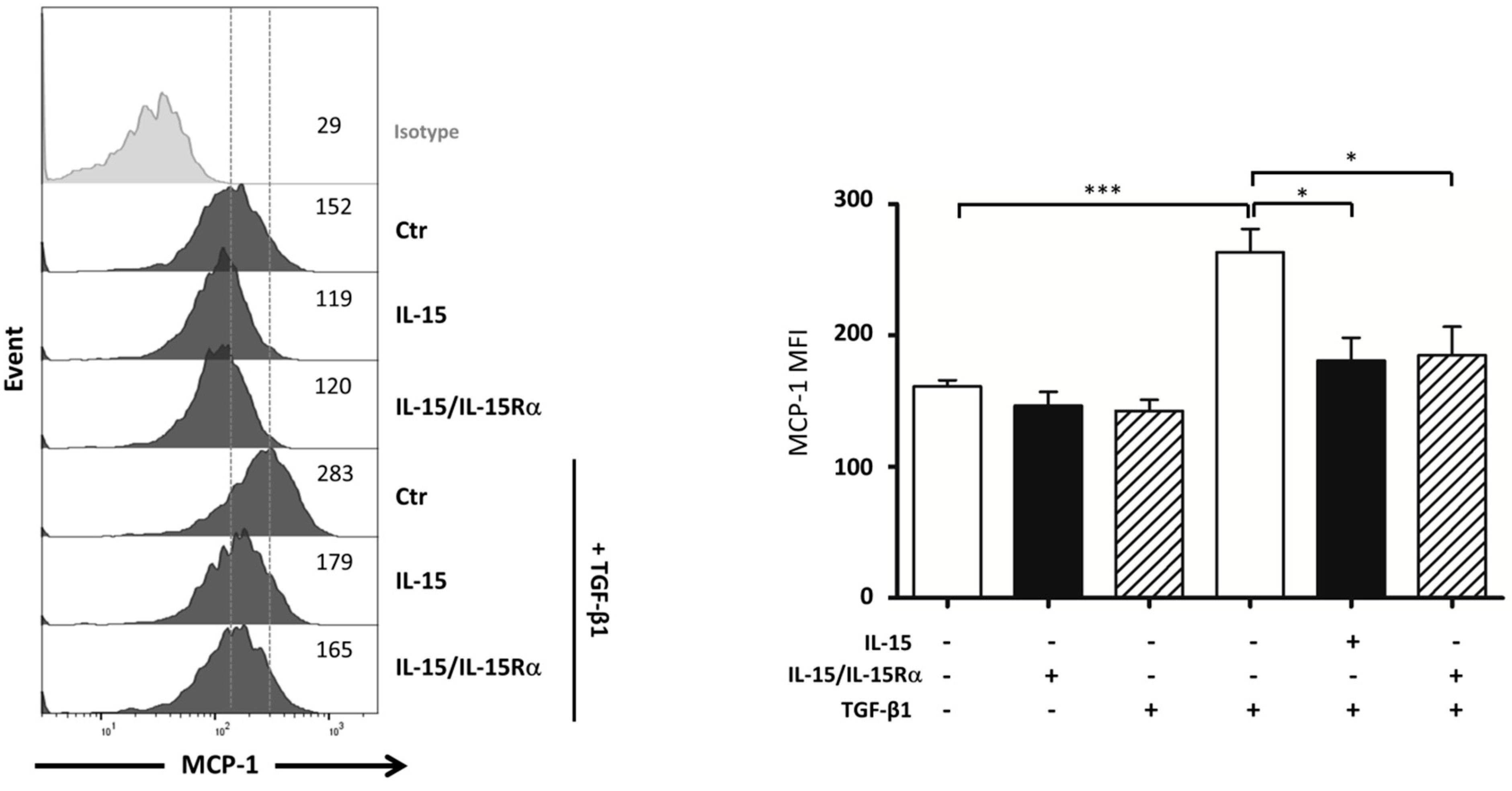
2.7. IL-15 Inhibits TGF-β1 Induced MCP-1 Production by Myofibroblasts
3. Discussion
4. Materials and Methods
4.1. Human Kidney Specimens
4.2. Animals
4.3. Unilateral Ureteral Obstruction (UUO)
4.4. Cell Culture
4.5. Immunohistochemistry
4.6. Histopathological Analysis of Renal Fibrosis
4.7. Quantification of Collagen Deposition in Obstructed Kidneys by Hydroxyproline Measurement
4.8. Quantification of Collagen Secretion by Myofibroblasts
4.9. Real-Time Quantitative PCR
4.10. Total Lysate from Kidneys and Cells
4.11. Western Blot Analysis
4.12. Flow Cytometric Analysis
4.13. Immunofluorescence Staining
4.14. Statistical Analyzes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host Responses in Tissue Repair and Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 241–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-edged Sword. J. Histochem. Cytochem. 2019, 67, 663–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- François, H.; Chatziantoniou, C. Renal fibrosis: Recent translational aspects. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 318–332. [Google Scholar] [CrossRef]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti–TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Piperidou, A.; Loutradis, C.; Sarafidis, P. SGLT-2 inhibitors and nephroprotection: Current evidence and future perspectives. J. Hum. Hypertens. 2021, 35, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Ewart, L.M.; Masterson, E.; Tennant, S.; Grebnev, G.; Prunotto, M.; Pomposiello, S.; Conde-Knape, K.; Martin, F.M.; Godson, C. BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 3095–3104. [Google Scholar] [CrossRef]
- Yu, M.-A.; Shin, K.-S.; Kim, J.H.; Kim, Y.-I.; Chung, S.S.; Park, S.-H.; Kang, D.-H.; Kim, Y.-L. HGF and BMP-7 Ameliorate High Glucose–Induced Epithelial-to-Mesenchymal Transition of Peritoneal Mesothelium. J. Am. Soc. Nephrol. 2009, 20, 567–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, K.A.; Guo, G.; Wozniak, M.; Martin, D.; Miller, S.; Liapis, H.; Loveday, K.; Klahr, S.; Sampath, T.K.; Morrissey, J. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am. J. Physiol.-Ren. Physiol. 2000, 279, F130–F143. [Google Scholar] [CrossRef]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho Inhibits Transforming Growth Factor-β1 (TGF-β1) Signaling and Suppresses Renal Fibrosis and Cancer Metastasis in Mice*. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateshaiah, S.U.; Niranjan, R.; Manohar, M.; Verma, A.K.; Kandikattu, H.K.; Lasky, J.A.; Mishra, A. Attenuation of Allergen-, IL-13–, and TGF-α–induced Lung Fibrosis after the Treatment of rIL-15 in Mice. Am. J. Respir. Cell Mol. Biol. 2019, 61, 97–109. [Google Scholar] [CrossRef]
- Manohar, M.; Kandikattu, H.K.; Verma, A.K.; Mishra, A. IL-15 regulates fibrosis and inflammation in a mouse model of chronic pancreatitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2018, 315, G954–G965. [Google Scholar] [CrossRef]
- Jiao, J.; Ooka, K.; Fey, H.; Fiel, M.I.; Rahmman, A.H.; Kojima, K.; Hoshida, Y.; Chen, X.; de Paula, T.; Vetter, D.; et al. Interleukin-15 receptor α on hepatic stellate cells regulates hepatic fibrogenesis in mice. J. Hepatol. 2016, 65, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Knudson, K.M.; Hodge, J.W.; Schlom, J.; Gameiro, S.R. Rationale for IL-15 superagonists in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 705–709. [Google Scholar] [CrossRef] [Green Version]
- Isvoranu, G.; Surcel, M.; Munteanu, A.N.; Bratu, O.G.; Ionita-Radu, F.; Neagu, M.T.; Chiritoiu-Butnaru, M. Therapeutic potential of interleukin-15 in cancer (Review). Exp. Ther. Med. 2021, 22, 1–6. [Google Scholar] [CrossRef]
- Wuttge, D.M.; Wildt, M.; Scheja, A.; Westergren-Thorsson, G. Interleukin-15 attenuates transforming growth factor-?1-induced myofibroblast differentiation in human fetal lung fibroblasts. Eur. Cytokine Netw. 2010, 21, 165–176. [Google Scholar] [CrossRef]
- Shinozaki, M.; Hirahashi, J.; Lebedeva, T.; Liew, F.Y.; Salant, D.J.; Maron, R.; Kelley, V.R. IL-15, a survival factor for kidney epithelial cells, counteracts apoptosis and inflammation during nephritis. J. Clin. Investig. 2002, 109, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Eini, H.; Tejman-Yarden, N.; Lewis, E.C.; Chaimovitz, C.; Zlotnik, M.; Douvdevani, A. Association Between Renal Injury and Reduced Interleukin-15 and Interleukin-15 Receptor Levels in Acute Kidney Injury. J. Interferon. Cytokine Res. 2010, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Giron-Michel, J.; Azzi, S.; Khawam, K.; Mortier, E.; Caignard, A.; Devocelle, A.; Ferrini, S.; Croce, M.; François, H.; Lecru, L.; et al. Interleukin-15 Plays a Central Role in Human Kidney Physiology and Cancer through the γc Signaling Pathway. PLoS ONE 2012, 7, e31624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devocelle, A.; Lecru, L.; François, H.; Desterke, C.; Gallerne, C.; Eid, P.; Estelle, O.; Azzarone, B.; Giron-Michel, J. Inhibition of TGF-β1 Signaling by IL-15: A Novel Role for IL-15 in the Control of Renal Epithelial-Mesenchymal Transition: IL-15 Counteracts TGF-β1-Induced EMT in Renal Fibrosis. Int. J. Cell Biol. 2019, 2019, 9151394. [Google Scholar] [CrossRef] [Green Version]
- Mesnard, L.; Keller, A.D.C.; Michel, M.-L.; Vandermeersch, S.; Rafat, C.; Letavernier, E.; Tillet, Y.; Rondeau, E.; Leite-De-Moraes, M.C. Invariant Natural Killer T Cells and TGF-β Attenuate Anti-GBM Glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 1282–1292. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.-H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a superagonist by binding to soluble IL-15R. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Luan, L.; Patil, N.K.; Sherwood, E.R. Immunobiology of the IL-15/IL-15Rα complex as an antitumor and antiviral agent. Cytokine Growth Factor Rev. 2017, 38, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Bascands, J.-L.; Schanstra, J.P. Obstructive nephropathy: Insights from genetically engineered animals. Kidney Int. 2005, 68, 925–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strutz, F.; Zeisberg, M. Renal Fibroblasts and Myofibroblasts in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2006, 17, 2992–2998. [Google Scholar] [CrossRef]
- Gewin, L. The many talents of transforming growth factor-β in the kidney. Curr. Opin. Nephrol. Hypertens. 2019, 28, 203–210. [Google Scholar] [CrossRef]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β signaling in the kidney: Profibrotic and protective effects. Am. J. Physiol.-Ren. Physiol. 2016, 310, F596–F606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelisetty, S.; Alford, C.; Reynolds, K.; Woodbury, L.; Nlandu-Khodo, S.; Yang, H.; Fogo, A.B.; Hao, C.-M.; Harris, R.C.; Zent, R.; et al. Renal fibrosis is not reduced by blocking transforming growth factor-β signaling in matrix-producing interstitial cells. Kidney Int. 2015, 88, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Chung, A.C.K.; Dong, Y.; Yang, W.; Zhong, X.; Lan, H.Y. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-β/Smad3-Azin1 pathway. Kidney Int. 2013, 84, 1129–1144. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.-X.; Li, Y.-Q.; Huang, X.R.; Wei, L.H.; Zhang, Y.; Feng, M.; Meng, X.-M.; Chen, H.Y.; Shi, Y.-J.; Lan, H.Y. Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin. Sci. 2014, 127, 195–208. [Google Scholar] [CrossRef]
- Chen, H.Y.; Zhong, X.; Huang, X.R.; Meng, X.-M.; You, Y.; Chung, A.C.; Lan, H.Y. MicroRNA-29b Inhibits Diabetic Nephropathy in db/db Mice. Mol. Ther. 2014, 22, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Manson, S.R.; Song, J.B.; Guo, Q.; Liapis, H.; Austin, P.F. Cell Type Specific Changes in BMP-7 Expression Contribute to the Progression of Kidney Disease in Patients with Obstructive Uropathy. J. Urol. 2015, 193, 1860–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, S.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor suppresses interstitial fibrosis in a mouse model of obstructive nephropathy. Kidney Int. 2001, 59, 1304–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzi, S.; Gallerne, C.; Romei, C.; LE Coz, V.; Gangemi, R.; Khawam, K.; Devocelle, A.; Gu, Y.; Bruno, S.; Ferrini, S.; et al. Human Renal Normal, Tumoral, and Cancer Stem Cells Express Membrane-Bound Interleukin-15 Isoforms Displaying Different Functions. Neoplasia 2015, 17, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Bergamaschi, C.; Rosati, M.; Jalah, R.; Valentin, A.; Kulkarni, V.; Alicea, C.; Zhang, G.-M.; Patel, V.; Felber, B.K.; Pavlakis, G.N. Intracellular Interaction of Interleukin-15 with Its Receptor α during Production Leads to Mutual Stabilization and Increased Bioactivity. J. Biol. Chem. 2008, 283, 4189–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, S.; Mariner, J.; Waldmann, T.A.; Tagaya, Y. IL-15Rα Recycles and Presents IL-15 In trans to Neighboring Cells. Immunity 2002, 17, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. α-Smooth Muscle Actin Is Crucial for Focal Adhesion Maturation in Myofibroblasts. Mol. Biol. Cell 2003, 14, 2508–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Pushpakumar, S.; Kundu, S.; Narayanan, N.; Sen, U. DNA hypermethylation in hyperhomocysteinemia contributes to abnormal extracellular matrix metabolism in the kidney. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4713–4725. [Google Scholar] [CrossRef] [Green Version]
- Eis, V. Chemokine Receptor CCR1 But Not CCR5 Mediates Leukocyte Recruitment and Subsequent Renal Fibrosis after Unilateral Ureteral Obstruction. J. Am. Soc. Nephrol. 2004, 15, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.-T.; Hughes, J.; Sethi, T. Galectin-3 Expression and Secretion Links Macrophages to the Promotion of Renal Fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.T.; Pérez-Barriocanal, F.; López-Novoa, J.M. Role of inflammation in túbulo-interstitial damage associated to obstructive nephropathy. J. Inflamm. 2010, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Tesch, G.H.; Schwarting, A.; Kinoshita, K.; Lan, H.Y.; Rollins, B.J.; Kelley, V.R. Monocyte chemoattractant protein-1 promotes macrophage-mediated tubular injury, but not glomerular injury, in nephrotoxic serum nephritis. J. Clin. Investig. 1999, 103, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Hilgers, K.F.; Hartner, A.; Porst, M.; Mai, M.; Wittmann, M.; Hugo, C.; Ganten, D.; Geiger, H.; Veelken, R.; Mann, J.F. Monocyte chemoattractant protein-1 and macrophage infiltration in hypertensive kidney injury. Kidney Int. 2000, 58, 2408–2419. [Google Scholar] [CrossRef] [Green Version]
- Titan, S.M.; Vieira, J.M.; Dominguez, W.V.; Moreira, S.R.S.; Pereira, A.B.; Barros, R.T.; Zatz, R. Urinary MCP-1 and RBP: Independent predictors of renal outcome in macroalbuminuric diabetic nephropathy. J. Diabetes Complicat. 2012, 26, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Vianna, H.R.; Soares, C.M.B.M.; Silveira, K.D.; Elmiro, G.S.; Mendes, P.M.; de Sousa Tavares, M.; Teixeira, M.M.; Miranda, D.; e Silva, A.C.S. Cytokines in chronic kidney disease: Potential link of MCP-1 and dyslipidemia in glomerular diseases. Pediatric Nephrol. 2012, 28, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sharkey, D.; Cantley, L.G. Tubular GM-CSF Promotes Late MCP-1/CCR2-Mediated Fibrosis and Inflammation after Ischemia/Reperfusion Injury. J. Am. Soc. Nephrol. 2019, 30, 1825–1840. [Google Scholar] [CrossRef]
- Lecru, L.; Desterke, C.; Grassin-Delyle, S.; Chatziantoniou, C.; Vandermeersch, S.; Devocelle, A.; Vernochet, A.; Ivanovski, N.; Ledent, C.; Ferlicot, S.; et al. Cannabinoid receptor 1 is a major mediator of renal fibrosis. Kidney Int. 2015, 88, 72–84. [Google Scholar] [CrossRef] [Green Version]
- dos Anjos Cassado, A. F4/80 as a Major Macrophage Marker: The Case of the Peritoneum and Spleen. In Macrophages; Kloc, M., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 161–179. [Google Scholar] [CrossRef]
- Gharaee-Kermani, M.; Denholm, E.M.; Phan, S.H. Costimulation of Fibroblast Collagen and Transforming Growth Factor β1 Gene Expression by Monocyte Chemoattractant Protein-1 via Specific Receptors. J. Biol. Chem. 1996, 271, 17779–17784. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.P.; Koya, D.; Kanasaki, K. MicroRNAs in Kidney Fibrosis and Diabetic Nephropathy: Roles on EMT and EndMT. BioMed Res. Int. 2013, 2013, 125469. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, I.; Wolf, G. Epithelial-to-Mesenchymal Transition in Diabetic Nephropathy: Fact or Fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Luque, Y.; Cathelin, D.; Vandermeersch, S.; Xu, X.; Sohier, J.; Placier, S.; Xu-Dubois, Y.-C.; Louis, K.; Hertig, A.; Bories, J.-C.; et al. Glomerular common gamma chain confers B- and T-cell–independent protection against glomerulonephritis. Kidney Int. 2017, 91, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Grimwood, L.; Masterson, R. Propagation and Culture of Renal Fibroblasts. Methods Mol. Biol. 2009, 466, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Devocelle, A.; Desterke, C.; de Souza, L.E.B.; Hadadi, É.; Acloque, H.; Foudi, A.; Xiang, Y.; Ballesta, A.; Chang, Y.; et al. BMAL1 Knockdown Leans Epithelial–Mesenchymal Balance toward Epithelial Properties and Decreases the Chemoresistance of Colon Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 5247. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devocelle, A.; Lecru, L.; Ferlicot, S.; Bessede, T.; Candelier, J.-J.; Giron-Michel, J.; François, H. IL-15 Prevents Renal Fibrosis by Inhibiting Collagen Synthesis: A New Pathway in Chronic Kidney Disease? Int. J. Mol. Sci. 2021, 22, 11698. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111698
Devocelle A, Lecru L, Ferlicot S, Bessede T, Candelier J-J, Giron-Michel J, François H. IL-15 Prevents Renal Fibrosis by Inhibiting Collagen Synthesis: A New Pathway in Chronic Kidney Disease? International Journal of Molecular Sciences. 2021; 22(21):11698. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111698
Chicago/Turabian StyleDevocelle, Aurore, Lola Lecru, Sophie Ferlicot, Thomas Bessede, Jean-Jacques Candelier, Julien Giron-Michel, and Hélène François. 2021. "IL-15 Prevents Renal Fibrosis by Inhibiting Collagen Synthesis: A New Pathway in Chronic Kidney Disease?" International Journal of Molecular Sciences 22, no. 21: 11698. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111698