
FLLL32 Triggers Caspase-Mediated Apoptotic Cell Death in Human Oral Cancer Cells by Regulating the p38 Pathway

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
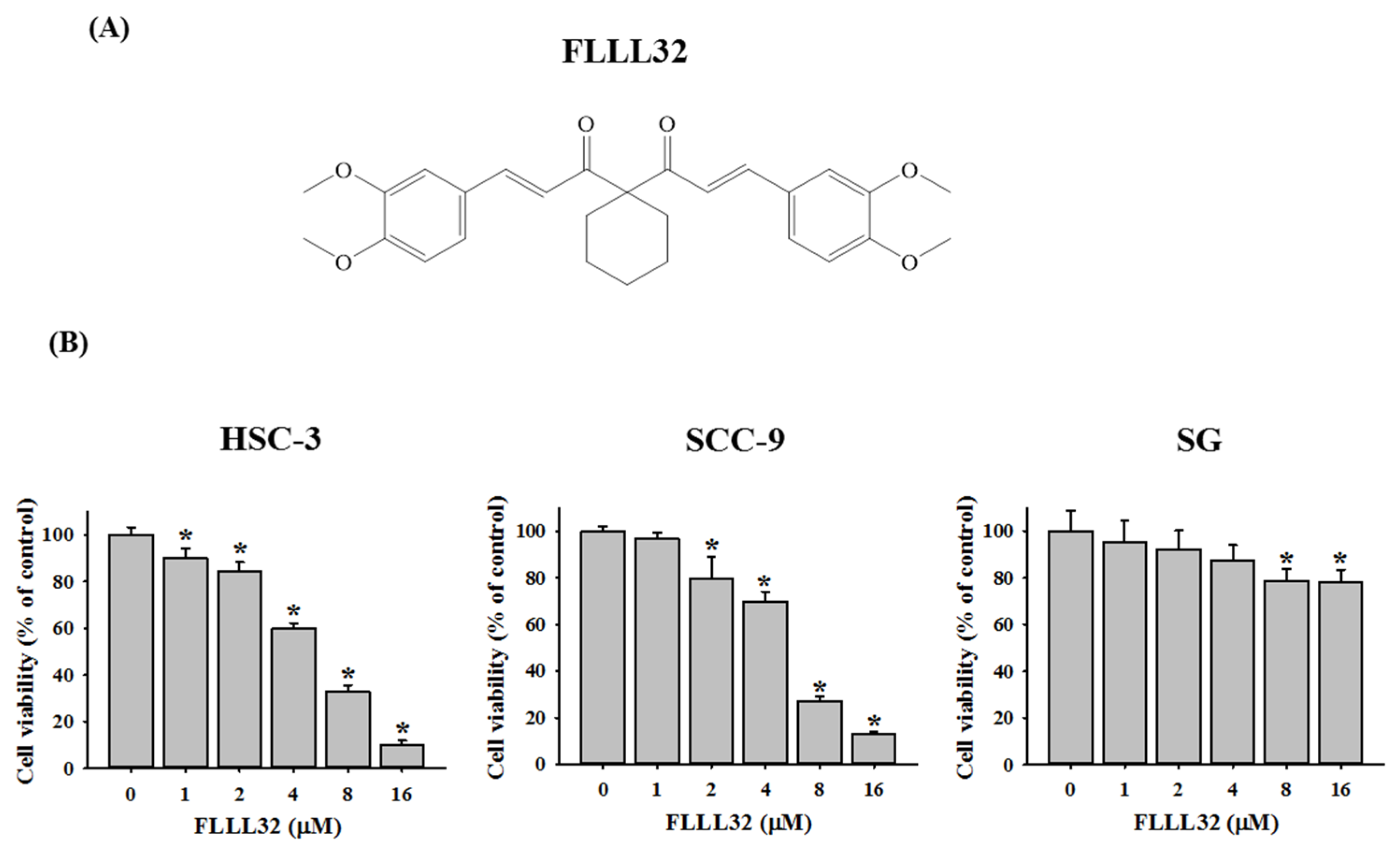
2.1. FLLL32 Inhibits Cell Viability in Oral Cancer Cells
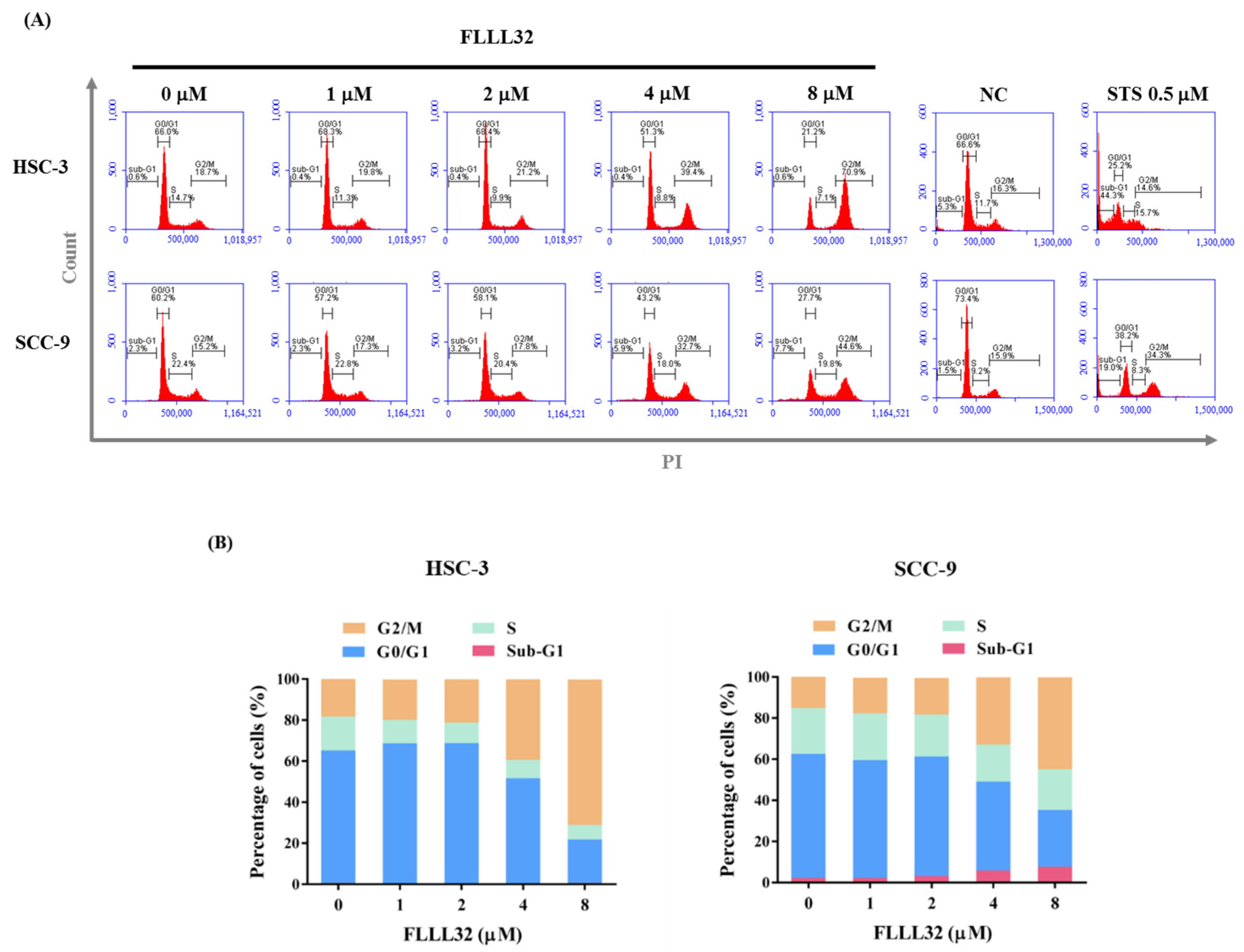
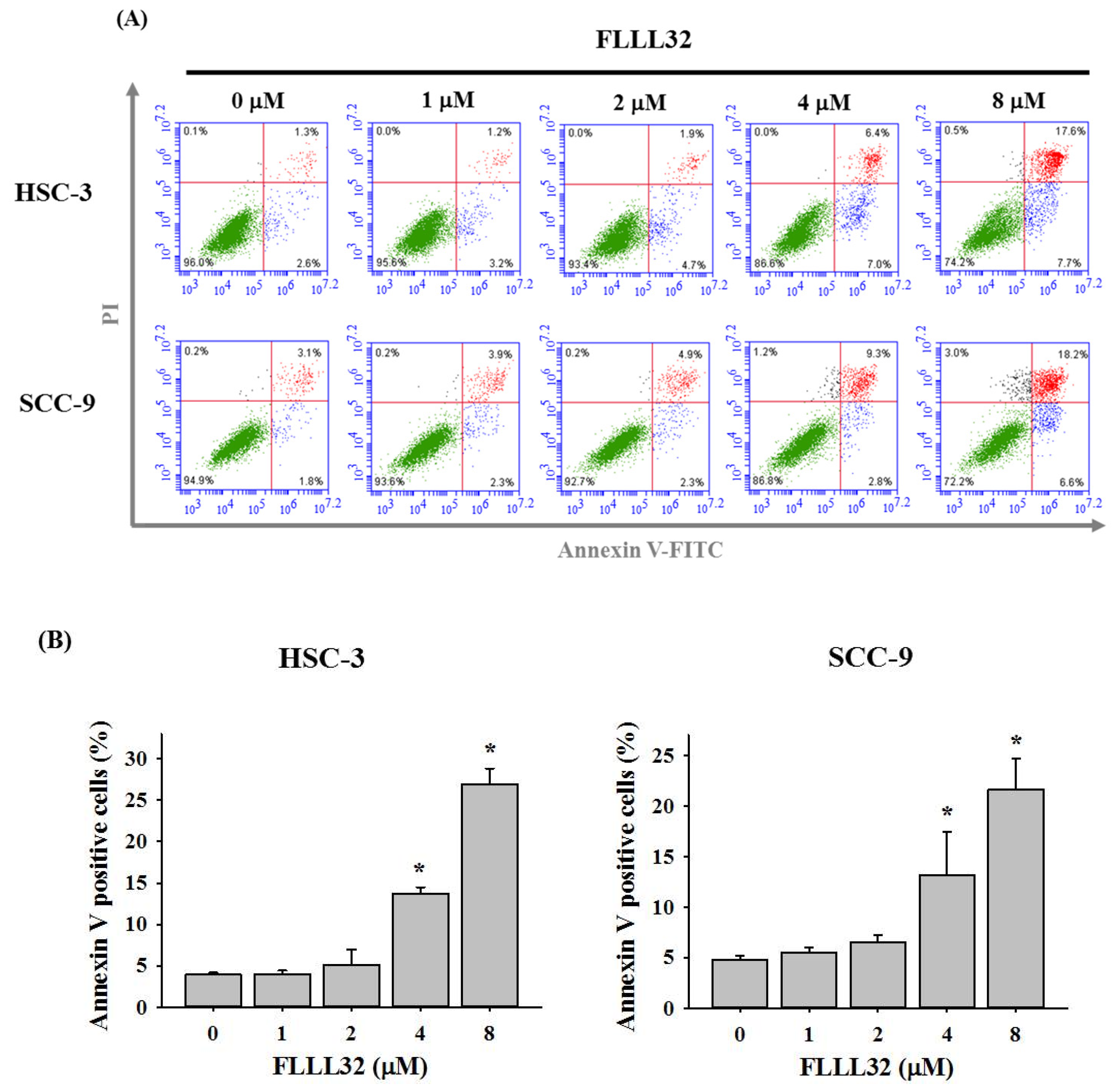
2.2. FLLL32 Causes G2/M Arrest and Induces Cell Apoptosis in Oral Cancer Cells
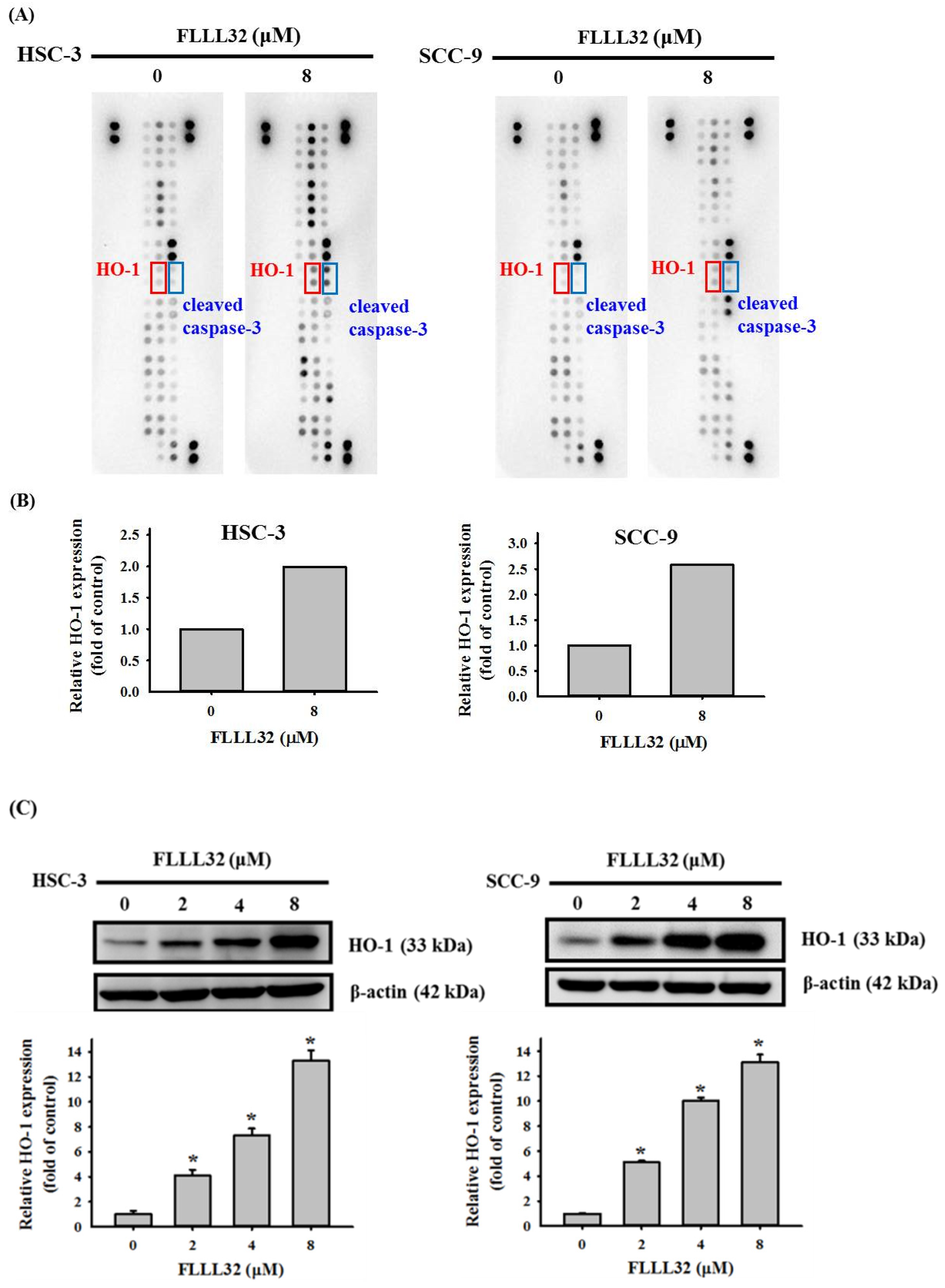
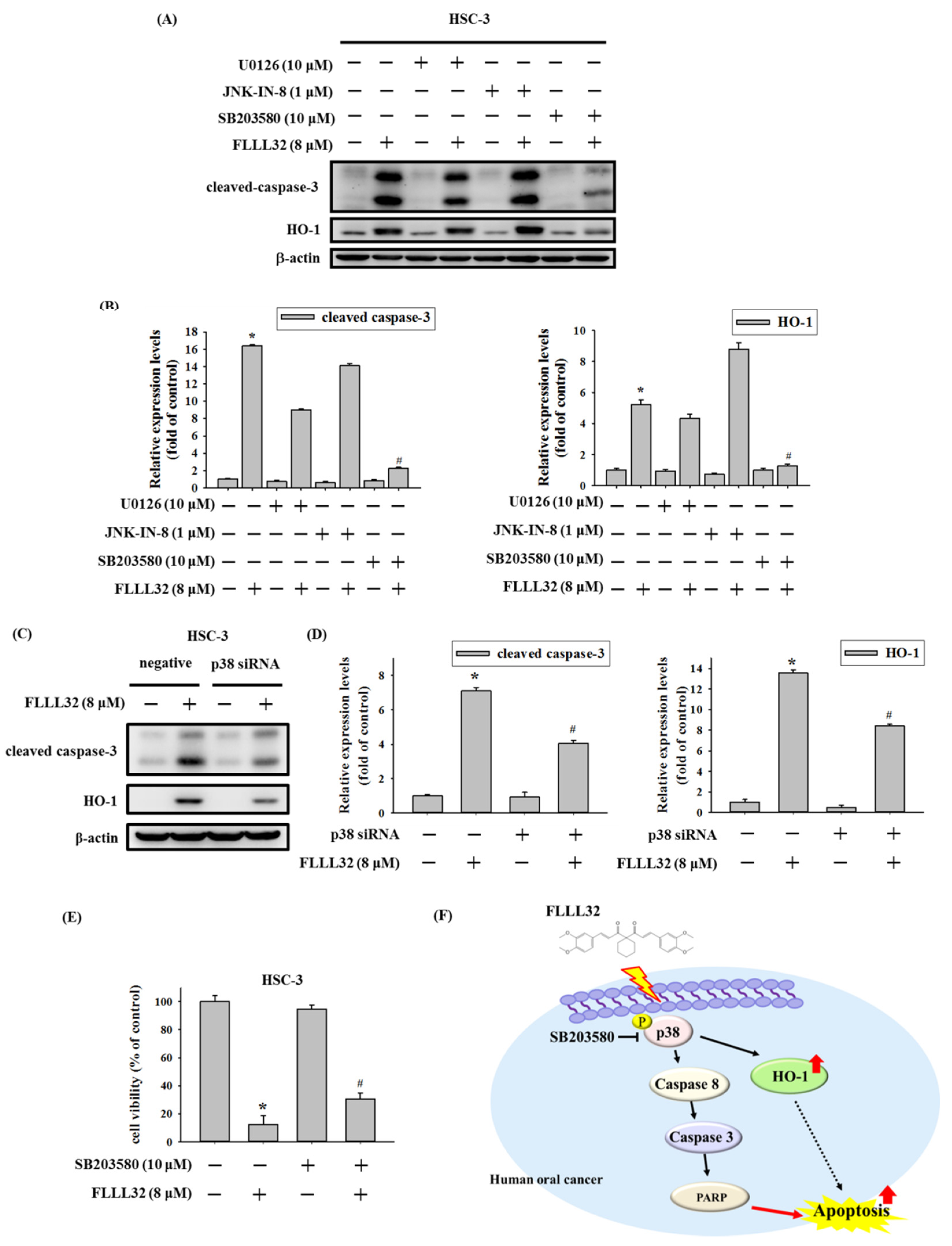
2.3. The Increase in HO-1 Is Involved in the Apoptosis Regulated by FLLL32
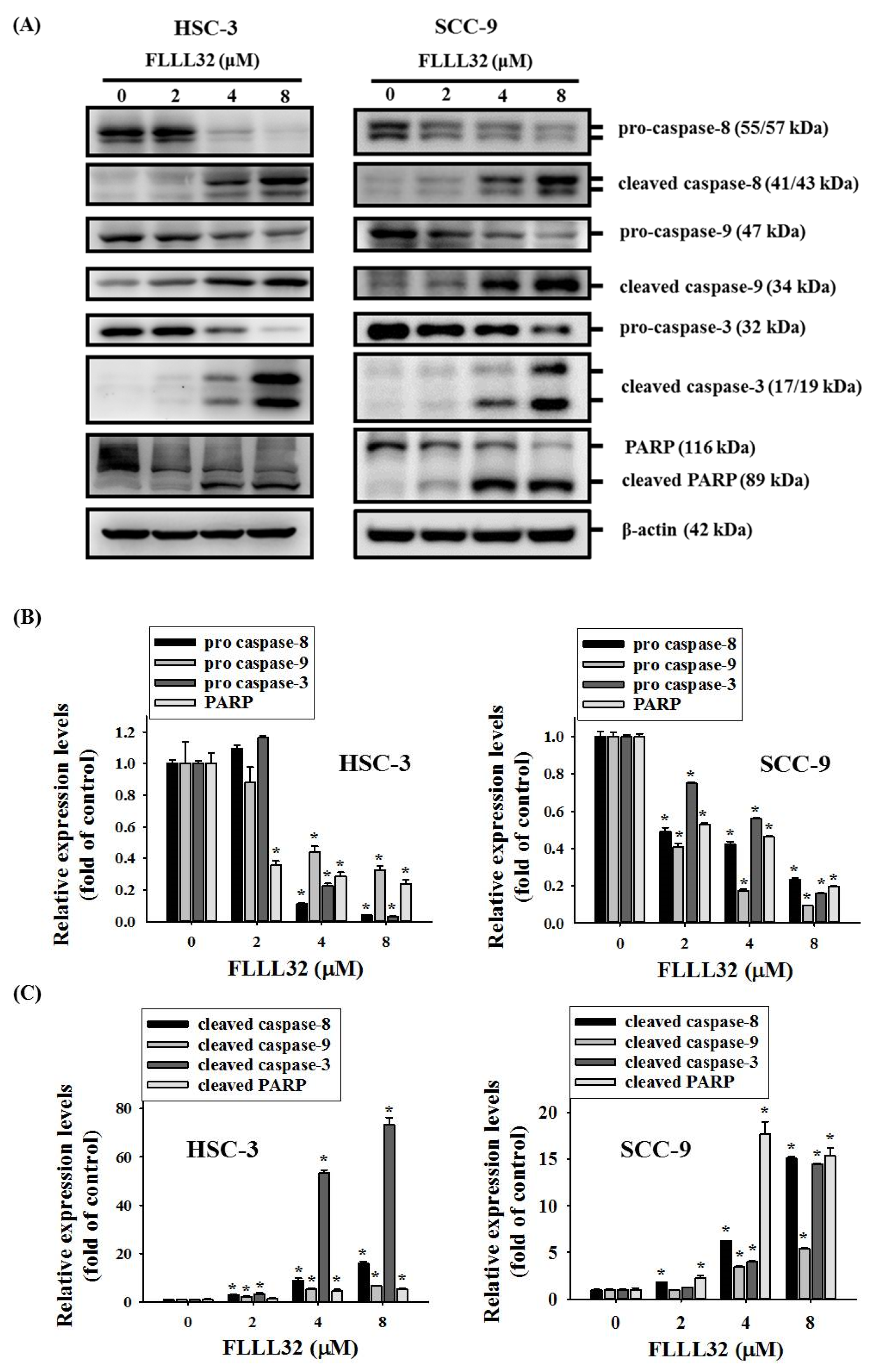
2.4. FLLL32-Induced Activation of Caspase-3, -8, -9, and PARP in Oral Cancer Cells
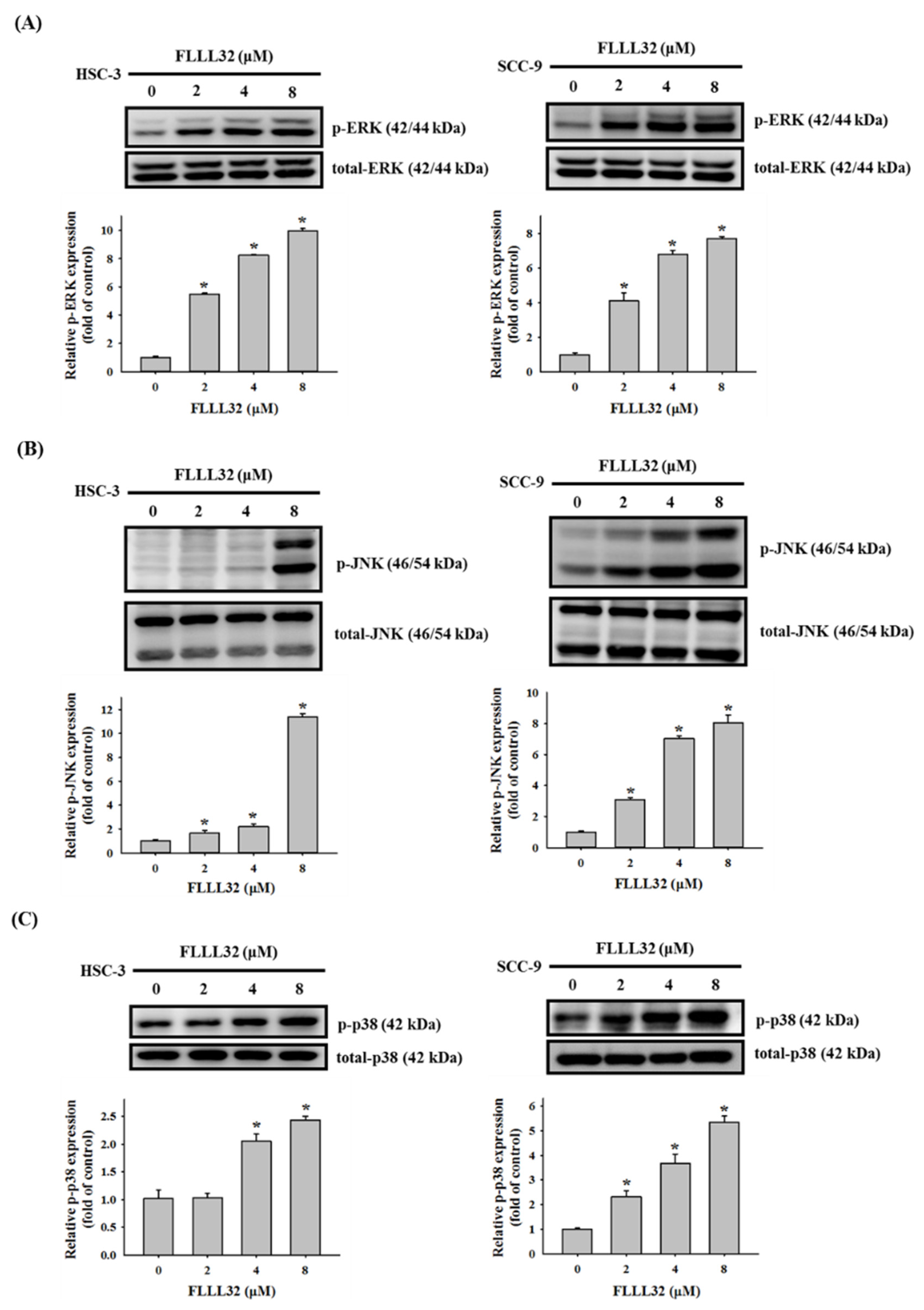
2.5. Involvement of p38 in FLLL32-Induced Activation of Caspase-3 and HO-1
3. Discussion
4. Materials and Methods
4.1. Cell and Cell Culture
4.2. Microculture Tetrazolium Colorimetric (MTT) Assay
4.3. Cell-Cycle Distribution Assay
4.4. Annexin V–FITC Apoptosis Staining Assay
4.5. Human Apoptosis Proteome Profiler Array
4.6. Protein Extraction and Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dauby, N. Head and neck cancer. N. Engl. J. Med. 2020, 382, e57. [Google Scholar]
- Noguti, J.; De Moura, C.F.; De Jesus, G.P.; Da Silva, V.H.; Hossaka, T.A.; Oshima, C.T.; Ribeiro, D.A. Metastasis from oral cancer: An overview. Cancer Genom. Proteom. 2012, 9, 329–335. [Google Scholar]
- Kumar, M.; Nanavati, R.; Modi, T.G.; Dobariya, C. Oral cancer: Etiology and risk factors: A review. J. Cancer Res. Ther. 2016, 12, 458–463. [Google Scholar] [CrossRef]
- Meurman, J.H. Infectious and dietary risk factors of oral cancer. Oral Oncol. 2010, 46, 411–413. [Google Scholar] [CrossRef]
- Rivera, C. Essentials of oral cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11884–11894. [Google Scholar]
- Kotha, R.R.; Luthria, D.L. Curcumin: Biological, pharmaceutical, nutraceutical, and analytical aspects. Molecules 2019, 24, 2930. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar]
- Dou, H.; Shen, R.; Tao, J.; Huang, L.; Shi, H.; Chen, H.; Wang, Y.; Wang, T. Curcumin suppresses the colon cancer proliferation by inhibiting wnt/beta-catenin pathways via mir-130a. Front. Pharmacol. 2017, 8, 877. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, Y.; Jia, Z.; Gao, Y.; Zhao, C.; Yao, Y. Curcumin inhibits bladder cancer progression via regulation of beta-catenin expression. Tumour Biol. 2017, 39, 1010428317702548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sun, L.; Lei, J.; Wu, Z.; Ma, Q.; Wang, Z. Curcumin inhibits pancreatic cancer cell invasion and emt by interfering with tumor-stromal crosstalk under hypoxic conditions via the il-6/erk/nf-κb axis. Oncol. Rep. 2020, 44, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, M.; Ghidoni, R. Dietary curcumin: Correlation between bioavailability and health potential. Nutrients 2019, 11, 2147. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Deangelis, S.; Foust, E.; Fuchs, J.; Li, C.; Li, P.K.; Schwartz, E.B.; Lesinski, G.B.; Benson, D.; Lu, J.; et al. A novel small molecule inhibits stat3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol. Cancer 2010, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Hutzen, B.; Zuo, M.; Ball, S.; Deangelis, S.; Foust, E.; Pandit, B.; Ihnat, M.A.; Shenoy, S.S.; Kulp, S.; et al. Novel stat3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 2010, 70, 2445–2454. [Google Scholar] [CrossRef] [Green Version]
- Jahangiri, A.; Dadmanesh, M. Stat3 inhibition reduced pd-l1 expression and enhanced antitumor immune responses. J. Cell. Physiol. 2020, 235, 9457–9463. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jiang, S.; Zhou, L.; Yu, F.; Ding, H.; Li, P.; Zhou, M.; Wang, K. Potential mechanisms of action of curcumin for cancer prevention: Focus on cellular signaling pathways and mirnas. Int. J. Biol. Sci. 2019, 15, 1200–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, C.D.; Thornton, T.M.; Sabio, G.; Davis, R.A.; Rincon, M. Nuclear localization of p38 mapk in response to DNA damage. Int. J. Biol. Sci. 2009, 5, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A review of curcumin and its derivatives as anticancer agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral bioavailability of curcumin: Problems and advancements. J. Drug Target. 2016, 24, 694–702. [Google Scholar] [CrossRef]
- Reid, J.M.; Buhrow, S.A.; Gilbert, J.A.; Jia, L.; Shoji, M.; Snyder, J.P.; Ames, M.M. Mouse pharmacokinetics and metabolism of the curcumin analog, 4-piperidinone,3,5-bis[(2-fluorophenyl)methylene]-acetate(3e,5e) (ef-24; nsc 716993). Cancer Chemother. Pharmacol. 2014, 73, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, P.C.; Chang, J.H.; Lee, W.J.; Ku, C.C.; Tsai, M.Y.; Yang, S.F.; Chien, M.H. The curcumin analogue, ef-24, triggers p38 mapk-mediated apoptotic cell death via inducing pp2a-modulated erk deactivation in human acute myeloid leukemia cells. Cancers 2020, 12, 2163. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Wu, H.H.; Lin, R.C.; Lin, Y.C.; Lu, P.W.; Yang, S.F.; Yang, J.S. Curcumin analogue l48h37 suppresses human osteosarcoma u2os and mg-63 cells’ migration and invasion in culture by inhibition of upa via the jak/stat signaling pathway. Molecules 2020, 26, 30. [Google Scholar] [CrossRef]
- Lin, L.; Fuchs, J.; Li, C.; Olson, V.; Bekaii-Saab, T.; Lin, J. Stat3 signaling pathway is necessary for cell survival and tumorsphere forming capacity in aldh+/cd133+ stem cell-like human colon cancer cells. Biochem. Biophys. Res. Commun. 2011, 416, 246–251. [Google Scholar] [CrossRef]
- Abuzeid, W.M.; Davis, S.; Tang, A.L.; Saunders, L.; Brenner, J.C.; Lin, J.; Fuchs, J.R.; Light, E.; Bradford, C.R.; Prince, M.E.; et al. Sensitization of head and neck cancer to cisplatin through the use of a novel curcumin analog. Arch. Otolaryngol.-Head Neck Surg. 2011, 137, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Bill, M.A.; Nicholas, C.; Mace, T.A.; Etter, J.P.; Li, C.; Schwartz, E.B.; Fuchs, J.R.; Young, G.S.; Lin, L.; Lin, J.; et al. Structurally modified curcumin analogs inhibit stat3 phosphorylation and promote apoptosis of human renal cell carcinoma and melanoma cell lines. PLoS ONE 2012, 7, e40724. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [PubMed] [Green Version]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Lee, M.K.; Kim, J.H. Curcumin induces cell cycle arrest and apoptosis in human osteosarcoma (hos) cells. Anticancer Res. 2009, 29, 5039–5044. [Google Scholar]
- Zhang, Z.; Lin, R.; Liu, Z.; Yan, T.; Xia, Y.; Zhao, L.; Lin, F.; Zhang, X.; Li, C.; Wang, Y. Curcumin analog, wz37, promotes g2/m arrest and apoptosis of hnscc cells through akt/mtor inhibition. Toxicol. In Vitro 2020, 65, 104754. [Google Scholar] [CrossRef]
- Chien, M.H.; Yang, W.E.; Yang, Y.C.; Ku, C.C.; Lee, W.J.; Tsai, M.Y.; Lin, C.W.; Yang, S.F. Dual targeting of the p38 mapk-ho-1 axis and ciap1/xiap by demethoxycurcumin triggers caspase-mediated apoptotic cell death in oral squamous cell carcinoma cells. Cancers 2020, 12, 703. [Google Scholar] [CrossRef] [Green Version]
- Waza, A.A.; Hamid, Z.; Ali, S.; Bhat, S.A.; Bhat, M.A. A review on heme oxygenase-1 induction: Is it a necessary evil. Inflamm. Res. 2018, 67, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.R.; Wang, H.M.; Liu, P.L.; Chen, Y.H.; Yang, M.C.; Chou, S.H.; Cheng, Y.J.; Yin, W.H.; Hwang, J.J.; Chong, I.W. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell. Oncol. 2012, 35, 461–471. [Google Scholar] [CrossRef]
- Ciesla, M.; Marona, P.; Kozakowska, M.; Jez, M.; Seczynska, M.; Loboda, A.; Bukowska-Strakova, K.; Szade, A.; Walawender, M.; Kusior, M.; et al. Heme oxygenase-1 controls an hdac4-mir-206 pathway of oxidative stress in rhabdomyosarcoma. Cancer Res. 2016, 76, 5707–5718. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.; Pereira, V.; Chauveau, C.; Zagani, R.; Remy, S.; Tesson, L.; Mazal, D.; Ubillos, L.; Brion, R.; Asghar, K.; et al. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: Mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J. 2005, 19, 1957–1968. [Google Scholar] [CrossRef]
- Tsuji, M.H.; Yanagawa, T.; Iwasa, S.; Tabuchi, K.; Onizawa, K.; Bannai, S.; Toyooka, H.; Yoshida, H. Heme oxygenase-1 expression in oral squamous cell carcinoma as involved in lymph node metastasis. Cancer Lett. 1999, 138, 53–59. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of mapk signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zhong, J.; Yan, L.; Li, J.; Wang, H.; Wen, Y.; Zhao, Y. Curcumin exerts antitumor effects in retinoblastoma cells by regulating the jnk and p38 mapk pathways. Int. J. Mol. Med. 2016, 38, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.S.; Lin, R.C.; Hsieh, Y.H.; Wu, H.H. Clefma activates the extrinsic and intrinsic apoptotic processes through jnk1/2 and p38 pathways in human osteosarcoma cells. Molecules 2019, 24, 3280. [Google Scholar] [CrossRef] [Green Version]
- Kang, N.; Wang, M.M.; Wang, Y.H.; Zhang, Z.N.; Cao, H.R.; Lv, Y.H.; Yang, Y.; Fan, P.H.; Qiu, F.; Gao, X.M. Tetrahydrocurcumin induces g2/m cell cycle arrest and apoptosis involving p38 mapk activation in human breast cancer cells. Food Chem. Toxicol. 2014, 67, 193–200. [Google Scholar] [CrossRef]
- Zhai, H.; Pan, T.; Yang, H.; Wang, H.; Wang, Y. Cadmium induces a549 cell migration and invasion by activating erk. Exp. Ther. Med. 2019, 18, 1793–1799. [Google Scholar]
- Sung, N.J.; Kim, N.H.; Bae, N.Y.; Jo, H.S.; Park, S.A. Dha inhibits gremlin-1-induced epithelial-to-mesenchymal transition via erk suppression in human breast cancer cells. Biosci. Rep. 2020, 40, BSR20200164. [Google Scholar] [CrossRef] [Green Version]
- Cagnol, S.; Chambard, J.C. Erk and cell death: Mechanisms of erk-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chen, P.N.; Lin, C.W.; Yang, W.E.; Ho, Y.T.; Yang, S.F. Cantharidic acid induces apoptosis in human nasopharyngeal carcinoma cells through p38-mediated upregulation of caspase activation. Environ. Toxicol. 2020, 35, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; López, J.M. Understanding mapk signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [Green Version]
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The novel curcumin analog flll32 decreases stat3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 2011, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Bill, M.A.; Fuchs, J.R.; Li, C.; Yui, J.; Bakan, C.; Benson, D.M., Jr.; Schwartz, E.B.; Abdelhamid, D.; Lin, J.; Hoyt, D.G.; et al. The small molecule curcumin analog flll32 induces apoptosis in melanoma cells via stat3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol. Cancer 2010, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Zhang, Z.G.; Tian, X.Q.; Sun, D.F.; Liang, Q.C.; Zhang, Y.J.; Lu, R.; Chen, Y.X.; Fang, J.Y. Inhibition of jak1, 2/stat3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia 2008, 10, 287–297. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.-W.; Chuang, C.-Y.; Chen, Y.-T.; Yang, W.-E.; Pan, Y.-P.; Lin, C.-W.; Yang, S.-F. FLLL32 Triggers Caspase-Mediated Apoptotic Cell Death in Human Oral Cancer Cells by Regulating the p38 Pathway. Int. J. Mol. Sci. 2021, 22, 11860. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111860
Su C-W, Chuang C-Y, Chen Y-T, Yang W-E, Pan Y-P, Lin C-W, Yang S-F. FLLL32 Triggers Caspase-Mediated Apoptotic Cell Death in Human Oral Cancer Cells by Regulating the p38 Pathway. International Journal of Molecular Sciences. 2021; 22(21):11860. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111860
Chicago/Turabian StyleSu, Chun-Wen, Chun-Yi Chuang, Yi-Tzu Chen, Wei-En Yang, Yi-Ping Pan, Chiao-Wen Lin, and Shun-Fa Yang. 2021. "FLLL32 Triggers Caspase-Mediated Apoptotic Cell Death in Human Oral Cancer Cells by Regulating the p38 Pathway" International Journal of Molecular Sciences 22, no. 21: 11860. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111860