
New Drug Targets to Prevent Death Due to Stroke: A Review Based on Results of Protein-Protein Interaction Network, Enrichment, and Annotation Analyses

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. DEP Analysis
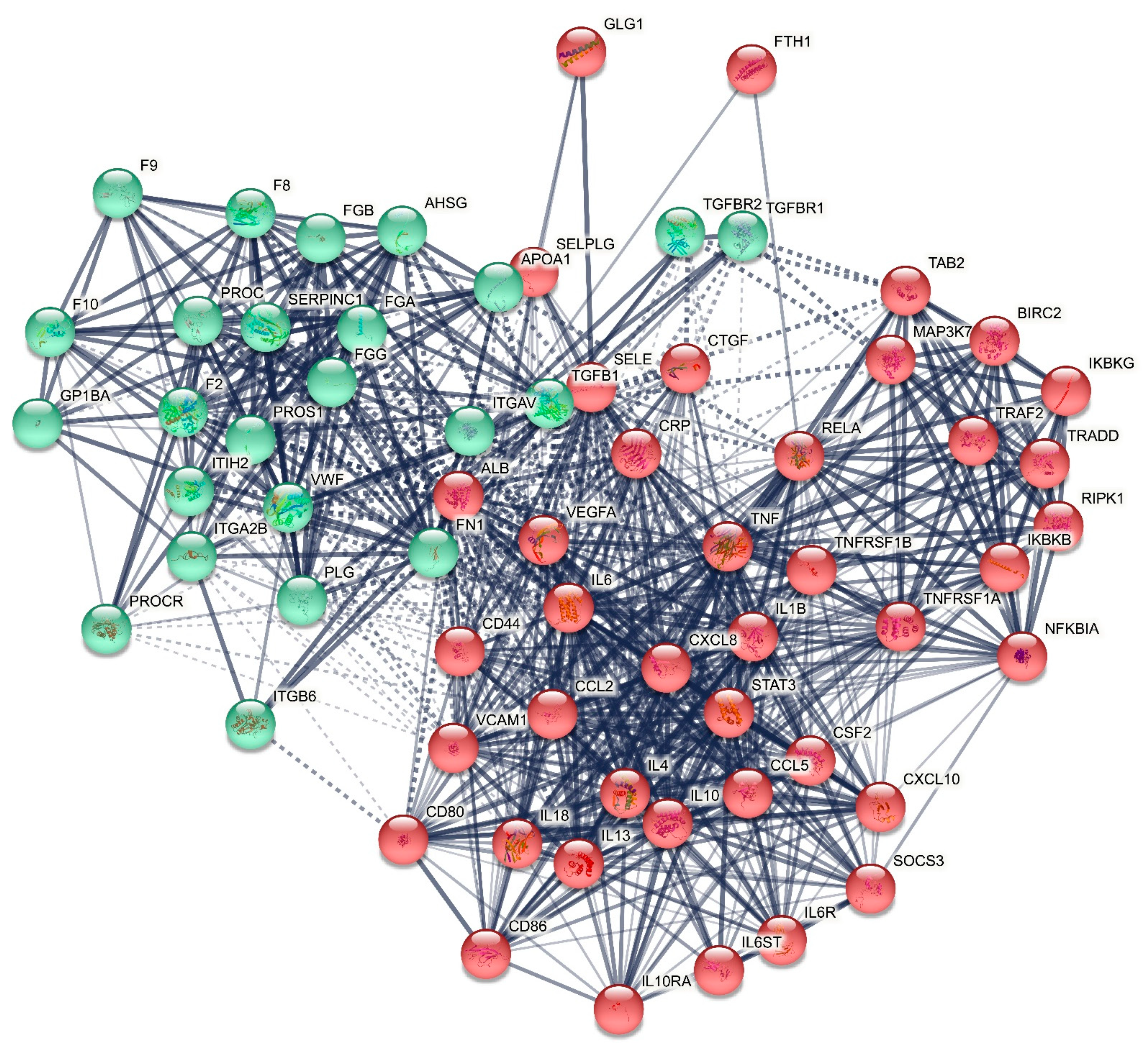
2.1.1. The DEP PPI Network Topography of Death Due to Ischemic Stroke
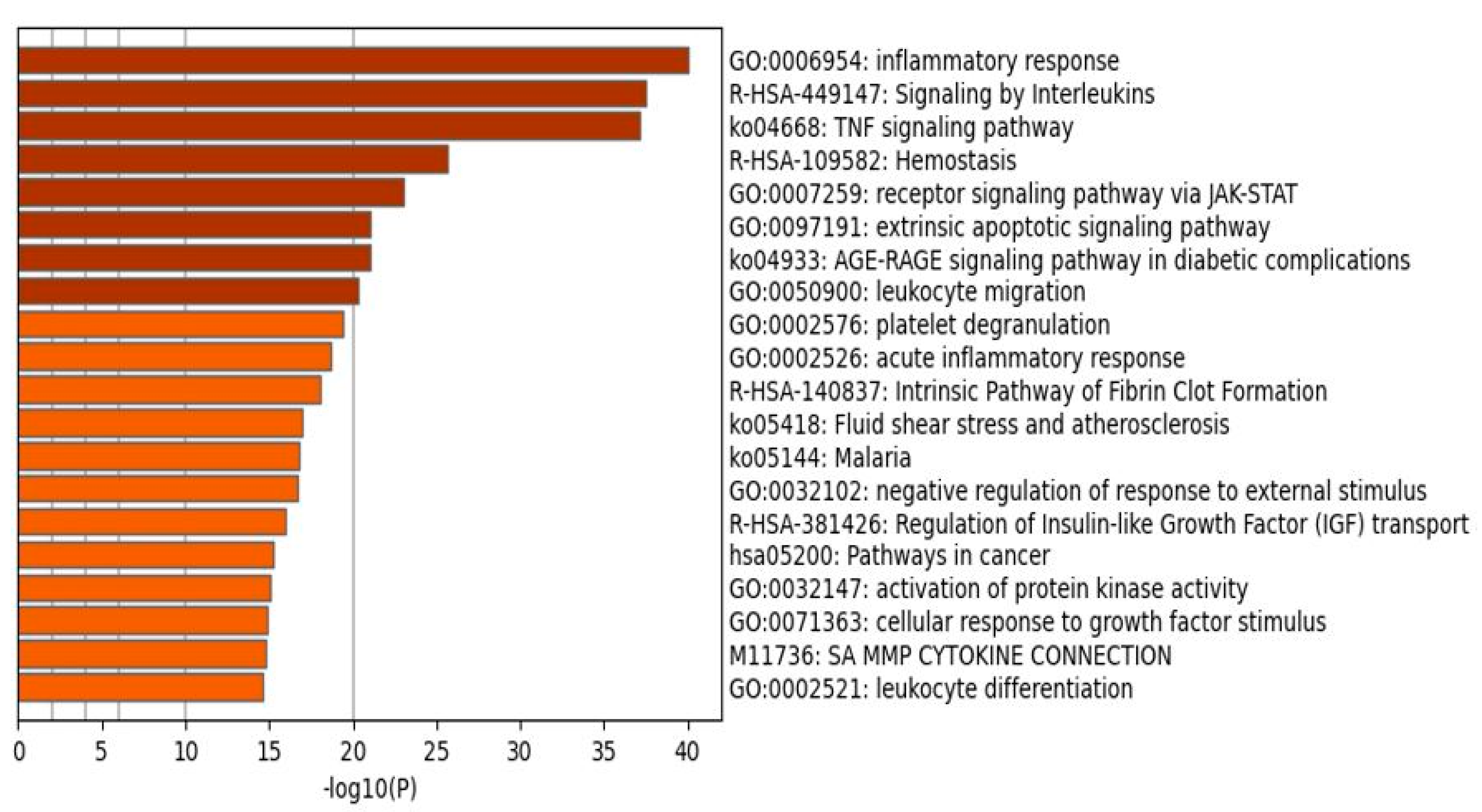
2.1.2. Enrichment Analysis in the DEPs of Death Due to Ischemic Stroke
2.1.3. Enrichment Analysis on Cluster One Genes of Death Due to Ischemic Stroke
2.1.4. Enrichment Analysis on Cluster Two Genes of Death Due to Ischemic Stroke
2.2. DEP + Gene Analysis
2.2.1. The DEP/Gene PPI Network Topography of Death Due to Ischemic Stroke
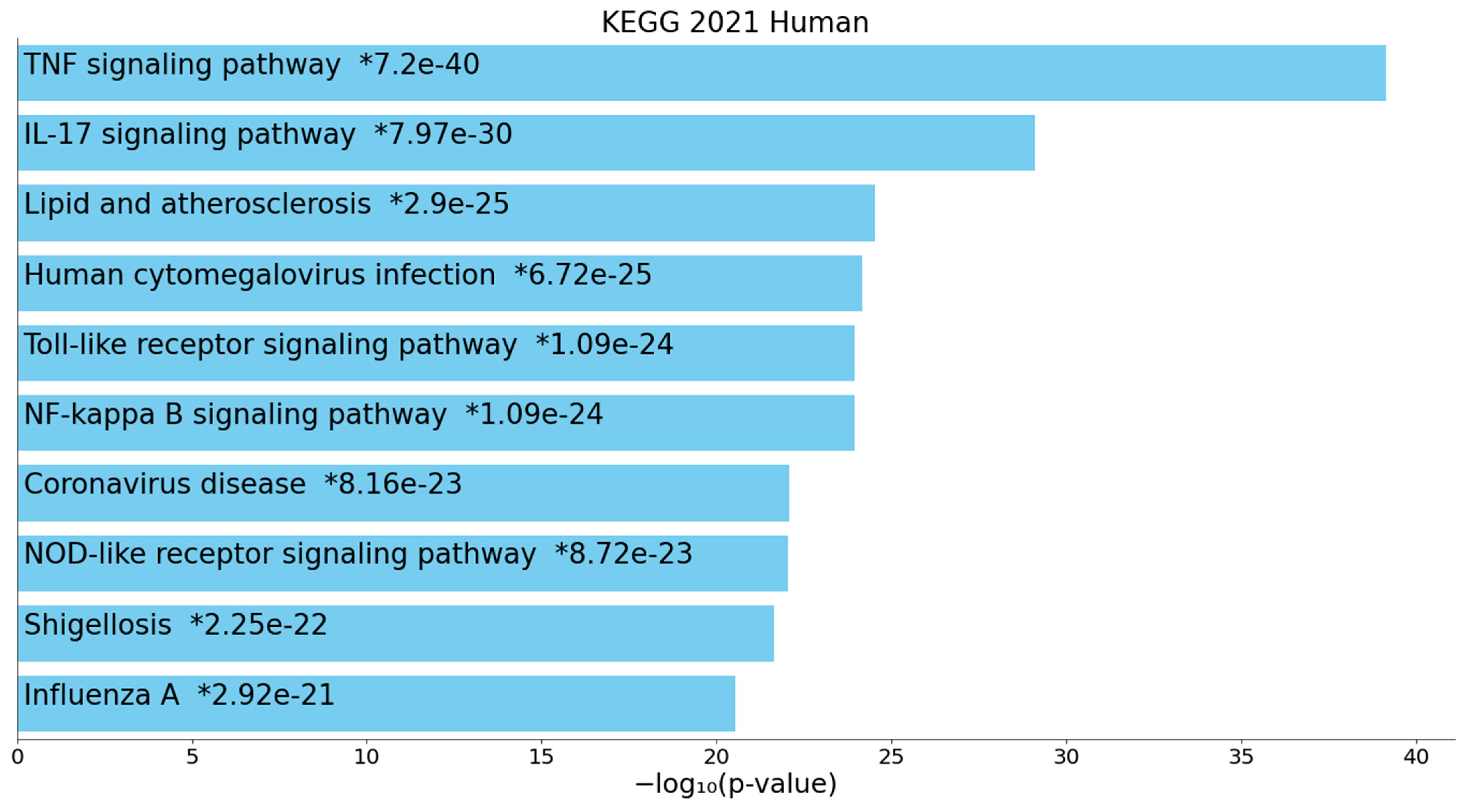
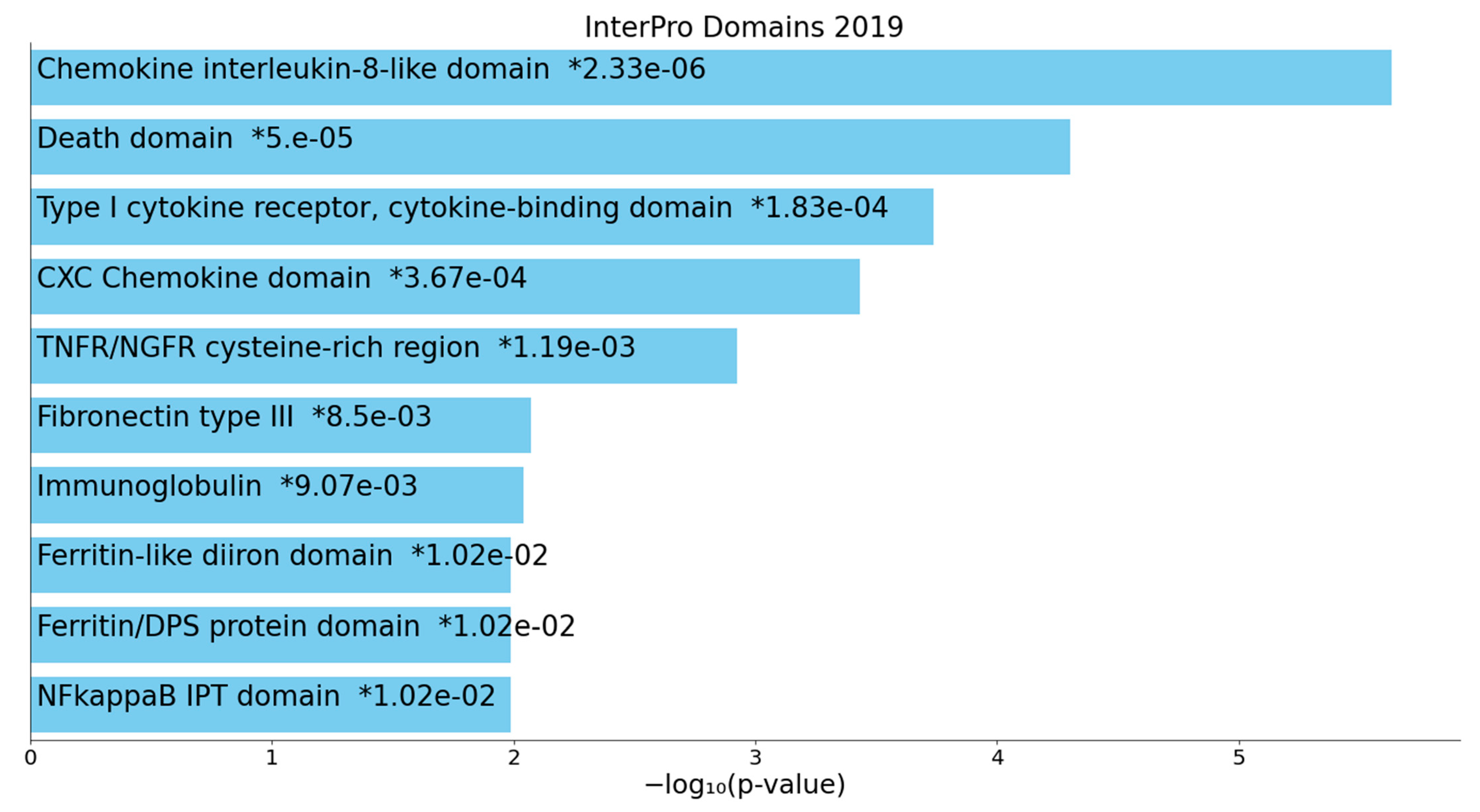
2.2.2. Enrichment/Annotation Analysis on Cluster One DEPs/Genes of Death Due to Ischemic Stroke
2.2.3. Enrichment/Annotation Analysis on Cluster Two DEPs/Genes of Death Due to Stroke
2.2.4. Building a Composite Network with MultiOmics Enrichment Analysis
3. Discussion
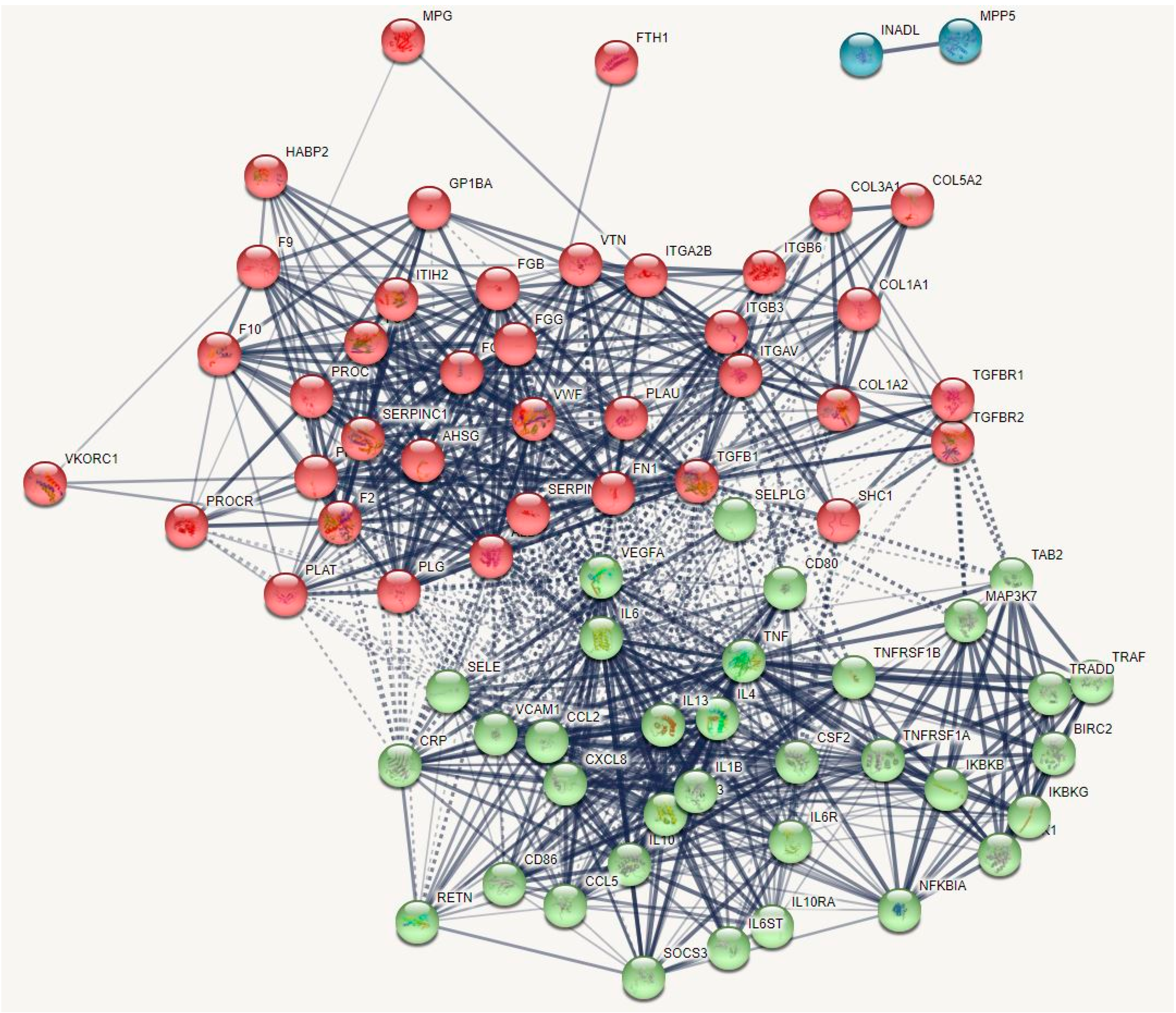
3.1. The Networks and Subnetworks of Death Due to Stroke
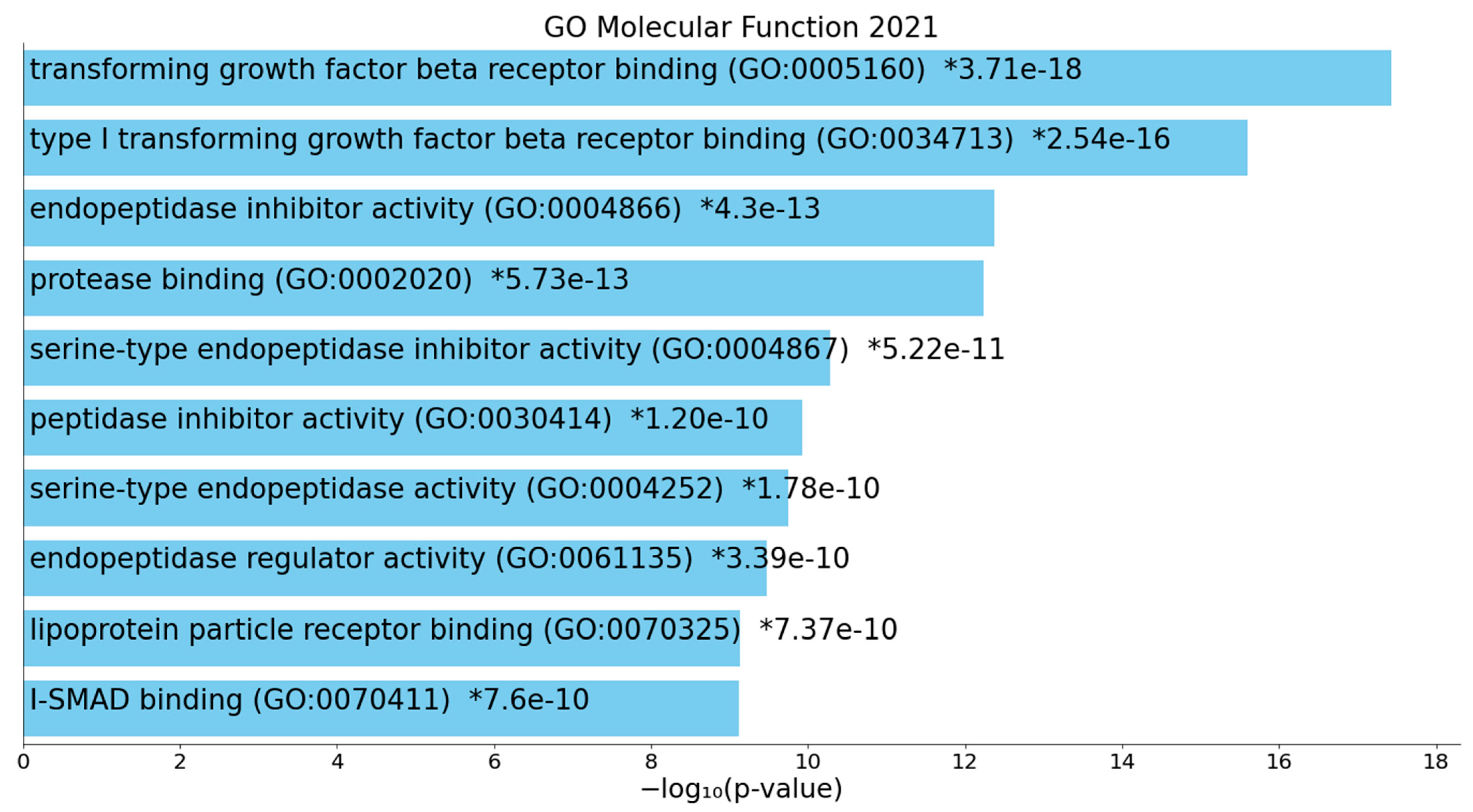
3.2. Terms Over-Represented in the Immune Subnetwork
3.3. Terms and Functions Over-Represented in the Hemostasis Subnetwork
3.4. Interactions, Pathways, and Functions Which Bridge the Immune and Hemostasis Subdomains
4. Methods
4.1. Selection of Seed Proteins, Genes, miRNA, and Metabolic Markers
4.2. PPI Network Construction, and Enrichment and Annotation Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609. [Google Scholar] [CrossRef]
- Sacco, R.L. Risk factors, outcomes, and stroke subtypes for ischemic stroke. Neurology 1997, 49, S39–S44. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, L.; Zhang, Y.; Yi, D.; Liu, L.; Rao, W.; Wu, Y.; Ma, D.; Liu, X.; Zhou, X.-H.A.; et al. Risk Factors of Stroke in Western and Asian Countries: A Systematic Review and Meta-analysis of Prospective Cohort Studies. BMC Public Health 2014, 14, 776. [Google Scholar] [CrossRef] [Green Version]
- Kleindorfer, D.O.; Towfighi, A.; Chaturvedi, S.; Cockroft, K.M.; Gutierrez, J.; Lombardi-Hill, D.; Kamel, H.; Kernan, W.N.; Kittner, S.J.; Leira, E.C.; et al. 2021 Guideline for the Prevention of Stroke in Patients With Stroke and Transient Ischemic Attack: A Guideline From the American Heart Association/American Stroke Association. Stroke 2021, 52, e364–e467, Erratum in 2021, 52, e483–e484. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, F.; Feng, X.; Wang, J.; Li, Z.; Zhan, Q.; Xia, J. Immunity and inflammation predictors for short-term outcome of stroke in young adults. Int. J. Neurosci. 2018, 128, 634–639. [Google Scholar] [CrossRef]
- Wiseman, S.; Marlborough, F.J.; Doubal, F.N.; Webb, D.J.; Wardlaw, J.M. Blood Markers of Coagulation, Fibrinolysis, Endothelial Dysfunction and Inflammation in Lacunar Stroke versus Non-Lacunar Stroke and Non-Stroke: Systematic Review and Meta-Analysis. Cerebrovasc. Dis. 2014, 37, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.L.C.F.; Alfieri, D.F.; de Araújo, M.C.M.; Trevisani, E.R.; Nagao, M.R.; Pesente, F.S.; Gelinski, J.R.; de Freitas, L.B.; Flauzino, T.; Lehmann, M.F.; et al. Immune-inflammatory, coagulation, adhesion, and imaging biomarkers combined in machine learning models improve the prediction of death 1 year after ischemic stroke. Clin. Exp. Med. 2021, 12. [Google Scholar] [CrossRef]
- Tzoulaki, I.; Murray, G.D.; Lee, A.J.; Rumley, A.; Lowe, G.D.; Fowkes, F.G.R. Relative Value of Inflammatory, Hemostatic, and Rheological Factors for Incident Myocardial Infarction and Stroke. Circulation 2007, 115, 2119–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alakbarzade, V.; Taylor, A.; Scully, M.; Simister, R.; Chandratheva, A. Utility of current thrombophilia screening in young patients with stroke and TIA. Stroke Vasc. Neurol. 2018, 3, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Hashem, S.S.; Helmy, S.M.; El-Fayomy, N.M.; Oraby, M.I.; Menshawy, M.; Dawood, N.A.; Hashem, H.S. Predictors of stroke outcome: The role of hemorheology, natural anticoagulants, and serum albumin. Egypt. J. Neurol. Psychiatry Neurosurg. 2018, 54, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Donkel, S.J.; Benaddi, B.; Dippel, D.W.; Cate, H.T.; De Maat, M.P. Prognostic Hemostasis Biomarkers in Acute Ischemic Stroke. Arter. Thromb. Vasc. Biol. 2019, 39, 360–372. [Google Scholar] [CrossRef]
- Lehmann, A.L.C.F.; Alfieri, D.F.; de Araújo, M.C.M.; Trevisani, E.R.; Nagao, M.R.; Pesente, F.S.; Gelinski, J.R.; de Freitas, L.B.; Flauzino, T.; Lehmann, M.F.; et al. Carotid intima media thickness measurements coupled with stroke severity strongly predict short-term outcome in patients with acute ischemic stroke: A machine learning study. Metab. Brain Dis. 2021, 36, 1747–1761. [Google Scholar] [CrossRef] [PubMed]
- Brott, T.; AdamsJr, H.P.; Olinger, C.P.; Marler, J.R.; Barsan, W.G.; Biller, J.; Spilker, J.; Holleran, R.; Eberle, R.; Hertzberg, V. Measurements of acute cerebral infarction: A clinical examination scale. Stroke 1989, 20, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonita, R.; Beaglehole, R. Recovery of motor function after stroke. Stroke 1988, 19, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Adams, H.P.; Davis, P.H.; Leira, E.C.; Chang, K.-C.; Bendixen, B.H.; Clarke, W.R.; Woolson, R.F.; Hansen, M.D. Baseline NIH Stroke Scale score strongly predicts outcome after stroke: A report of the Trial of Org 10172 in Acute Stroke Treatment (TOAST). Neurology 1999, 53, 126. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.C.; Wagner, D.P.; Haley, J.E.C.; Connors, A.F. Combined Clinical and Imaging Information as an Early Stroke Outcome Measure. Stroke 2002, 33, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Weimar, C.; König, I.; Kraywinkel, K.; Ziegler, A.; Diener, H. Age and National Institutes of Health Stroke Scale Score Within 6 Hours After Onset Are Accurate Predictors of Outcome After Cerebral Ischemia. Stroke 2004, 35, 158–162. [Google Scholar] [CrossRef]
- Bustamante, A.; Simats, A.; Vilar-Bergua, A.; García-Berrocoso, T.; Montaner, J. Blood/Brain Biomarkers of Inflammation After Stroke and Their Association with Outcome: From C-Reactive Protein to Damage-Associated Molecular Patterns. Neurotherapeutics 2016, 13, 671–684. [Google Scholar] [CrossRef]
- Johnson, C.O.; Nguyen, M.; A Roth, G.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Donkor, E.S. Stroke in the21stCentury: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 27, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Heller, R.F.; Langhorne, P.; James, E. Improving stroke outcome: The benefits of increasing availability of technology. Bull. World Health Organ. 2000, 78, 1337–1343. [Google Scholar] [PubMed]
- Brønnum-Hansen, H.; Davidsen, M.; Thorvaldsen, P. Long-Term Survival and Causes of Death After Stroke. Stroke 2001, 32, 2131–2136. [Google Scholar] [CrossRef]
- Medicover Hospitals. What is the Life Expectancy after a Brain Stroke? 2021. Available online: https://www.medicoverhospitals.in/what-is-the-life-expectancy-after-a-brain-stroke/ (accessed on 26 August 2021).
- Petty, G.W.; Brown, R.D.; Whisnant, J.P.; Sicks, J.D.; O’Fallon, W.M.; Wiebers, D.O. Survival and recurrence after first cerebral infarction. Neurology 1998, 50, 208–216. [Google Scholar] [CrossRef]
- Vernino, S.; Brown, R.D., Jr.; Sejvar, J.J.; Sicks, J.D.; Petty, G.W.; O’Fallon, W.M. Cause specific mortality after first cerebral infarction: A population-based study. Stroke 2003, 34, 1828–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koton, S.; Tanne, D.; Green, M.S.; Bornstein, N.M. Mortality and predictors of death 1 month and 3 years after first-ever ischemic stroke: Data from the first national acute stroke Israeli survey (NASIS 2004). Neuroepidemiology 2010, 34, 90–96. [Google Scholar] [CrossRef]
- Jia, Q.; Liu, G.; Zheng, H.; Zhao, X.; Wang, C.; Wang, Y.; Wang, Y. Impaired Glucose Regulation Predicted 1-Year Mortality of Chinese Patients with Ischemic Stroke: Data from Abnormal Glucose Regulation in Patients with Acute Stroke across China. Stroke 2014, 45, 1498–1500. [Google Scholar]
- Tziomalos, K.; Spanou, M.; Bouziana, S.D.; Papadopoulou, M.; Giampatzis, V.; Kostaki, S.; Dourliou, V.; Tsopozidi, M.; Savopoulos, C.; Hatzitolios, A.I. Type 2 diabetes is associated with a worse functional outcome of ischemic stroke. World J. Diabetes 2014, 5, 939–944. [Google Scholar] [CrossRef]
- Carter, A.M.; Catto, A.J.; Bamford, J.M.; Grant, P.J. Platelet GP IIIa Pl A and GP Ib Variable Number Tandem Repeat Polymorphisms and Markers of Platelet Activation in Acute Stroke. Arter. Thromb. Vasc. Biol. 1998, 18, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, T.; Geleijnse, J.M.; Kluft, C.; Giltay, E.J.; Kok, F.J.; Schouten, E.G. 4G/4G Genotype of PAI-1 Gene Is Associated with Reduced Risk of Stroke in Elderly. Stroke 2003, 34, 2822–2828. [Google Scholar] [CrossRef] [Green Version]
- Reiner, A.P.; Carty, C.L.; Jenny, N.S.; Nievergelt, C.; Cushman, M.; Stearns-Kurosawa, D.J.; Kurosawa, S.; Kuller, L.H.; Lange, L.A. PROC, PROCR and PROS1 polymorphisms, plasma anticoagulant phenotypes, and risk of cardiovascular disease and mortality in older adults: The Cardiovascular Health Study. J. Thromb. Haemost. 2008, 6, 1625–1632. [Google Scholar] [CrossRef] [Green Version]
- Trompet, S.; Pons, D.; Kanse, S.M.; de Craen, A.J.M.; Ikram, M.A.; Verschuren, J.J.W.; Zwinderman, A.H.; Doevendans, P.A.F.M.; Tio, R.A.; de Winter, R.J.; et al. Factor VII Activating Protease Polymorphism (G534E) Is Associated with Increased Risk for Stroke and Mortality. Stroke Res. Treat. 2011, 2011, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.; Lin, Y.; Song, W.; Sun, K.; Yu, H.; Zhang, Y.; Zhang, C.; Li, L.; Suo, M.; Hui, R.; et al. Variants of COL3A1 Are Associated with the Risk of Stroke Recurrence and Prognosis in the Chinese Population: A Prospective Study. J. Mol. Neurosci. 2014, 53, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Martiskainen, M.; Oksala, N.; Pohjasvaara, T.; Kaste, M.; Oksala, A.; Karhunen, P.J.; Erkinjuntti, T. Βeta-fibrinogen gene promoter A −455 allele associated with poor longterm survival among 55–71 years old Caucasian women in Finnish stroke cohort. BMC Neurol. 2014, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Shemirani, A.-H.; Antalfi, B.; Pongrácz, E.; Mezei, Z.A.; Bereczky, Z.; Csiki, Z. Factor XIII-A subunit Val34Leu polymorphism in fatal atherothrombotic ischemic stroke. Blood Coagul. Fibrinolysis 2014, 25, 364–368. [Google Scholar] [CrossRef]
- Bouziana, S.D.; Tziomalos, K.; Goulas, A.; Vyzantiadis, T.-A.; Panderi, A.; Hatzitolios, A. Major Adipokines and the −420C>G Resistin Gene Polymorphism as Predictors of Acute Ischemic Stroke Severity and In-Hospital Outcome. J. Stroke Cerebrovasc. Dis. 2018, 27, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Yang, Y.; Yang, S.; Tu, X.; Wang, Y.; Song, Y.; Hui, R.; Zhang, W. Effect of gene–gene and gene–environment interaction on the risk of first-ever stroke and poststroke death. Mol. Genet. Genom. Med. 2019, 7, e846. [Google Scholar] [CrossRef] [Green Version]
- Mola-Caminal, M.; Carrera, C.; Soriano-Tárraga, C.; Giralt-Steinhauer, E.; Díaz-Navarro, R.M.; Tur, S.; Jiménez, C.; Medina-Dols, A.; Cullell, N.; Torres-Aguila, N.P.; et al. PATJ Low Frequency Variants Are Associated with Worse Ischemic Stroke Functional Outcome. Circ. Res. 2019, 124, 114–120. [Google Scholar] [CrossRef]
- Ryu, C.S.; Bae, J.; Kim, I.J.; Kim, J.; Oh, S.H.; Kim, O.J.; Kim, N.K. MPG and NPRL3 Polymorphisms are Associated with Ischemic Stroke Susceptibility and Post-Stroke Mortality. Diagnostics 2020, 10, 947. [Google Scholar] [CrossRef]
- Ryu, C.S.; Oh, S.H.; Lee, K.O.; Park, H.S.; An, H.J.; Lee, J.Y.; Ko, E.J.; Park, H.W.; Kim, O.J.; Kim, N.K. MiR-10a, 27a, 34b/c, and 300 Polymorphisms are Associated with Ischemic Stroke Susceptibility and Post-Stroke Mortality. Life 2020, 10, 309. [Google Scholar] [CrossRef]
- Song, L.; Li, H.; Suo, M.; Sun, Y.; Su, M.; Song, Y.; Xiao, N.; Hui, R.; Qin, C.; Chen, J. A functional variant of the long noncoding RNA AL110200 is associated with the risk of ischaemic stroke recurrence. Eur. J. Neurol. 2021, 28, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, C.; Chimowitz, M.I. Stroke Caused by Atherosclerosis of the Major Intracranial Arteries. Circ. Res. 2017, 120, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Jüttler, E.; Unterberg, A.; Woitzik, J.; Bösel, J.; Amiri, H.; Sakowitz, O.W.; Gondan, M.; Hill, M.; Limprecht, R.; Luntz, S.; et al. Hemicraniectomy in Older Patients with Extensive Middle-Cerebral-Artery Stroke. N. Engl. J. Med. 2014, 370, 1091–1100. [Google Scholar] [CrossRef]
- Norrving, B.; Barrick, J.; Davalos, A.; Dichgans, M.; Cordonnier, C.; Guekht, A.; Kutluk, K.; Mikulik, R.; Wardlaw, J.; Richard, E.; et al. Action Plan for Stroke in Europe 2018–2030. Eur. Stroke J. 2018, 3, 309–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Liu, S.; Liu, X.; Zhuang, W. Bioinformatics Gene Analysis of Potential Biomarkers and Therapeutic Targets for Unstable Atherosclerotic Plaque-Related Stroke. J. Mol. Neurosci. 2021, 71, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef]
- Steffen, B.J.; Breier, G.; Butcher, E.C.; Schulz, M.; Engelhardt, B. ICAM-1, VCAM-1, and MAdCAM-1 are expressed on choroid plexus epithelium but not endothelium and mediate binding of lymphocytes in vitro. Am. J. Pathol. 1996, 148, 1819–1838. [Google Scholar]
- Tang, S.-C.; Arumugam, T.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.-G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [Green Version]
- Nurmi, A.; Lindsberg, P.; Koistinaho, M.; Zhang, W.; Juettler, E.; Karjalainen-Lindsberg, M.-L.; Weih, F.; Frank, N.; Schwaninger, M.; Koistinaho, J. Nuclear Factor-κB Contributes to Infarction After Permanent Focal Ischemia. Stroke 2004, 35, 987–991. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Shah, F.A.; Zeb, A.; Malik, I.; Alvi, A.M.; AlKury, L.T.; Rashid, S.; Hussain, I.; Ullah, N.; Khan, A.U.; et al. NF-κB Inhibitors Attenuate MCAO Induced Neurodegeneration and Oxidative Stress—A Reprofiling Approach. Front. Mol. Neurosci. 2020, 13, 33. [Google Scholar] [CrossRef]
- Kim, S.K.; Jang, H.M.; Kim, D.-Y. The promoter polymorphism of NFKB1 gene contributes to susceptibility of ischemic stroke in Korean population. J. Exerc. Rehabilitation 2018, 14, 1096–1100. [Google Scholar] [CrossRef]
- Lanzillotta, A.; Sarnico, I.; Ingrassia, R.; Boroni, F.; Branca, C.; Benarese, M.; Faraco, G.; Blasi, F.; Chiarugi, A.; Spano, P.; et al. The acetylation of RelA in Lys310 dictates the NF-κB-dependent response in post-ischemic injury. Cell Death Dis. 2010, 1, e96, Erratum in 2010, 1, e107. [Google Scholar] [CrossRef] [Green Version]
- Inta, I.; Paxian, S.; Maegele, I.; Zhang, W.; Pizzi, M.; Spano, P.; Sarnico, I.; Muhammad, S.; Herrmann, O.; Inta, D.; et al. Bim and Noxa Are Candidates to Mediate the Deleterious Effect of the NF- B Subunit RelA in Cerebral Ischemia. J. Neurosci. 2006, 26, 12896–12903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajalli-Nezhad, S.; Karimian, M.; Beyer, C.; Atlasi, M.A.; Tameh, A.A. The regulatory role of Toll-like receptors after ischemic stroke: Neurosteroids as TLR modulators with the focus on TLR2/4. Experientia 2019, 76, 523–537. [Google Scholar] [CrossRef]
- Brea, D.; Blanco, M.; Ramos-Cabrer, P.; Moldes, O.; Arias, S.; Pérez-Mato, M.; Leira, R.; Sobrino, T.; Castillo, J. Toll-like receptors 2 and 4 in ischemic stroke: Outcome and therapeutic values. J. Cereb. Blood Flow Metab. 2011, 31, 1424–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.-X.; Yang, Q.-W.; Lv, F.-L.; Cui, J.; Fu, H.-B.; Wang, J.-Z. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem. Biophys. Res. Commun. 2007, 353, 509–514. [Google Scholar] [CrossRef]
- Caso, J.R.; Pradillo, J.M.; Hurtado, O.; Leza, J.C.; Moro, M.A.; Lizasoain, I. Toll-Like Receptor 4 Is Involved in Subacute Stress–Induced Neuroinflammation and in the Worsening of Experimental Stroke. Stroke 2008, 39, 1314–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Chang, Y.-M.; Yu, J.-M.; Yen, J.-H.; Chang, J.-G.; Hu, C.-J. Toll-like receptor 4 gene C119A but not Asp299Gly polymorphism is associated with ischemic stroke among ethnic Chinese in Taiwan. Atherosclerosis 2005, 180, 305–309. [Google Scholar] [CrossRef]
- Zaremba, J.; Losy, J. Early TNF-α levels correlate with ischaemic stroke severity. Acta Neurol. Scand. 2001, 104, 288–295. [Google Scholar] [CrossRef]
- Doll, D.N.; Rellick, S.L.; Barr, T.L.; Ren, X.; Simpkins, J.W. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J. Neurochem. 2015, 132, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Zhang, X.; Wang, J.-H.; Liu, Q.-W.; Wang, X.-Q.; Wu, Z.-Q.; Wang, J.; Zhang, C.; Qun, S. Gene polymorphisms and circulating levels of the TNF-alpha are associated with ischemic stroke: A meta-analysis based on 19,873 individuals. Int. Immunopharmacol. 2019, 75, 105827. [Google Scholar] [CrossRef]
- Tian, J.; Bai, Y.; You, A.; Shen, R.; Yan, J.; Deng, W.; Wen, L.; Li, M.; Teng, J. Interleukin-17 receptor C gene polymorphism reduces treatment effect and promotes poor prognosis of ischemic stroke. Biosci. Rep. 2019, 39, BSR20190435, Erratum in 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liao, Y.; Liu, Z.; Dai, Y.; Li, Y.; Li, Y.; Tang, Y. Interleukin-17 and ischaemic stroke. Immunology 2021, 162, 179–193. [Google Scholar] [CrossRef]
- Planas, A.M.; Soriano, M.A.; Berruezo, M.; Justicia, C.; Estrada, A.; Pitarch, S.; Ferrer, I. Induction of Stat3, a Signal Transducer and Transcription Factor, in Reactive Microglia following Transient Focal Cerebral Ischaemia. Eur. J. Neurosci. 1996, 8, 2612–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Tanaka, K.; Nogawa, S.; Dembo, T.; Kosakai, A.; Fukuuchi, Y. Phosphorylation of Signal Transducer and Activator of Transcription-3 (Stat3) after Focal Cerebral Ischemia in Rats. Exp. Neurol. 2001, 170, 63–71. [Google Scholar] [CrossRef]
- Choi, J.-S.; Kim, S.Y.; Cha, J.-H.; Choi, Y.-S.; Sung, K.-W.; Oh, S.T.; Kim, O.N.; Chung, J.-W.; Chun, M.-H.; Lee, S.B.; et al. Upregulation of gp130 and STAT3 activation in the rat hippocampus following transient forebrain ischemia. Glia 2003, 41, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Satriotomo, I.; Bowen, K.K.; Vemuganti, R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J. Neurochem. 2006, 98, 1353–1368. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, J.; Xu, J.; Zheng, W.; Chen, Q.; Jiao, D. Study on the mechanism of JAK2/STAT3 signaling pathway-mediated inflammatory reaction after cerebral ischemia. Mol. Med. Rep. 2018, 17, 5007–5012. [Google Scholar] [CrossRef]
- Wang, X.-L.; Qiao, C.-M.; Liu, J.-O.; Li, C.-Y. Inhibition of the SOCS1-JAK2-STAT3 Signaling Pathway Confers Neuroprotection in Rats with Ischemic Stroke. Cell. Physiol. Biochem. 2017, 44, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geddes, K.; Magalhães, J.G.; Girardin, S.E. Unleashing the therapeutic potential of NOD-like receptors. Nat. Rev. Drug Discov. 2009, 8, 465–479. [Google Scholar] [CrossRef]
- Liu, H.; Wei, X.; Kong, L.; Liu, X.; Cheng, L.; Yan, S.; Zhang, X.; Chen, L. NOD2 is Involved in the Inflammatory Response after Cerebral Ischemia-Reperfusion Injury and Triggers NADPH Oxidase 2-Derived Reactive Oxygen Species. Int. J. Biol. Sci. 2015, 11, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, N.; Castillo, J.; Dávalos, A.; Chamorro, A. Proinflammatory Cytokines and Early Neurological Worsening in Ischemic Stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef] [Green Version]
- Lv, K.; Yang, Y. Relationship between interleukin-10 polymorphisms and susceptibility to ischemic stroke: A Meta-analysis. Scand. J. Clin. Lab. Investig. 2020, 80, 20–24. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, H.; Sun, G.; Zhang, J.; Edwards, N.J.; Aronowski, J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J. Neurosci. 2015, 35, 11281–11291. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Barreto, G.E.; Xu, L.; Ouyang, Y.B.; Xie, X.S.; Giffard, R.G. Increased Brain Injury and Worsened Neurological Outcome in Interleukin-4 Knockout Mice After Transient Focal Cerebral Ischemia. Stroke 2011, 42, 2026–2032. [Google Scholar] [CrossRef] [Green Version]
- Taj, S.H.; Le Blon, D.; Hoornaert, C.; Daans, J.; Quarta, A.; Praet, J.; Van Der Linden, A.; Ponsaerts, P.; Hoehn, M. Targeted intracerebral delivery of the anti-inflammatory cytokine IL13 promotes alternative activation of both microglia and macrophages after stroke. J. Neuroinflam. 2018, 15, 1–17. [Google Scholar] [CrossRef]
- Couper, K.; Blount, D.G.; Riley, E.M. IL-10: The Master Regulator of Immunity to Infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Pera, J.; Zawadzka, M.; Kaminska, B.; Szczudlik, A. Influence of chemical and ischemic preconditioning on cytokine expression after focal brain ischemia. J. Neurosci. Res. 2004, 78, 132–140. [Google Scholar] [CrossRef]
- Bonni, S.; Bonni, A. SnoN signaling in proliferating cells and postmitotic neurons. FEBS Lett. 2012, 586, 1977–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caraci, F.; Battaglia, G.; Sortino, M.A.; Spampinato, S.F.; Molinaro, G.; Copani, A.; Nicoletti, F.; Bruno, V.M.G. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: Still a hot topic? Neurochem. Int. 2012, 61, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Kumar, P.; Misra, S.; Kumar, A.; Faruq, M.; Shakya, S.; Vardhan, G.; Vivekanandhan, S.; Srivastava, A.K. Transforming growth factor-β1 (C509T, G800A, and T869C) gene polymorphisms and risk of ischemic stroke in North Indian population: A hospital-based case-control study. Ann. Indian Acad. Neurol. 2017, 20, 5–12. [Google Scholar] [CrossRef]
- Konkel, J.E.; Chen, W. Balancing acts: The role of TGF-β in the mucosal immune system. Trends Mol. Med. 2011, 17, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, P.; Somanathan, R. Cell signaling and receptors in toxicity of advanced glycation end products (AGEs): α-dicarbonyls, radicals, oxidative stress, and antioxidants. J. Recept. Signal Transduct. 2011, 31, 332–339. [Google Scholar] [CrossRef]
- Song, L.; Liu, F.-F.; Liu, C.-Y.; Li, X.-P.; Zheng, S.-Z.; Li, Q.-Q.; Liu, Q. Neuroprotective effects of SMADs in a rat model of cerebral ischemia/reperfusion. Neural Regen. Res. 2015, 10, 438–444. [Google Scholar] [CrossRef]
- Zhu, H.; Gui, Q.; Hui, X.; Wang, X.; Jiang, J.; Ding, L.; Sun, X.; Wang, Y.; Chen, H. TGF-β1/Smad3 Signaling Pathway Suppresses Cell Apoptosis in Cerebral Ischemic Stroke Rats. Med. Sci. Monit. 2017, 23, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Supanc, V.; Biloglav, Z.; Kes, V.B.; Demarin, V. Role of cell adhesion molecules in acute ischemic stroke. Ann. Saudi Med. 2011, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [Green Version]
- Kivisäkk, P.; Mahad, D.J.; Callahan, M.K.; Trebst, C.; Tucky, B.; Wei, T.; Wu, L.; Baekkevold, E.S.; Lassmann, H.; Staugaitis, S.M.; et al. Human cerebrospinal fluid central memory CD4+T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA 2003, 100, 8389–8394. [Google Scholar] [CrossRef] [Green Version]
- Maglinger, B.; Sands, M.; Frank, J.A.; McLouth, C.J.; Trout, A.L.; Roberts, J.M.; Grupke, S.; Turchan-Cholewo, J.; Stowe, A.M.; Fraser, J.F.; et al. Intracranial VCAM1 at time of mechanical thrombectomy predicts ischemic stroke severity. J. Neuroinflam. 2021, 18, 1–13. [Google Scholar] [CrossRef]
- Frijns, C.; Kappelle, L. Inflammatory Cell Adhesion Molecules in Ischemic Cerebrovascular Disease. Stroke 2002, 33, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Álvarez-Sabín, J.; Martínez-Vila, E.; Montaner, J.; Sobrino, T.; Vivancos, J. Inflammation markers and prediction of post-stroke vascular disease recurrence: The MITICO study. J. Neurol. 2009, 256, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.R.; Brey, R.L.; Tilley, B.C.; Thompson, J.L.P.; Sacco, R.L.; Sciacca, R.R.; Murphy, A.; Lu, Y.; Costigan, T.M.; Rhine, C.; et al. Antiphospholipid Antibodies and Subsequent Thrombo-occlusive Events in Patients with Ischemic Stroke. JAMA 2004, 291, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, C.; Siddiqi, Z.; Morgenlander, J.C.; Goldstein, L.B. Use of specialized coagulation testing in the evaluation of patients with acute ischemic stroke. Neurology 2001, 56, 624–627. [Google Scholar] [CrossRef]
- Bushnell, C.D.; Siddiqi, Z.; Goldstein, L.B. Improving patient selection for coagulopathy testing in the setting of acute ischemic stroke. Neurology 2001, 57, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.L.; Sachan, R.; Seth, G. Combined deficiency of proteins C and S: Ischaemic stroke in young individuals. BMJ Case Rep. 2013, 2013, bcr2012008016. [Google Scholar] [CrossRef]
- Luo, G.-P.; Ni, B.; Yang, X.; Wu, Y.-Z. von Willebrand Factor: More Than a Regulator of Hemostasis and Thrombosis. Acta Haematol. 2012, 128, 158–169. [Google Scholar] [CrossRef]
- Gragnano, F.; Sperlongano, S.; Golia, E.; Natale, F.; Bianchi, R.; Crisci, M.; Fimiani, F.; Pariggiano, I.; Diana, V.; Carbone, A.; et al. The Role of von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediat. Inflamm. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Sidelmann, J.J.; Gram, J.B.; Jespersen, J.; Kluft, C. Fibrin Clot Formation and Lysis: Basic Mechanisms. Semin. Thromb. Hemost. 2000, 26, 605–618. [Google Scholar] [CrossRef]
- Kozuka, K.; Kohriyama, T.; Nomura, E.; Ikeda, J.; Kajikawa, H.; Nakamura, S. Endothelial markers and adhesion molecules in acute ischemic stroke—sequential change and differences in stroke subtype. Atherosclerosis 2002, 161, 161–168. [Google Scholar] [CrossRef]
- Hanson, E.; Jood, K.; Karlsson, S.; Nilsson, S.; Blomstrand, C.; Jern, C. Plasma levels of von Willebrand factor in the etiologic subtypes of ischemic stroke. J. Thromb. Haemost. 2011, 9, 275–281. [Google Scholar] [CrossRef]
- Ancedy, Y.; Berthelot, E.; Lang, S.; Ederhy, S.; Boyer-Chatenet, L.; Di Angelantonio, E.; Dufour, L.S.; Etienney, A.; Adavane-Scheublé, S.; Boccara, F.; et al. Is von Willebrand factor associated with stroke and death at mid-term in patients with non-valvular atrial fibrillation? Arch. Cardiovasc. Dis. 2018, 111, 357–369. [Google Scholar] [CrossRef]
- Tóth, N.K.; Székely, E.G.; Czuriga-Kovács, K.R.; Sarkady, F.; Nagy, O.; Lánczi, L.I.; Berényi, E.; Fekete, K.; Fekete, I.; Csiba, L.; et al. Elevated Factor VIII and von Willebrand Factor Levels Predict Unfavorable Outcome in Stroke Patients Treated with Intravenous Thrombolysis. Front. Neurol. 2018, 8, 721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirks, M.; Grosse, G.M.; Böckmann, M.; Goetz, F.; Pasedag, T.; Bode-Böger, S.M.; Martens-Lobenhoffer, J.; Budde, U.; Lanfermann, H.; Lichtinghagen, R.; et al. ADAMTS-13 Activity Predicts Outcome in Acute Ischaemic Stroke Patients Undergoing Endovascular Treatment. Thromb. Haemost. 2018, 47, 758–767. [Google Scholar] [CrossRef]
- Dai, K.; Gao, W.; Ruan, C. The Sma I Polymorphism in the von Willebrand Factor Gene Associated with Acute Ischemic Stroke. Thromb. Res. 2001, 104, 389–395. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, S.F.; Stoll, G.; Wagner, D.D.; Kleinschnitz, C. von Willebrand Factor. Stroke 2012, 43, 599–606. [Google Scholar] [CrossRef]
- Drouet, L. Fibrinogen: A Treatable Risk Factor? Cerebrovasc. Dis. 1996, 6, 2–6. [Google Scholar] [CrossRef]
- Pieters, M.; Wolberg, A.S. Fibrinogen and fibrin: An illustrated review. Res. Pr. Thromb. Haemost. 2019, 3, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Rallidis, L.S.; Vikelis, M.; Panagiotakos, D.B.; Liakos, G.K.; Krania, E.; Kremastinos, D.T. Usefulness of inflammatory and haemostatic markers to predict short-term risk for death in middle-aged ischaemic stroke patients. Acta Neurol. Scand. 2008, 117, 415–420. [Google Scholar] [CrossRef]
- Swarowska, M.; Polczak, A.; Pera, J.; Klimkowicz-Mrowiec, A.; Slowik, A.; Dziedzic, T. Hyperfibrinogenemia predicts long-term risk of death after ischemic stroke. J. Thromb. Thrombolysis 2014, 38, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Di Napoli, M.; Papa, F. Inflammation, Hemostatic Markers, and Antithrombotic Agents in Relation to Long-Term Risk of New Cardiovascular Events in First-Ever Ischemic Stroke Patients. Stroke 2002, 33, 1763–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikija, S.; Trkulja, V.; Mutzenbach, J.S.; McCoy, M.R.; Ganger, P.; Sellner, J. Fibrinogen consumption is related to intracranial clot burden in acute ischemic stroke: A retrospective hyperdense artery study. J. Transl. Med. 2016, 14, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beamer, N.B.; Coull, B.M.; Clark, W.M.; Hazel, J.S.; Silberger, J.R. Interleukin-6 and interleukin-1 receptor antagonist in acute stroke. Ann. Neurol. 1995, 37, 800–805. [Google Scholar]
- Di Napoli, M.; Papa, F.; Bocola, V. Prognostic Influence of Increased C-Reactive Protein and Fibrinogen Levels in Ischemic Stroke. Stroke 2001, 32, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, M.; Singh, P. Is Plasma Fibrinogen Useful in Evaluating Ischemic Stroke Patients? Stroke 2009, 40, 1549–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, W.M.; Erickson, L.P.; Bruck, D.; Kittelson, J. Hemostatic Markers in Acute Ischemic Stroke. Stroke 1996, 27, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.; Langhorne, P.; Rumley, A.; Lowe, G.D.; Stott, D.J. D-dimer predicts early clinical progression. Stroke 2004, 35, 1421–1425. [Google Scholar]
- Carter, A.M.; Catto, A.J.; Mansfield, M.; Bamford, J.M.; Grant, P.J. Predictive Variables for Mortality After Acute Ischemic Stroke. Stroke 2007, 38, 1873–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaner, J.; Molina, C.A.; Arenillas, J.F.; Santamarina, E.; Alvarez-Sabín, J.; Ribo, M. Admission fibrinolytic profile predicts clot lysis resistance in stroke patients treated with tissue plasminogen activator. Thromb. Haemost. 2004, 91, 1146–1151. [Google Scholar] [CrossRef]
- Merino, P.; Díaz, A.; Yepes, M. Urokinase-type Plasminogen Activator (uPA) and its Receptor (uPAR) Promote Neurorepair in the Ischemic Brain. Recept. Clin. Investig. 2017, 4, e1552. [Google Scholar] [CrossRef]
- Diaz, A.; Merino, P.; Manrique, L.G.; Cheng, L.; Yepes, M. Urokinase-type plasminogen activator (uPA) protects the tripartite synapse in the ischemic brain via ezrin-mediated formation of peripheral astrocytic processes. Br. J. Pharmacol. 2019, 39, 2157–2171. [Google Scholar] [CrossRef]
- Menshikov, M.; Plekhanova, O.; Cai, H.; Chalupsky, K.; Parfyonova, Y.; Bashtrikov, P.; Tkachuk, V.; Berk, B.C. Urokinase Plasminogen Activator Stimulates Vascular Smooth Muscle Cell Proliferation Via Redox-Dependent Pathways. Arter. Thromb. Vasc. Biol. 2006, 26, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan, and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guell, K.; Bix, G.J. Brain endothelial cell specific integrins and ischemic stroke. Expert Rev. Neurother. 2014, 14, 1287–1292. [Google Scholar] [CrossRef]
- Edwards, D.N.; Bix, G.J. Roles of blood-brain barrier integrins and extracellular matrix in stroke. Am. J. Physiol. Physiol. 2019, 316, C252–C263. [Google Scholar] [CrossRef]
- Edwards, D.N.; Bix, G.J. The Inflammatory Response After Ischemic Stroke: Targeting β2 and β1 Integrins. Front. Neurosci. 2019, 13, 540. [Google Scholar] [CrossRef]
- Baeten, K.M.; Akassoglou, K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev. Neurobiol. 2011, 71, 1018–1039. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Kusindarta, D.; Wihadmadyatami, H. The Role of Extracellular Matrix in Tissue Regeneration. IntechOpen Book Series. Open access peer-reviewed paper. 2018. Available online: https://www.intechopen.com/chapters/60312 (accessed on 19 September 2021).
- Paar, M.; Rossmann, C.; Nusshold, C.; Wagner, T.; Schlagenhauf, A.; Leschnik, B.; Oettl, K.; Koestenberger, M.; Cvirn, G.; Hallström, S. Anticoagulant action of low, physiologic, and high albumin levels in whole blood. PLoS ONE 2017, 12, e0182997. [Google Scholar] [CrossRef]
- Rogers, K. Serum Albumin. In Encyclopaedia Britannica; Encyclopaedia Britannica, Inc.: London, UK, 2018; Available online: www.britannica.cpm/science/serum-albumin (accessed on 30 August 2021).
- Greenberg, D.A.; Jin, K. Vascular endothelial growth factors (VEGFs) and stroke. Cell. Mol. Life Sci. 2013, 70, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.J.; Hong, S.H.; Oh, S.H.; Kim, T.G.; Min, K.T.; Oh, D.; Kim, N.K. Association Between VEGF Polymorphisms and Homocysteine Levels in Patients with Ischemic Stroke and Silent Brain Infarction. Stroke 2011, 42, 2393–2402. [Google Scholar] [CrossRef] [Green Version]
- Vallance, T.M.; Zeuner, M.-T.; Williams, H.F.; Widera, D.; Vaiyapuri, S. Toll-Like Receptor 4 Signalling and Its Impact on Platelet Function, Thrombosis, and Haemostasis. Mediat. Inflamm. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhang, S.; Hu, L.; Zhai, L.; Xue, R.; Ye, J.; Chen, L.; Cheng, G.; Mruk, J.; Kunapuli, S.P.; et al. Nucleotide-Binding Oligomerization Domain 2 Receptor Is Expressed in Platelets and Enhances Platelet Activation and Thrombosis. Circulation 2015, 131, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Van der Poll, T.; Jansen, P.M.; Van Zee, K.J.; Welborn, M.B., 3rd; de Jong, I.; Hack, C.E.; Loetscher, H.; Lesslauer, W.; Lowry, S.F.; Moldawer, L.L. Tumor necrosis factor-alpha induces activation of coagulation and fibrinolysis in baboons through an exclusive effect on the p55 receptor. Blood 1996, 88, 922–927. [Google Scholar] [PubMed]
- Fateh-Moghadam, S.; Li, Z.; Ersel, S.; Reuter, T.; Htun, P.; Plöckinger, U.; Bocksch, W.; Dietz, R.; Gawaz, M. Platelet Degranulation Is Associated with Progression of Intima-Media Thickness of the Common Carotid Artery in Patients with Diabetes Mellitus Type 2. Arter. Thromb. Vasc. Biol. 2005, 25, 1299–1303. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Snyder, M.; Dinesh-Kumar, S.P. Discovery of Novel Human Gene Regulatory Modules from Gene Co-expression, and Promoter Motif Analysis. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- DeGracia, D.J. Regulation of mRNA following brain ischemia and reperfusion. Wiley Interdiscip. Rev. RNA 2017, 8, e1415. [Google Scholar] [CrossRef]
- DeGracia, N.J. Disease of mRNA Regulation: Relevance for Ischemic Brain Injury. Transl. Stroke Res. 2018, 9, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Khajavi, M.; Inoue, K.; Lupski, J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006, 14, 1074–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Li, Z.; Xu, R.; Zhu, X.; Hu, R.; Xue, Y.; Xu, W. miR-16-5p Suppresses Progression and Invasion of Osteosarcoma via Targeting at Smad3. Front. Pharmacol. 2020, 26, 11. [Google Scholar] [CrossRef]
- Zhou, R.; Li, X.; Hu, G.; Gong, A.-Y.; Drescher, K.M.; Chen, X.-M. miR-16 Targets Transcriptional Corepressor SMRT and Modulates NF-kappaB-Regulated Transactivation of Interleukin-8 Gene. PLoS ONE 2012, 7, e30772. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhang, J.; Ma, Y.; Gu, J.; Jing, X.; Lu, S.; Chen, X.; Yang, W.; Bian, Y.; Fu, S. The Identification and Verification of Key Long Noncoding RNAs in Ischemic Stroke. BioMed Res. Int. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Su, Z.-F.; Sun, Z.-W.; Zhang, Y.; Wang, S.; Yu, Q.-G.; Wu, Z.-B. Regulatory effects of miR-146a/b on the function of endothelial progenitor cells in acute ischemic stroke in mice. Kaohsiung J. Med. Sci. 2017, 33, 369–378. [Google Scholar] [CrossRef]
- Koutsis, G.; Siasos, G.; Spengos, K. The emerging role of microRNA in stroke. Curr. Top. Med. Chem. 2013, 13, 1573–1588. [Google Scholar] [CrossRef]
- Wu, J.; Du, K.; Lu, X. Elevated expressions of serum miR-15a, miR-16, and miR-17-5p are associated with acute ischemic stroke. Int. J. Clin. Exp. Med. 2015, 8, 21071–21079. [Google Scholar]
- Reiche, E.M.V.; Gelinksi, J.R.; Alfieri, D.F.; Flauzino, T.; Lehmann, M.F.; De Araújo, M.C.M.; Lozovoy, M.A.B.; Simão, A.N.C.; De Almeida, E.R.D.; Maes, M. Immune-inflammatory, oxidative stress and biochemical biomarkers predict short-term acute ischemic stroke death. Metab. Brain Dis. 2019, 34, 789–804. [Google Scholar] [CrossRef]
- Maes, M.; Plaimas, K.; Suratanee, A.; Noto, C.; Kanchanatawan, B. The Protein-Protein Interaction Network of First Episode Psychosis and Schizophrenia Reveals Possible Trigger Factors and New Drug Targets among Intracellular Signal Transduction Pathways and Neurotoxicity Processes. Preprints 2021, 2021070596. [Google Scholar] [CrossRef]
- Martin, R.H.; Yeatts, S.D.; Hill, M.; Moy, C.S.; Ginsberg, M.D.; Palesch, Y.Y. ALIAS (Albumin in Acute Ischemic Stroke) Trials: Analysis of the Combined Data from Parts 1 and 2. Stroke 2016, 47, 2355–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, O.-N. Targeting von Willebrand factor as a novel anti-platelet therapy; Application of ARC1779, an Anti-vWF aptamer, against thrombotic risk. Arch. Pharmacal Res. 2012, 35, 1693–1699. [Google Scholar] [CrossRef]
- Drakeford, C.; O’Donnell, J.S. Targeting von Willebrand Factor–Mediated Inflammation. Arter. Thromb. Vasc. Biol. 2017, 37, 1590–1591. [Google Scholar] [CrossRef] [Green Version]
- Buchtele, N.; Schwameis, M.; Gilbert, J.C.; Schoergenhofer, C.; Jilma, B. Targeting von Willebrand Factor in Ischaemic Stroke: Focus on Clinical Evidence. Thromb. Haemost. 2018, 118, 959–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef] [Green Version]
- Buisson, A.; Lesne, S.; Docagne, F.; Ali, C.; Nicole, O.; MacKenzie, E.T.; Vivien, D. Transforming Growth Factor-β and Ischemic Brain Injury. Cell. Mol. Neurobiol. 2003, 23, 539–550. [Google Scholar] [CrossRef]
- Walimbe, T.; Panitch, A. Proteoglycans in Biomedicine: Resurgence of an Underexploited Class of ECM Molecules. Front. Pharmacol. 2020, 10, 1661. [Google Scholar] [CrossRef]
- Lee, B.; Clarke, D.; Alahmad, A.; Kahle, M.; Parham, C.; Auckland, L.; Shaw, C.; Fidanboylu, M.; Orr, A.; Ogunshola, O.; et al. Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J. Clin. Investig. 2011, 121, 3005–3023, Erratum in 2012, 122, 777. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Jian, Z.; Zhong, Y.; Ye, Y.; Zhang, Y.; Hu, X.; Pu, B.; Gu, L.; Xiong, X. Janus Kinase Inhibition Ameliorates Ischemic Stroke Injury and Neuroinflammation Through Reducing NLRP3 Inflammasome Activation via JAK2/STAT3 Pathway Inhibition. Front. Immunol. 2021, 12, 714943. [Google Scholar] [CrossRef]
- Raible, D.J.; Frey, L.C.; Brooks-Kayal, A.R. Effects of JAK2-STAT3 signaling after cerebral insults. JAK STAT 2014, 3, e29510. [Google Scholar] [CrossRef] [Green Version]
- Hong, P.; Gu, R.-N.; Li, F.-X.; Xiong, X.-X.; Liang, W.-B.; You, Z.-J.; Zhang, H.-F. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J. Neuroinflam. 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Azam, S.; Jakaria, M.; Kim, I.-S.; Kim, J.; Haque, E.; Choi, D.-K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Hattori, K.; Hamanaka, J.; Murase, T.; Egashira, Y.; Mishiro, K.; Ishiguro, M.; Tsuruma, K.; Hirose, Y.; Tanaka, H.; et al. Pharmacological inhibition of TLR4-NOX4 signal protects against neuronal death in transient focal ischemia. Sci. Rep. 2012, 2, 896. [Google Scholar] [CrossRef] [PubMed]
- Gesuete, R.; Kohama, S.G.; Stenzel-Poore, M.P. Toll-Like Receptors and Ischemic Brain Injury. J. Neuropathol. Exp. Neurol. 2014, 73, 378–386. [Google Scholar] [CrossRef]
- Howell, J.A.; Bidwell, G.L. Targeting the NF-κB pathway for therapy of ischemic stroke. Ther. Deliv. 2020, 11, 113–123. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCODE Components | GO ID | Biological Term | Log10 (p) |
|---|---|---|---|
| All DEPs, MCODE1 | R-HSA-449147 | Signaling by Interleukins | −42.1 |
| GO:0006954 | Inflammatory response | −40.9 | |
| ko04668 | Cytokine signaling in immune system | −40.1 | |
| All DEPs, MCODE2 | R-HSA-140877 | Formation of fibrin clot (Clotting Cascade) | −19.4 |
| R-HSA-140875 | Common pathway of fibrin clot formation | −17.5 | |
| GO:0050819 | Negative regulation of coagulation | −15.1 | |
| All DEPs, MCODE3 | R-HSA-109582 | Hemostasis | −6.6 |
| GO:0007596 | Blood coagulation | −5.1 | |
| GO:0007599 | Hemostasis | −5.1 | |
| Cluster Two DEPs, MCODE2 | M174 | PID UPA UPAR PATHWAY | −21.5 |
| R-HSA-1566948 | Elastic fiber formation | −20.2 | |
| GO:0009611 | Response to wounding | −20.2 | |
| Cluster Two DEPs, MCODE3 | GO:0043691 | Reverse cholesterol transport | −9.5 |
| GO:0071827 | Plasma lipoprotein particle organization | −8.5 | |
| GO:0071825 | Protein-lipid complex subunit organization | −8.1 |
| Path ID | Pathway Names in Cluster One | Found | Total | Ratio | p Value | pFDR |
|---|---|---|---|---|---|---|
| R-HSA-5357956 | TNFR1-induced NF-kappa B signaling pathway | 11 | 30 | 0.002063 | 1.11 × 10−16 | 6.77 × 10−15 |
| R-HSA-6783783 | Interleukin-10 signaling | 31 | 86 | 0.005914 | 1.11 × 10−16 | 6.77 × 10−15 |
| R-HSA-6785807 | Interleukin-4 and interleukin-13 signaling | 28 | 211 | 0.01451 | 1.11 × 10−16 | 6.77 × 10−15 |
| R-HSA-449147 | Signaling by interleukins | 55 | 643 | 0.044217 | 1.11 × 10−16 | 6.77 × 10−15 |
| R-HSA-1280215 | Cytokine signaling in immune system | 62 | 1092 | 0.075093 | 1.11 × 10−16 | 6.77 × 10−15 |
| R-HSA-168256 | Immune system | 67 | 2681 | 0.184363 | 1.11 × 10−16 | 6.77 × 10−15 |
| R-HSA-75893 | TNF signaling | 11 | 51 | 0.003507 | 1.67 × 10−14 | 8.66 × 10−13 |
| R-HSA-1059683 | Interleukin-6 signaling | 8 | 17 | 0.001169 | 1.55 × 10−13 | 7.13 × 10−12 |
| R-HSA-446652 | Interleukin-1 family signaling | 14 | 167 | 0.011484 | 1.23 × 10−12 | 5.04 × 10−11 |
| R-HSA-5357905 | Regulation of TNFR1 signaling | 9 | 41 | 0.002819 | 3.82 × 10−12 | 1.38 × 10−10 |
| R-HSA-73887 | Death receptor signaling | 13 | 158 | 0.010865 | 1.05 × 10−11 | 3.46 × 10−10 |
| R-HSA-6783589 | Interleukin-6 family signaling | 8 | 30 | 0.002063 | 1.37 × 10−11 | 4.11 × 10−10 |
| R-HSA-168164 | Toll-Like Receptor 3 (TLR3) cascade | 10 | 102 | 0.007014 | 5.59 × 10−10 | 1.56 × 10−08 |
| R-HSA-937061 | TRIF(TICAM1)-mediated TLR4 signaling | 10 | 107 | 0.007358 | 8.81 × 10−10 | 2.11 × 10−08 |
| Path ID | Pathway Names in Cluster Two | Found | Total | Ratio | p | pFDR |
| R-HSA-140875 | Common pathway of fibrin clot formation | 16 | 25 | 0.001719 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-8957275 | Post-translational protein phosphorylation | 17 | 109 | 0.007496 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-216083 | Integrin cell-surface interactions | 15 | 86 | 0.005914 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-140877 | Formation of fibrin clot (Clotting Cascade) | 24 | 43 | 0.002957 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-76009 | Platelet aggregation (Plug Formation) | 13 | 53 | 0.003645 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-140837 | Intrinsic pathway of fibrin clot formation | 14 | 26 | 0.001788 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-109582 | Hemostasis | 49 | 801 | 0.055082 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-114608 | Platelet degranulation | 24 | 139 | 0.009559 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-76002 | Platelet activation, signaling and aggregation | 31 | 291 | 0.020011 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-76005 | Response to elevated platelet cytosolic Ca2+ | 24 | 146 | 0.01004 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-381426 | Regulation of insulin-like growth factor (IGF) transport and uptake by insulin-like growth factor binding proteins (IGFBPs) | 19 | 127 | 0.008733 | 1.11 × 10−16 | 4.33 × 10−15 |
| R-HSA-1566948 | Elastic fiber formation | 12 | 46 | 0.003163 | 8.88 × 10−16 | 3.20 × 10−14 |
| R-HSA-2129379 | Molecules associated with elastic fibers | 11 | 38 | 0.002613 | 5.00 × 10−15 | 1.65 × 10−13 |
| MCODE Components | GO ID | Biological Term | Log10 (p) Value |
|---|---|---|---|
| All DEPs/genes, cluster one, MCODE2 | R-HSA-3000178 | ECM proteoglycans | −7.1 |
| WP306 | Focal adhesion | −5.8 | |
| R-HSA-1474244 | Extracellular matrix organization | −5.3 | |
| All DEPs/genes, cluster two, MCODE1 | ko04668 | TNF signaling pathway | −40.4 |
| hsa04668 | TNF signaling pathway | −39.8 | |
| ko04657 | IL-17 signaling pathway | −28.7 |
| Path ID | Pathway Names Associated with Cluster One | Observed | Background | Strength | pFDR |
|---|---|---|---|---|---|
| hsa04610 | Complement and coagulation cascades | 27 | 78 | 1.89 | 4.62 × 10−39 |
| hsa04510 | Focal adhesion | 24 | 197 | 1.43 | 4.40 × 10−25 |
| hsa05205 | Proteoglycans in cancer | 23 | 195 | 1.42 | 7.70 × 10−24 |
| hsa04151 | PI3K-Akt signaling pathway | 25 | 348 | 1.2 | 3.09 × 10−21 |
| hsa04611 | Platelet activation | 18 | 123 | 1.51 | 3.93 × 10−20 |
| hsa04512 | ECM-receptor interaction | 16 | 81 | 1.64 | 9.21 × 10−20 |
| hsa04933 | AGE-RAGE signaling pathway in diabetic complications | 16 | 98 | 1.56 | 1.21 × 10−18 |
| hsa04926 | Relaxin signaling pathway | 16 | 130 | 1.44 | 6.38 × 10−17 |
| hsa04810 | Regulation of actin cytoskeleton | 14 | 205 | 1.18 | 9.32 × 10−12 |
| hsa04068 | FoxO signaling pathway | 12 | 130 | 1.31 | 1.52 × 10−11 |
| Path ID | Pathway Names Associated with Cluster Two | Observed | Background | Strength | pFDR |
| hsa04668 | TNF signaling pathway | 30 | 108 | 1.81 | 4.79 × 10−42 |
| hsa04060 | Cytokine-cytokine receptor interaction | 35 | 263 | 1.49 | 5.35 × 10−40 |
| hsa04620 | Toll-like receptor signaling pathway | 23 | 102 | 1.72 | 3.08 × 10−30 |
| hsa04657 | IL-17 signaling pathway | 22 | 92 | 1.75 | 1.44 × 10−29 |
| hsa04064 | NF-kappa B signaling pathway | 22 | 93 | 1.74 | 1.48 × 10−29 |
| hsa04621 | NOD-like receptor signaling pathway | 25 | 166 | 1.55 | 1.84 × 10−29 |
| hsa04630 | Jak-STAT signaling pathway | 22 | 160 | 1.51 | 3.66 × 10−25 |
| hsa04380 | Osteoclast differentiation | 20 | 124 | 1.57 | 4.30 × 10−24 |
| hsa04659 | Th17 cell differentiation | 19 | 102 | 1.64 | 5.84 × 10−24 |
| hsa04622 | RIG-I-like receptor signaling pathway | 17 | 70 | 1.75 | 3.72 × 10−23 |
| DOID ID | Disease | Size | Overlap | Enrichment | p-Value |
|---|---|---|---|---|---|
| DOID:0060032 | Autoimmune disease of the musculoskeletal system | 645 | 62/267 | 7.20 | 1.8 × 10−35 |
| DOID:1247 | Blood coagulation disease | 238 | 42/267 | 13.22 | 7.3 × 10−35 |
| DOID:612 | Primary immunodeficiency syndrome | 1.3k | 83/267 | 4.67 | 5.0 × 10−34 |
| DOID:74 | Hematopoietic disease | 1.6k | 91/267 | 4.20 | 5.4 × 10−34 |
| DOID:2349 | Atherosclerosis | 363 | 48/247 | 9.90 | 9.4 × 10−34 |
| DOID:7148 | Rheumatoid arthritis | 313 | 45/247 | 10.77 | 2.9 × 10−33 |
| DOID:2348 | Atherosclerotic cardiovascular disease | 352 | 47/247 | 10.00 | 3.0 × 10−33 |
| DOID:417 | Autoimmune disease | 1.1k | 74/246 | 5.20 | 3.3 × 10−33 |
| DOID:0060903 | Thrombosis | 108 | 31/247 | 21.50 | 5.9 × 10−33 |
| DOID:2941 | Immune system disease | 1.9k | 95/267 | 3.75 | 1.2 × 10−31 |
| REACTOME Pathways | Total | Expected | Hits | p | pFDR |
|---|---|---|---|---|---|
| Metabolism of mRNA | 317 | 30.3 | 95 | 4.15 × 10−26 | 5.82 × 10−23 |
| Metabolism of RNA | 339 | 32.4 | 98 | 1.39 × 10−25 | 9.73 × 10−23 |
| 3′-UTR-mediated translational regulation | 201 | 19.2 | 65 | 6.16 × 10−20 | 1.73 × 10−17 |
| L13a-mediated translational silencing of ceruloplasmin expression | 201 | 19.2 | 65 | 6.16 × 10−20 | 1.73 × 10−17 |
| Translation | 249 | 23.8 | 73 | 1.63 × 10−19 | 3.81 × 10−17 |
| Nonsense-mediated decay independent of the exon junction complex | 184 | 17.6 | 61 | 2.27 × 10−19 | 4.54 × 10−17 |
| GTP hydrolysis and joining of the 60S ribosomal subunit | 201 | 19.2 | 64 | 3.02 × 10−19 | 5.30 × 10−17 |
| Eukaryotic translation elongation | 186 | 17.8 | 61 | 4.19 × 10−19 | 6.53 × 10−17 |
| Nonsense-mediated decay enhanced by the exon junction complex | 203 | 19.4 | 64 | 5.39 × 10−19 | 6.73 × 10−17 |
| Nonsense-mediated decay | 203 | 19.4 | 64 | 5.39 × 10−19 | 6.73 × 10−17 |
| PANTHER biological processes | |||||
| Translation | 315 | 21.2 | 93 | 3.74 × 10−36 | 7.25 × 10−34 |
| MRNA splicing, via spliceosome | 236 | 15.9 | 52 | 1.26 × 10−14 | 8.12 × 10−13 |
| RNA splicing | 289 | 19.5 | 55 | 1.33 × 10−12 | 6.44 × 10−11 |
| RNA metabolic process | 47 | 3.17 | 20 | 5.08 × 10−12 | 1.97 × 10−10 |
| Protein folding | 157 | 10.6 | 37 | 1.09 × 10−11 | 3.52 × 10−10 |
| MRNA processing | 370 | 24.9 | 61 | 4.62 × 10−11 | 1.28 × 10−09 |
| Rhythmic process | 124 | 8.35 | 31 | 1.05 × 10−10 | 2.43 × 10−09 |
| Blood coagulation | 193 | 13 | 40 | 1.13 × 10−10 | 2.43 × 10−09 |
| Cell-matrix adhesion | 91 | 6.13 | 25 | 8.03 × 10−10 | 1.56 × 10−08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maes, M.; Nikiforov, N.G.; Plaimas, K.; Suratanee, A.; Alfieri, D.F.; Vissoci Reiche, E.M. New Drug Targets to Prevent Death Due to Stroke: A Review Based on Results of Protein-Protein Interaction Network, Enrichment, and Annotation Analyses. Int. J. Mol. Sci. 2021, 22, 12108. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212108
Maes M, Nikiforov NG, Plaimas K, Suratanee A, Alfieri DF, Vissoci Reiche EM. New Drug Targets to Prevent Death Due to Stroke: A Review Based on Results of Protein-Protein Interaction Network, Enrichment, and Annotation Analyses. International Journal of Molecular Sciences. 2021; 22(22):12108. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212108
Chicago/Turabian StyleMaes, Michael, Nikita G. Nikiforov, Kitiporn Plaimas, Apichat Suratanee, Daniela Frizon Alfieri, and Edna Maria Vissoci Reiche. 2021. "New Drug Targets to Prevent Death Due to Stroke: A Review Based on Results of Protein-Protein Interaction Network, Enrichment, and Annotation Analyses" International Journal of Molecular Sciences 22, no. 22: 12108. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212108