
Overcoming Microenvironment-Mediated Chemoprotection through Stromal Galectin-3 Inhibition in Acute Lymphoblastic Leukemia

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Fluorescence Activated Cell Sorting (FACS)
2.3. Cell Cycle and Apoptosis
2.4. Viability and Proliferation, Drug Treatment
2.5. Adhesion
2.6. Migration
2.7. Plasmids and CRISPR/Cas9
2.8. Western Immunoblot
2.9. Statistical Analysis
3. Results
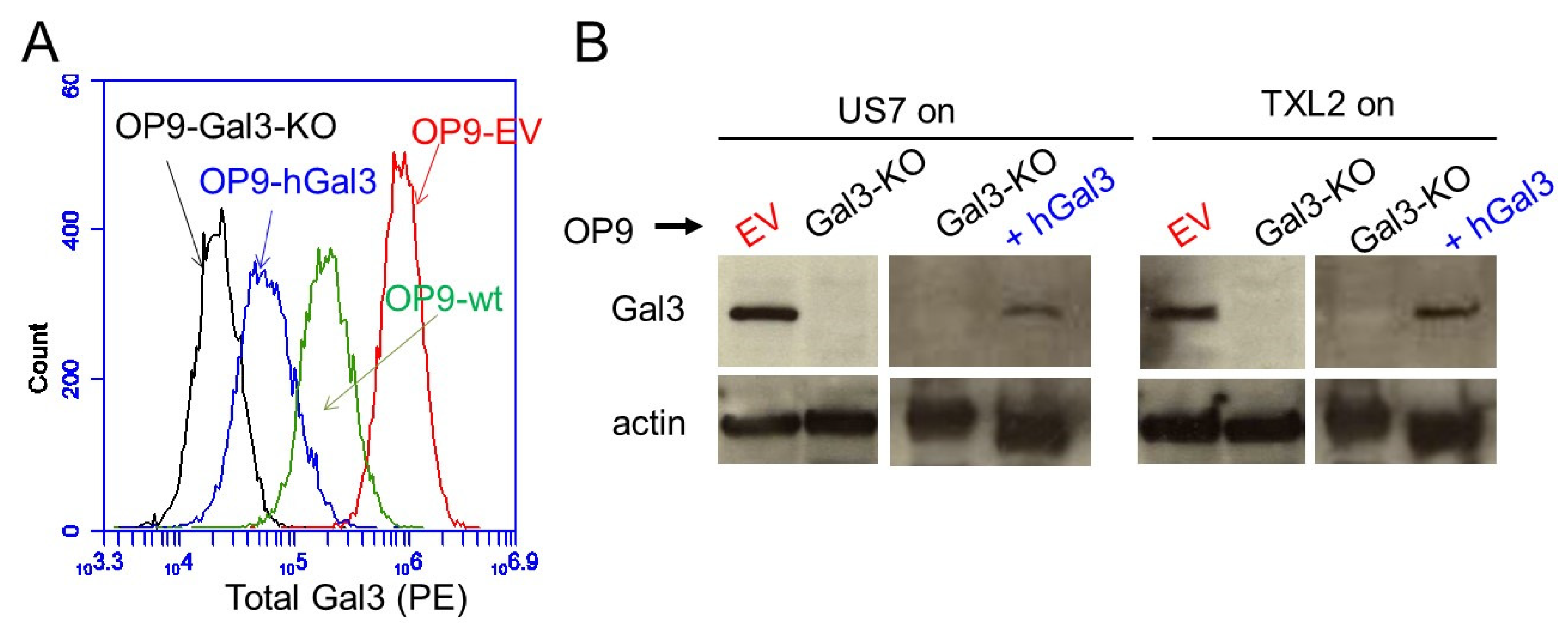
3.1. Bone Marrow Stromal Cells as Source of Galectin-3
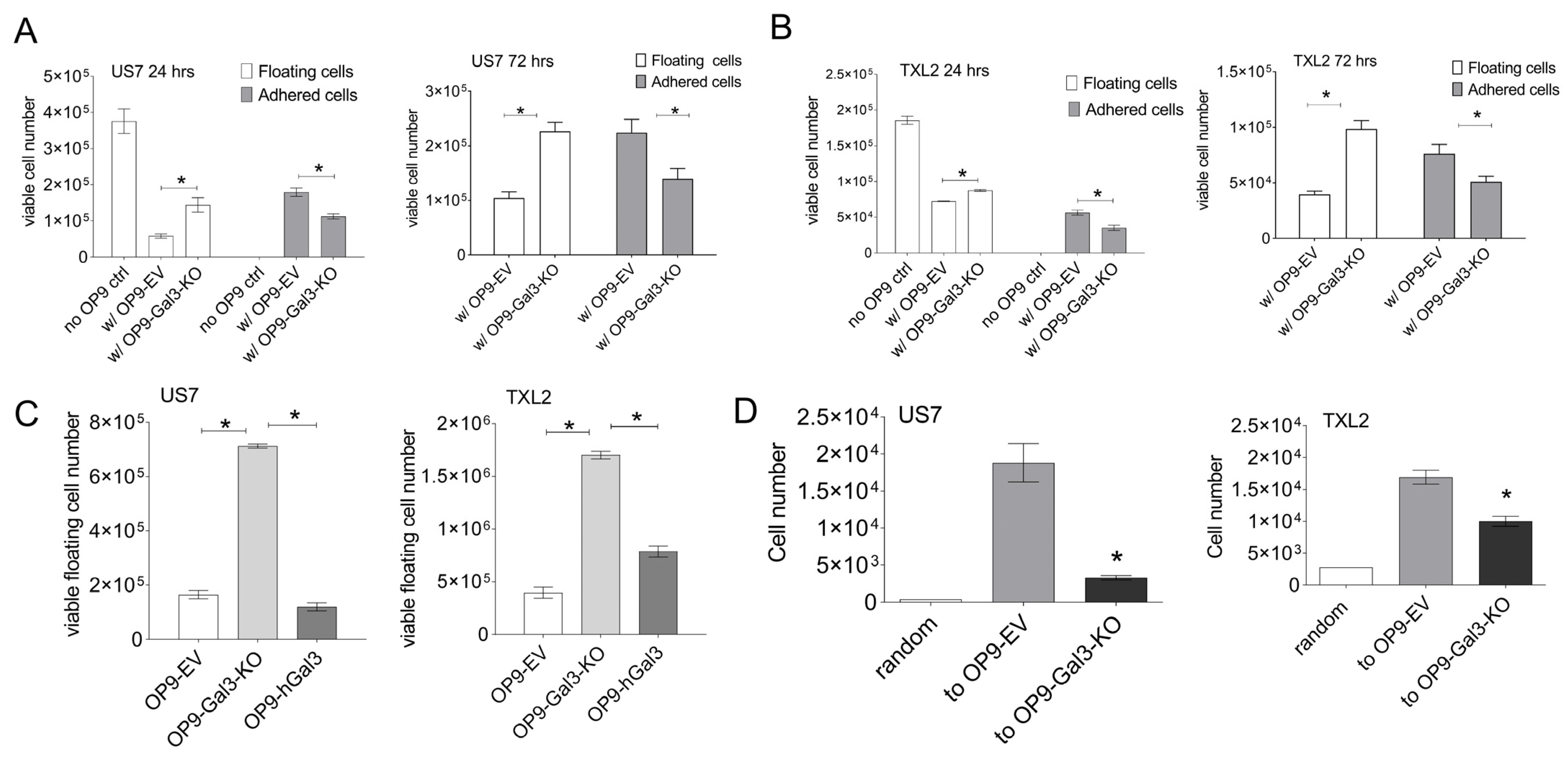
3.2. Gal3 Made by Stroma Is a Major Source for BCP-ALL Cells
3.3. Effect of Gal3 Loss on BCP-ALL Homeostasis
3.4. Depletion of Gal3 Increases BCP-ALL Cells Response to Drug Treatment
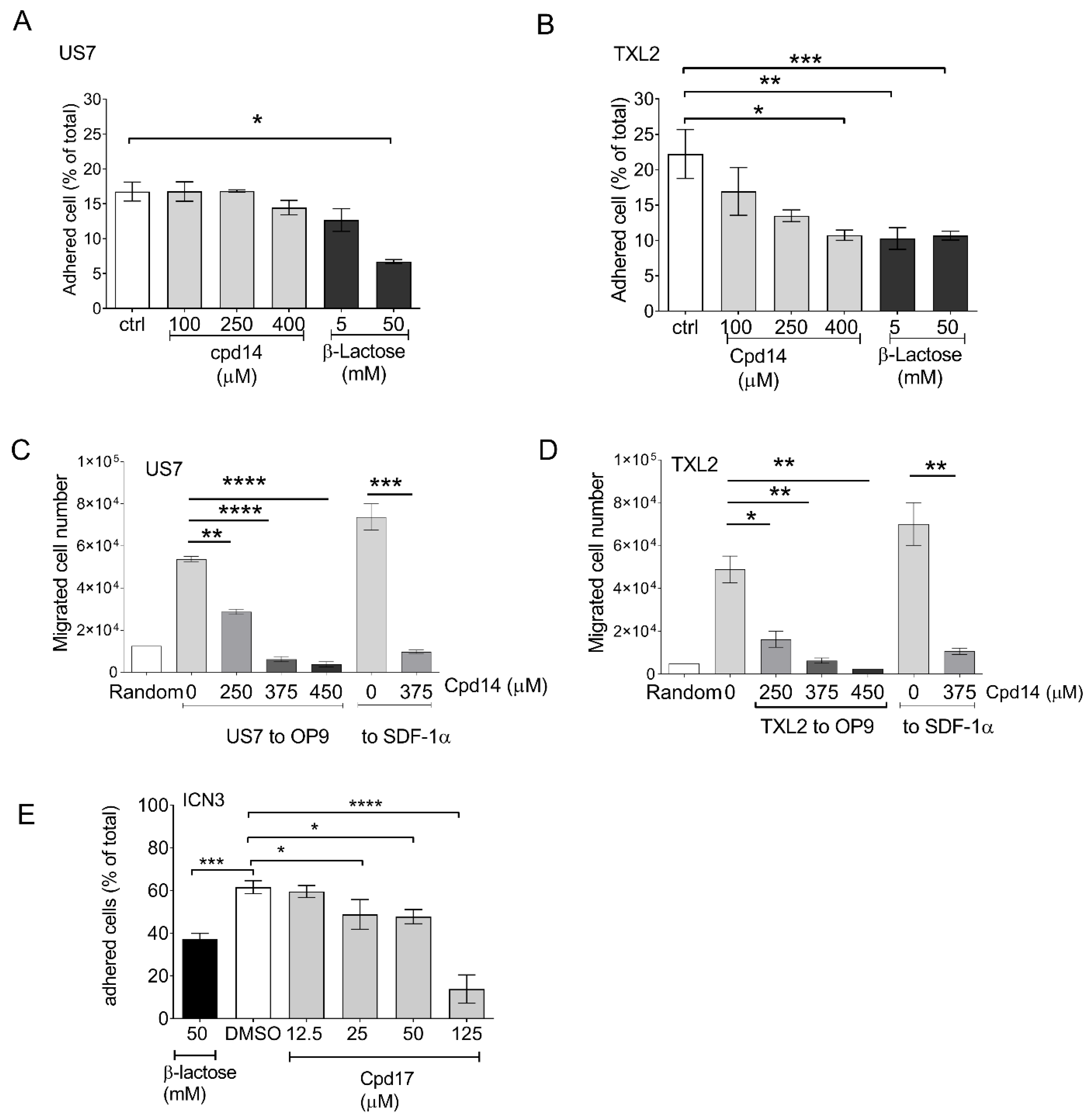
3.5. Novel Galectin-3 Inhibitors-Effect on BCP-ALL Cell Function
3.6. Chemotherapeutic Treatment of BCP-ALL Combined with Cpd17
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, S.; Henderson, M.J.; Kwan, E.; Beesley, A.H.; Sutton, R.; Bahar, A.Y.; Giles, J.; Venn, N.C.; Pozza, L.D.; Baker, D.L.; et al. Relapse in children with acute lymphoblastic leukemia involving selection of a preexisting drug-resistant subclone. Blood 2007, 110, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, F.; Lonetti, A.; Evangelisti, C.; Buontempo, F.; Orsini, E.; Evangelisti, C.; Cappellini, A.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta 2016, 1863, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, M.A.; DiPersio, J.F. Mobilization of hematopoietic stem and leukemia cells. J. Leukoc. Biol. 2012, 91, 47–57. [Google Scholar] [CrossRef]
- Stamatopoulos, B.; Meuleman, N.; De Bruyn, C.; Pieters, K.; Mineur, P.; Le Roy, C.; Saint-Georges, S.; Varin-Blank, N.; Cymbalista, F.; Bron, D.; et al. AMD3100 disrupts the cross-talk between chronic lymphocytic leukemia cells and a mesenchymal stromal or nurse-like cell-based microenvironment: Pre-clinical evidence for its association with chronic lymphocytic leukemia treatments. Haematologica 2012, 97, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, R.; Yu, M.; Lim, M.; Groffen, J.; Heisterkamp, N. Combination of drug therapy in acute lymphoblastic leukemia with a CXCR4 antagonist. Leukemia 2011, 25, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Gang, E.J.; Parameswaran, R.; Stoddart, S.; Fei, F.; Schmidhuber, S.; Park, E.; Hsieh, Y.T.; Yang, A.S.; Groffen, J.; et al. AMD3100 sensitizes acute lymphoblastic leukemia cells to chemotherapy in vivo. Blood Cancer J. 2011, 1, e14. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.S.F.; da Silva, K.N.; Silva, D.N.; Rocha, V.P.C.; Paredes, B.D.; Azevedo, C.M.; Nonaka, C.K.; Carvalho, G.B.; Vasconcelos, J.F.; Dos Santos, R.R.; et al. Galectin-3 Knockdown Impairs Survival, Migration, and Immunomodulatory Actions of Mesenchymal Stromal Cells in a Mouse Model of Chagas Disease Cardiomyopathy. Stem. Cells Int. 2017, 2017, 3282656. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Xu, Y.; Li, Y.; Wang, L.H.; Liu, Y.J.; Zhu, D. Secreted galectin-3 as a possible biomarker for the immunomodulatory potential of human umbilical cord mesenchymal stromal cells. Cytotherapy 2013, 15, 1208–1217. [Google Scholar] [CrossRef]
- Sioud, M.; Mobergslien, A.; Boudabous, A.; Floisand, Y. Evidence for the involvement of galectin-3 in mesenchymal stem cell suppression of allogeneic T-cell proliferation. Scand J. Immunol. 2010, 71, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Abdel-Azim, H.; Lim, M.; Arutyunyan, A.; von Itzstein, M.; Groffen, J.; Heisterkamp, N. Galectin-3 in pre-B acute lymphoblastic leukemia. Leukemia 2013, 27, 2385–2388. [Google Scholar] [CrossRef] [Green Version]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Balan, V.; Tai, G.; Raz, A. Galectin-3 induces cell migration via a calcium-sensitive MAPK/ERK1/2 pathway. Oncotarget 2014, 5, 2077–2084. [Google Scholar] [CrossRef]
- Ruvolo, P.P. Galectin 3 as a guardian of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1863, 427–437. [Google Scholar] [CrossRef]
- Newlaczyl, A.U.; Yu, L.G. Galectin-3—A jack-of-all-trades in cancer. Cancer Lett. 2011, 313, 123–128. [Google Scholar] [CrossRef]
- Ochieng, J.; Furtak, V.; Lukyanov, P. Extracellular functions of galectin-3. Glycoconj. J. 2002, 19, 527–535. [Google Scholar] [CrossRef]
- George, A.A.; Paz, H.; Fei, F.; Kirzner, J.; Kim, Y.M.; Heisterkamp, N.; Abdel-Azim, H. Phosphoflow-Based Evaluation of Mek Inhibitors as Small-Molecule Therapeutics for B-Cell Precursor Acute Lymphoblastic Leukemia. PLoS ONE 2015, 10, e0137917. [Google Scholar] [CrossRef]
- Parameswaran, R.; Muschen, M.; Kim, Y.M.; Groffen, J.; Heisterkamp, N. A functional receptor for B-cell-activating factor is expressed on human acute lymphoblastic leukemias. Cancer Res. 2010, 70, 4346–4356. [Google Scholar] [CrossRef] [Green Version]
- Gang, E.J.; Bosnakovski, D.; Figueiredo, C.A.; Visser, J.W.; Perlingeiro, R.C. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood 2007, 109, 1743–1751. [Google Scholar] [CrossRef]
- Beckenkamp, L.R.; Souza, L.E.B.; Melo, F.U.F.; Thome, C.H.; Magalhaes, D.A.R.; Palma, P.V.B.; Covas, D.T. Comparative characterization of CD271(+) and CD271(-) subpopulations of CD34(+) human adipose-derived stromal cells. J. Cell Biochem. 2018, 119, 3873–3884. [Google Scholar] [CrossRef]
- Bum-Erdene, K. Structure-Based Development of Galectin-Specific Inhibitors. Ph.D. Thesis, Griffith University, Southport, Australia, 2015. Available online: https://research-repository.griffith.edu.au/handle/10072/368150 (accessed on 11 September 2021).
- Schraw, J.M.; Junco, J.J.; Brown, A.L.; Scheurer, M.E.; Rabin, K.R.; Lupo, P.J. Metabolomic profiling identifies pathways associated with minimal residual disease in childhood acute lymphoblastic leukaemia. EBioMedicine 2019, 48, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeoh, A.E.; Li, Z.; Dong, D.; Lu, Y.; Jiang, N.; Trka, J.; Tan, A.M.; Lin, H.P.; Quah, T.C.; Ariffin, H.; et al. Effective Response Metric: A novel tool to predict relapse in childhood acute lymphoblastic leukaemia using time-series gene expression profiling. Br. J. Haematol. 2018, 181, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dander, E.; Palmi, C.; D’Amico, G.; Cazzaniga, G. The Bone Marrow Niche in B-Cell Acute Lymphoblastic Leukemia: The Role of Microenvironment from Pre-Leukemia to Overt Leukemia. Int. J. Mol. Sci. 2021, 22, 4426. [Google Scholar] [CrossRef] [PubMed]
- Delahaye, M.C.; Salem, K.I.; Pelletier, J.; Aurrand-Lions, M.; Mancini, S.J.C. Toward Therapeutic Targeting of Bone Marrow Leukemic Niche Protective Signals in B-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2020, 10, 606540. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Ruvolo, V.R.; Burks, J.K.; Qiu, Y.; Wang, R.Y.; Shpall, E.J.; Mirandola, L.; Hail, N., Jr.; Zeng, Z.; McQueen, T.; et al. Role of MSC-derived galectin 3 in the AML microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Kornblau, S.M.; Ruvolo, P.P.; Wang, R.Y.; Battula, V.L.; Shpall, E.J.; Ruvolo, V.R.; McQueen, T.; Qui, Y.; Zeng, Z.; Pierce, S.; et al. Distinct protein signatures of acute myeloid leukemia bone marrow-derived stromal cells are prognostic for patient survival. Haematologica 2018, 103, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, D.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.N.; Ruan, Y.; Ogana, H.; Kim, Y.M. Cadherins, Selectins, and Integrins in CAM-DR in Leukemia. Front. Oncol. 2020, 10, 592733. [Google Scholar] [CrossRef] [PubMed]
- Bum-Erdene, K.C.P.; Hugo, M.W.; Tarighat, S.S.; Fei, F.; Kishor, C.; Leffler, H.; Nilsson, U.J.; Groffen, J.; Grice, I.D.; Heisterkamp, N.; et al. Novel selective galectin-3 antagonists are cytotoxic to acute lymphoblastic leukaemia. J. Med. Chem. 2021. submitted. [Google Scholar]
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One Molecule for an Alphabet of Diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379. [Google Scholar] [CrossRef] [Green Version]
- Coppin, L.; Jannin, A.; Ait Yahya, E.; Thuillier, C.; Villenet, C.; Tardivel, M.; Bongiovanni, A.; Gaston, C.; de Beco, S.; Barois, N.; et al. Galectin-3 modulates epithelial cell adaptation to stress at the ER-mitochondria interface. Cell Death Dis. 2020, 11, 360. [Google Scholar] [CrossRef] [PubMed]
- Coppin, L.; Leclerc, J.; Vincent, A.; Porchet, N.; Pigny, P. Messenger RNA Life-Cycle in Cancer Cells: Emerging Role of Conventional and Non-Conventional RNA-Binding Proteins? Int. J. Mol. Sci. 2018, 19, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppin, L.; Vincent, A.; Frenois, F.; Duchene, B.; Lahdaoui, F.; Stechly, L.; Renaud, F.; Villenet, C.; Van Seuningen, I.; Leteurtre, E.; et al. Galectin-3 is a non-classic RNA binding protein that stabilizes the mucin MUC4 mRNA in the cytoplasm of cancer cells. Sci. Rep. 2017, 7, 43927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumic, J.; Dabelic, S.; Flogel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Claude-Taupin, A.; Gu, Y.; Choi, S.W.; Peters, R.; Bissa, B.; Mudd, M.H.; Allers, L.; Pallikkuth, S.; Lidke, K.A.; et al. Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev. Cell 2020, 52, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Magescas, J.; Sengmanivong, L.; Viau, A.; Mayeux, A.; Dang, T.; Burtin, M.; Nilsson, U.J.; Leffler, H.; Poirier, F.; Terzi, F.; et al. Spindle pole cohesion requires glycosylation-mediated localization of NuMA. Sci. Rep. 2017, 7, 1474. [Google Scholar] [CrossRef] [Green Version]
- Joeh, E.; O’Leary, T.; Li, W.; Hawkins, R.; Hung, J.R.; Parker, C.G.; Huang, M.L. Mapping glycan-mediated galectin-3 interactions by live cell proximity labeling. Proc. Natl. Acad. Sci. USA 2020, 117, 27329–27338. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.; Qattan, M.; Mutti, L.; Demonacos, C.; Krstic-Demonacos, M. The role of microenvironment and immunity in drug response in leukemia. Biochim. Biophys. Acta 2016, 1863, 414–426. [Google Scholar] [CrossRef]
- Parameswaran, R.; Yu, M.; Lyu, M.A.; Lim, M.; Rosenblum, M.G.; Groffen, J.; Heisterkamp, N. Treatment of acute lymphoblastic leukemia with an rGel/BLyS fusion toxin. Leukemia 2012, 26, 1786–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.T.; Gang, E.J.; Shishido, S.N.; Kim, H.N.; Pham, J.; Khazal, S.; Osborne, A.; Esguerra, Z.A.; Kwok, E.; Jang, J.; et al. Effects of the small-molecule inhibitor of integrin alpha4, TBC3486, on pre-B-ALL cells. Leukemia 2014, 28, 2101–2104. [Google Scholar] [CrossRef] [Green Version]
- Gang, E.J.; Kim, H.N.; Hsieh, Y.T.; Ruan, Y.; Ogana, H.A.; Lee, S.; Pham, J.; Geng, H.; Park, E.; Klemm, L.; et al. Integrin alpha6 mediates the drug resistance of acute lymphoblastic B-cell leukemia. Blood 2020, 136, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.A.; Dalton, W.S. Mechanisms associated with cell adhesion mediated drug resistance (CAM-DR) in hematopoietic malignancies. Cancer Metastasis Rev. 2001, 20, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sedlar, A.; Travnickova, M.; Bojarova, P.; Vlachova, M.; Slamova, K.; Kren, V.; Bacakova, L. Interaction between Galectin-3 and Integrins Mediates Cell-Matrix Adhesion in Endothelial Cells and Mesenchymal Stem Cells. Int. J. Mol. Sci. 2021, 22, 5144. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Joshi, B.; Nabi, I.R. Galectin-3 Overrides PTRF/Cavin-1 Reduction of PC3 Prostate Cancer Cell Migration. PLoS ONE 2015, 10, e0126056. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Kho, D.H.; Yanagawa, T.; Harazono, Y.; Hogan, V.; Chen, W.; Ali-Fehmi, R.; Mehra, R.; Raz, A. Galectin-3 Cleavage Alters Bone Remodeling: Different Outcomes in Breast and Prostate Cancer Skeletal Metastasis. Cancer Res. 2016, 76, 1391–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, P.P.; Ruvolo, V.R.; Benton, C.B.; AlRawi, A.; Burks, J.K.; Schober, W.; Rolke, J.; Tidmarsh, G.; Hail, N., Jr.; Davis, R.E.; et al. Combination of galectin inhibitor GCS-100 and BH3 mimetics eliminates both p53 wild type and p53 null AML cells. Biochim. Biophys. Acta 2016, 1863, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.L.; Hou, H.A.; Lee, M.C.; Liu, C.Y.; Jhuang, J.Y.; Lai, Y.J.; Lin, C.W.; Chen, H.Y.; Liu, F.T.; Chou, W.C.; et al. Higher bone marrow LGALS3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood 2013, 121, 3172–3180. [Google Scholar] [CrossRef]
- Yamamoto-Sugitani, M.; Kuroda, J.; Ashihara, E.; Nagoshi, H.; Kobayashi, T.; Matsumoto, Y.; Sasaki, N.; Shimura, Y.; Kiyota, M.; Nakayama, R.; et al. Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 2011, 108, 17468–17473. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Gu, Y.; Lou, L.; Liu, L.; Hu, Y.; Wang, B.; Luo, Y.; Shi, J.; Yu, X.; Huang, H. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/beta-catenin signaling pathway. J. Hematol. Oncol. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, B.B.; Funkhouser, A.T.; Goodwin, J.L.; Strigenz, A.M.; Chaballout, B.H.; Martin, J.C.; Arthur, C.M.; Funk, C.R.; Edenfield, W.J.; Blenda, A.V. Increased Circulating Levels of Galectin Proteins in Patients with Breast, Colon, and Lung Cancer. Cancers 2021, 13, 4819. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarighat, S.S.; Fei, F.; Joo, E.J.; Abdel-Azim, H.; Yang, L.; Geng, H.; Bum-Erdene, K.; Grice, I.D.; von Itzstein, M.; Blanchard, H.; et al. Overcoming Microenvironment-Mediated Chemoprotection through Stromal Galectin-3 Inhibition in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 12167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212167
Tarighat SS, Fei F, Joo EJ, Abdel-Azim H, Yang L, Geng H, Bum-Erdene K, Grice ID, von Itzstein M, Blanchard H, et al. Overcoming Microenvironment-Mediated Chemoprotection through Stromal Galectin-3 Inhibition in Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2021; 22(22):12167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212167
Chicago/Turabian StyleTarighat, Somayeh S., Fei Fei, Eun Ji Joo, Hisham Abdel-Azim, Lu Yang, Huimin Geng, Khuchtumur Bum-Erdene, I. Darren Grice, Mark von Itzstein, Helen Blanchard, and et al. 2021. "Overcoming Microenvironment-Mediated Chemoprotection through Stromal Galectin-3 Inhibition in Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 22, no. 22: 12167. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212167