
CD93 Signaling via Rho Proteins Drives Cytoskeletal Remodeling in Spreading Endothelial Cells

 , , and
, , and 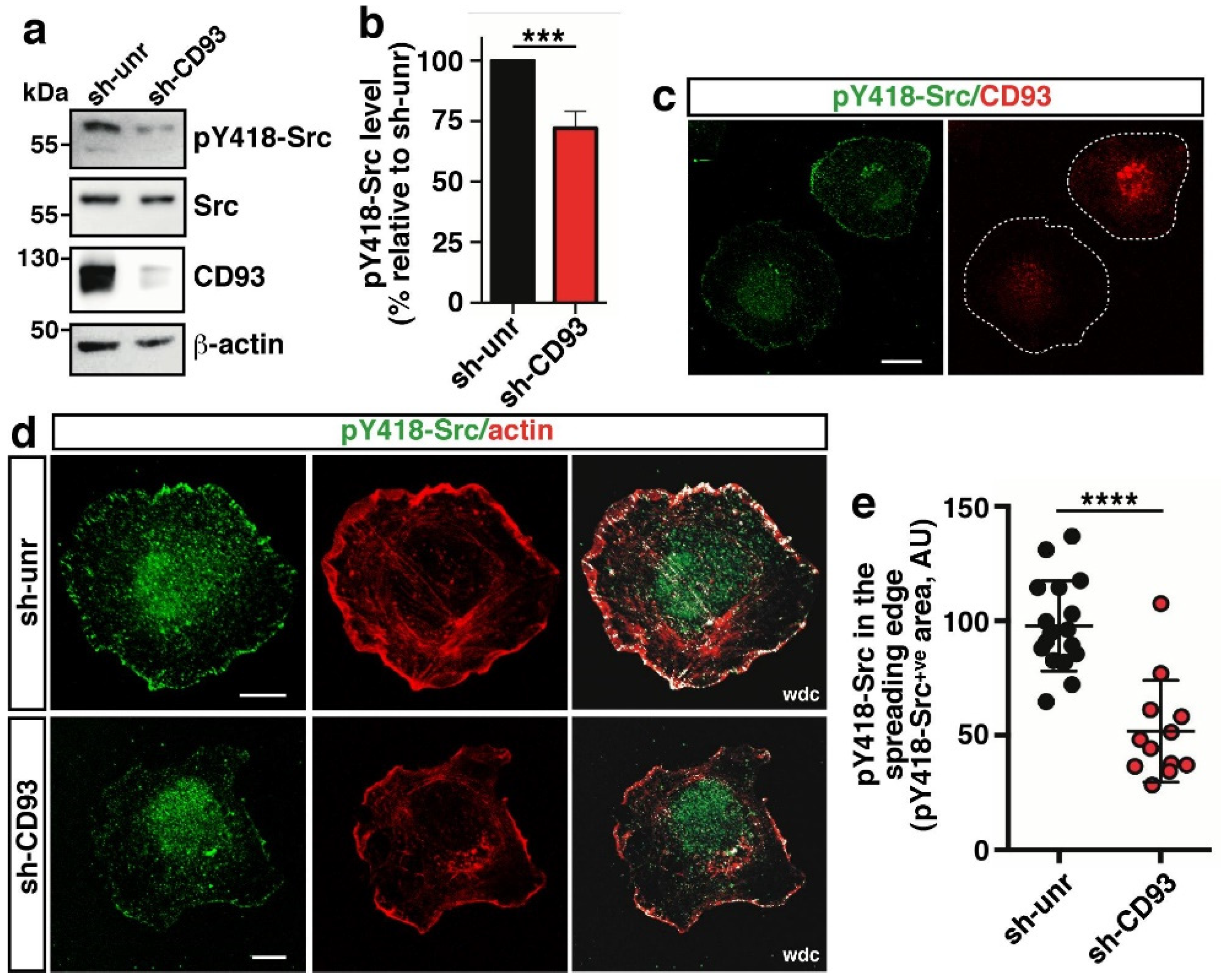{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. CD93 Modulates Activation and Localization of Src Kinase
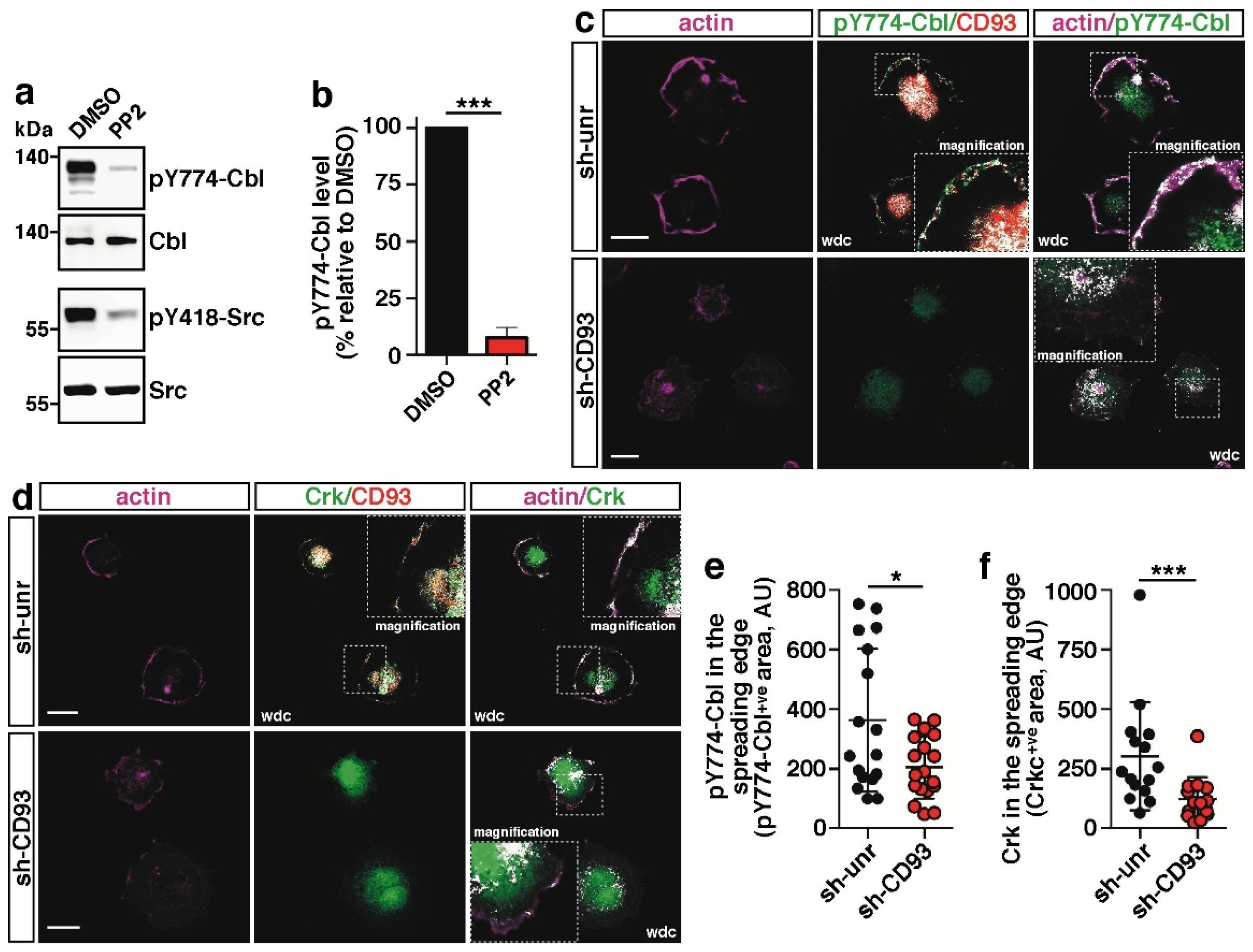
2.2. CD93 Controls Phosphorylation and Cellular Localization of Cbl
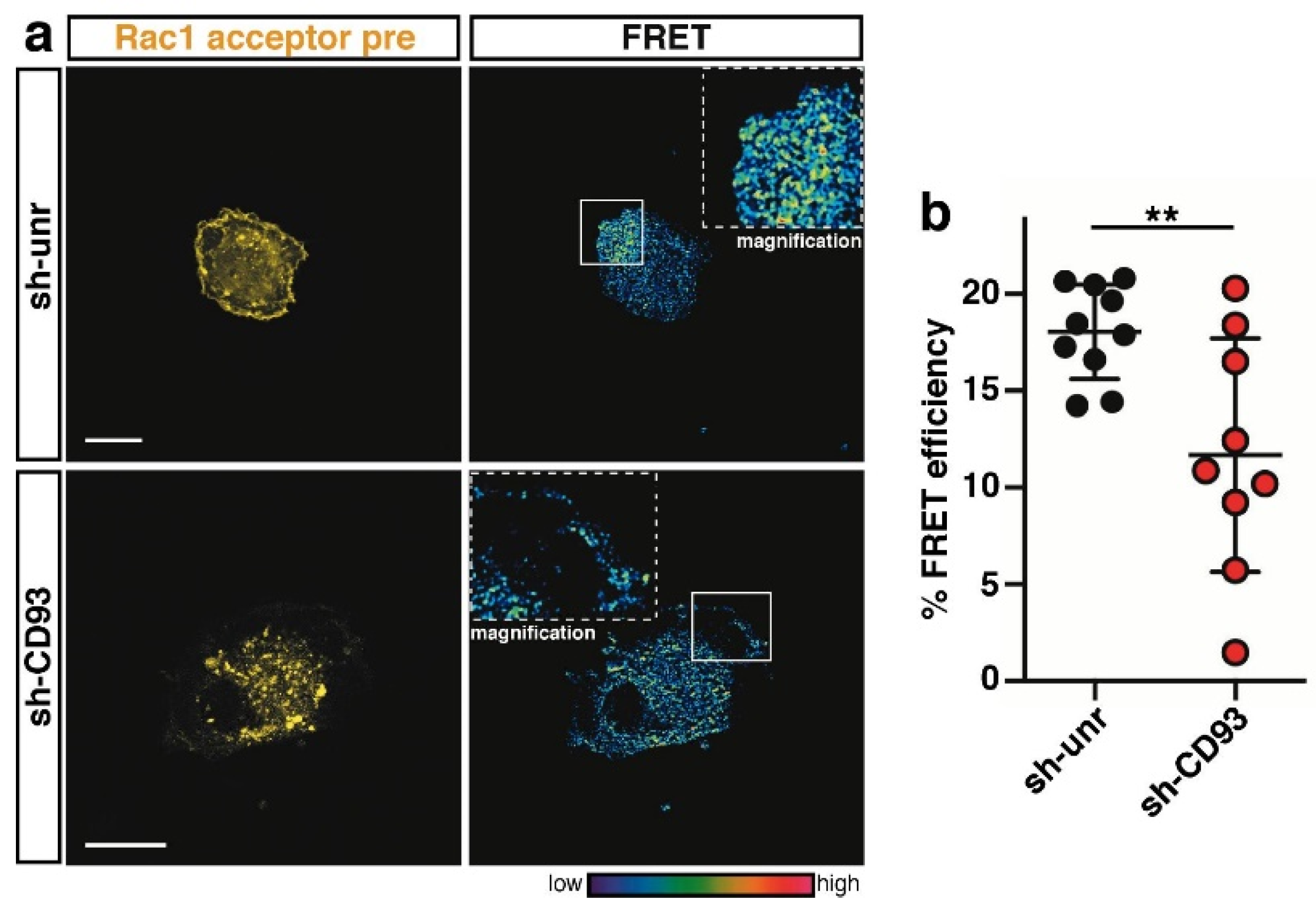
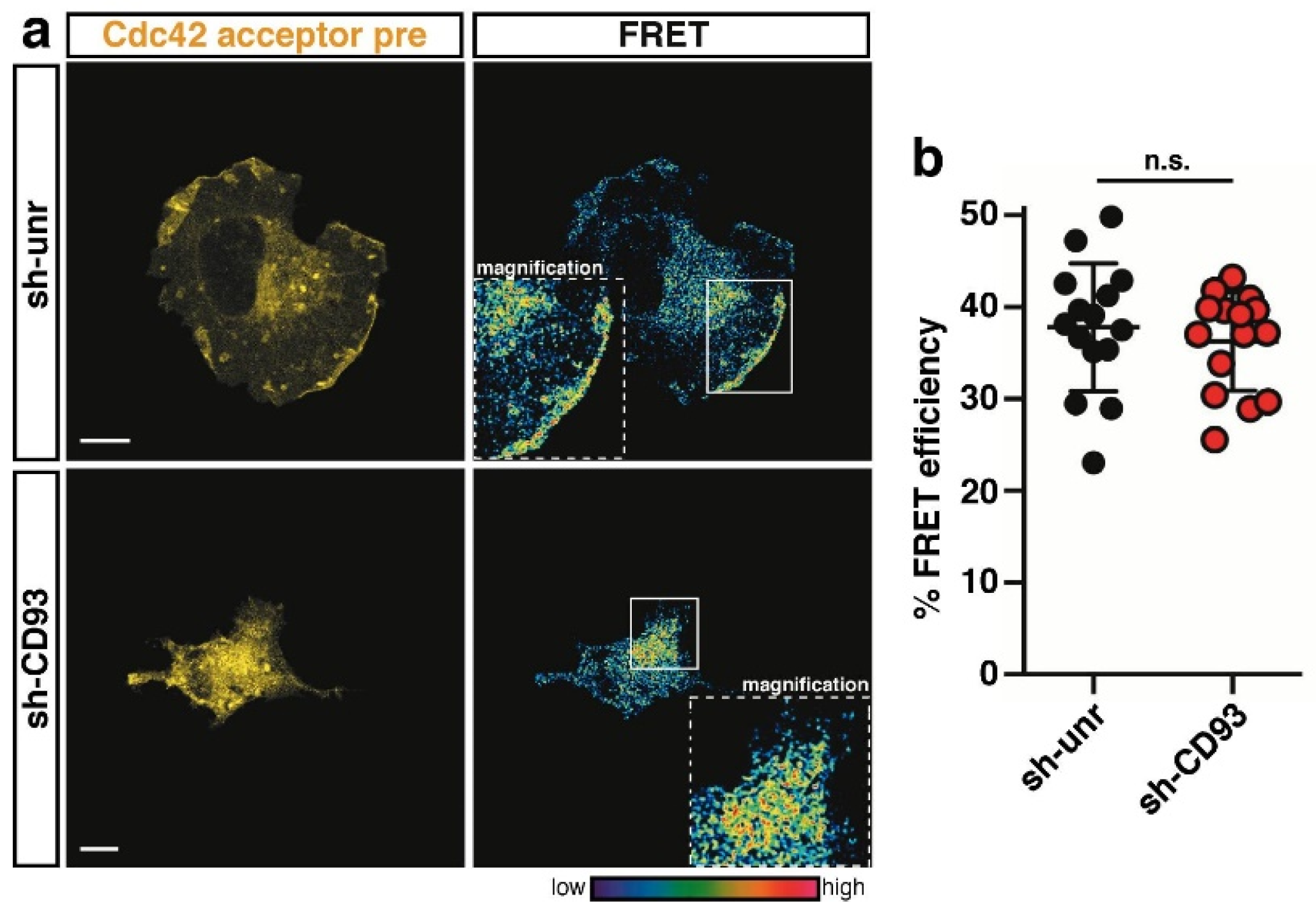
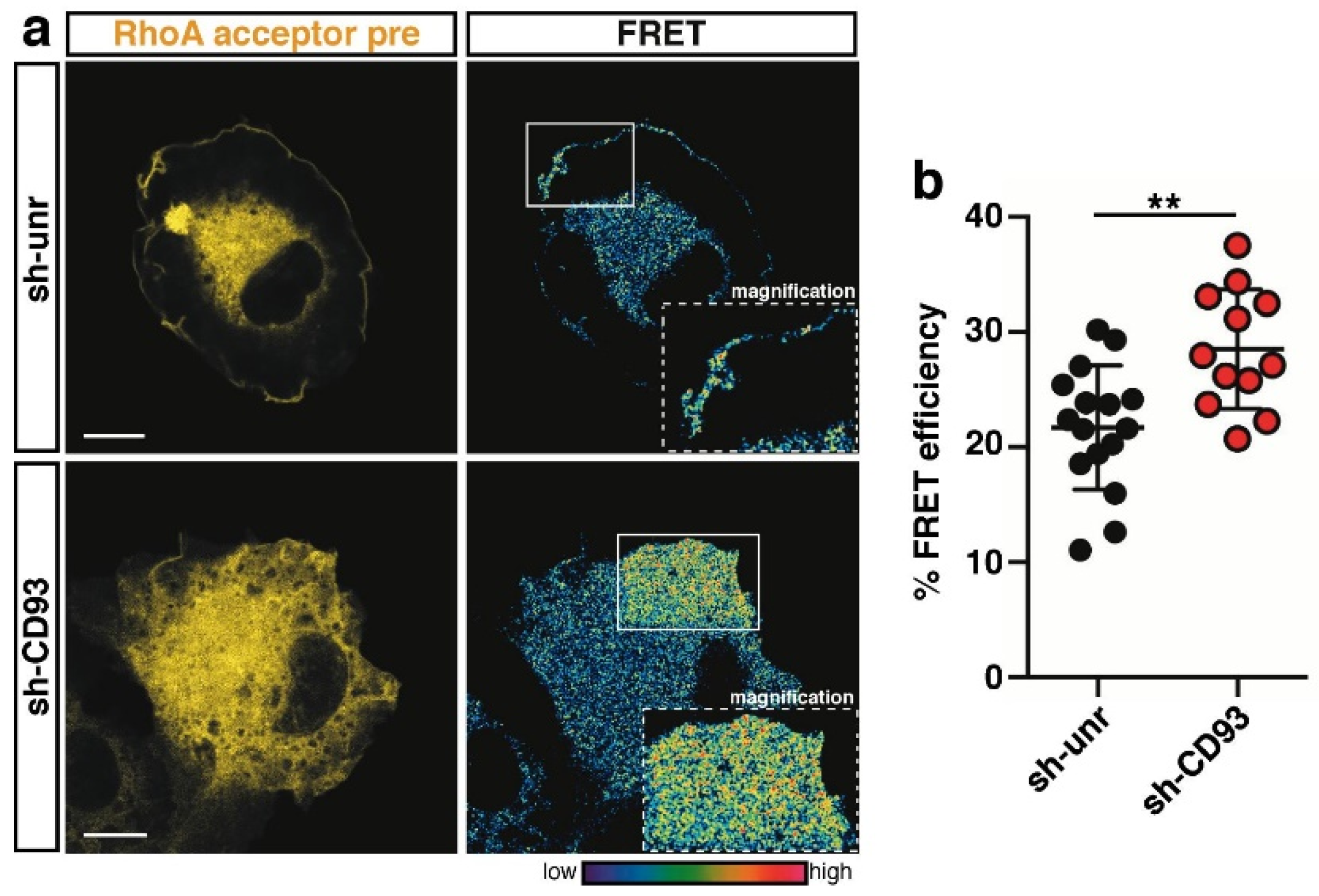
2.3. CD93 Regulates Activity of Rho-Proteins along the Cell Edge of Spreading ECs
3. Discussion
4. Materials and Methods
4.1. DNA Constructs and RNA Interference
4.2. Cell Culture
4.3. Antibodies and Reagents
4.4. Western Blotting Analyses
4.5. Immunofluorescent and FRET Analyses
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Schmidt, S.; Friedl, P. Interstitial cell migration: Integrin-dependent and alternative adhesion mechanisms. Cell Tissue Res. 2010, 339, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandini, M.; Galvagni, F.; Bardelli, M.; Rocchigiani, M.; Lentucci, C.; Anselmi, F.; Zippo, A.; Bini, L.; Oliviero, S. The characterization of a novel monoclonal antibody against CD93 unveils a new antiangiogenic target. Oncotarget 2014, 5, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Langenkamp, E.; Zhang, L.; Lugano, R.; Huang, H.; Elhassan, T.E.A.; Georganaki, M.; Bazzar, W.; Lööf, J.; Trendelenburg, G.; Essand, M.; et al. Elevated expression of the C-type lectin CD93 in the glioblastoma vasculature regulates cytoskeletal rearrangements that enhance vessel function and reduce host survival. Cancer Res. 2015, 75, 4504–4516. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.A.; McMurray, J.L.; Mohammed, F.; Bicknell, R. C-type lectin domain group 14 proteins in vascular biology, cancer and inflammation. FEBS J. 2019, 286, 3299–3332. [Google Scholar] [CrossRef]
- Galvagni, F.; Nardi, F.; Spiga, O.; Trezza, A.; Tarticchio, G.; Pellicani, R.; Andreuzzi, E.; Caldi, E.; Toti, P.; Tosi, G.M.; et al. Dissecting the CD93-Multimerin 2 interaction involved in cell adhesion and migration of the activated endothelium. Matrix Biol. 2017, 64, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Vemuri, K.; Yu, D.; Bergqvist, M.; Smits, A.; Essand, M.; Johansson, S.; Dejana, E.; Dimberg, A. CD93 promotes β1 integrin activation and fibronectin fibrillogenesis during tumor angiogenesis. J. Clin. Investig. 2018, 128, 3280–3297. [Google Scholar] [CrossRef] [Green Version]
- Galvagni, F.; Nardi, F.; Maida, M.; Bernardini, G.; Vannuccini, S.; Petraglia, F.; Santucci, A.; Orlandini, M. CD93 and dystroglycan cooperation in human endothelial cell adhesion and migration. Oncotarget 2016, 7, 10090–10103. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, H.; Ninomiya, H.; Kitamura, Y.; Fujiwara, K.; Masaki, T. Vascular endothelial cells that express dystroglycan are involved in angiogenesis. J. Cell Sci. 2002, 115, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Pellicani, R.; Poletto, E.; Andreuzzi, E.; Paulitti, A.; Doliana, R.; Bizzotto, D.; Braghetta, P.; Colladel, R.; Tarticchio, G.; Sabatelli, P.; et al. Multimerin-2 maintains vascular stability and permeability. Matrix Biol. 2020, 87, 11–25. [Google Scholar] [CrossRef]
- Tosi, G.M.; Neri, G.; Barbera, S.; Mundo, L.; Parolini, B.; Lazzi, S.; Lugano, R.; Poletto, E.; Leoncini, L.; Pertile, G.; et al. The binding of CD93 to Multimerin-2 promotes choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2020, 61, 30. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, W.; Torphy, R.J.; Yao, S.; Zhu, G.; Lin, R.; Lugano, R.; Miller, E.N.; Fujiwara, Y.; Bian, L.; et al. Blockade of the CD93 pathway normalizes tumor vasculature to facilitate drug delivery and immunotherapy. Sci. Transl. Med. 2021, 13, eabc8922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Bohlson, S.S.; Dy, M.; Tenner, A.J. Modulated interaction of the ERM protein, moesin, with CD93. Immunology 2005, 115, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Barbera, S.; Nardi, F.; Elia, I.; Realini, G.; Lugano, R.; Santucci, A.; Tosi, G.M.; Dimberg, A.; Galvagni, F.; Orlandini, M. The small GTPase Rab5c is a key regulator of trafficking of the CD93/Multimerin-2/β1 integrin complex in endothelial cell adhesion and migration. Cell Commun. Signal. 2019, 17, 55. [Google Scholar] [CrossRef] [Green Version]
- Barbera, S.; Lugano, R.; Pedalina, A.; Mongiat, M.; Santucci, A.; Tosi, G.M.; Dimberg, A.; Galvagni, F.; Orlandini, M. The C-type lectin CD93 controls endothelial cell migration via activation of the Rho family of small GTPases. Matrix Biol. 2021, 99, 1–17. [Google Scholar] [CrossRef]
- Lawson, C.D.; Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014, 5, e27958. [Google Scholar] [CrossRef] [Green Version]
- Kraynov, V.S.; Chamberlain, C.; Bokoch, G.M.; Schwartz, M.A.; Slabaugh, S.; Hahn, K.M. Localized Rac activation dynamics visualized in living cells. Science 2000, 290, 333–337. [Google Scholar] [CrossRef] [Green Version]
- Nalbant, P.; Hodgson, L.; Kraynov, V.; Toutchkine, A.; Hahn, K.M. Activation of endogenous Cdc42 visualized in living cells. Science 2004, 305, 1615–1619. [Google Scholar] [CrossRef] [Green Version]
- Pertz, O.; Hodgson, L.; Klemke, R.L.; Hahn, K.M. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 2006, 440, 1069–1072. [Google Scholar] [CrossRef]
- Mack, N.A.; Georgiou, M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Machacek, M.; Hodgson, L.; Welch, C.; Elliott, H.; Pertz, O.; Nalbant, P.; Abell, A.; Johnson, G.L.; Hahn, K.M.; Danuser, G. Coordination of Rho GTPase activities during cell protrusion. Nature 2009, 461, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.M.; Caldi, E.; Parolini, B.; Toti, P.; Neri, G.; Nardi, F.; Traversi, C.; Cevenini, G.; Marigliani, D.; Nuti, E.; et al. CD93 as a potential target in neovascular age-related macular degeneration. J. Cell. Physiol. 2017, 232, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Pugacheva, E.N.; Roegiers, F.; Golemis, E.A. Interdependence of cell attachment and cell cycle signaling. Curr. Opin. Cell Biol. 2006, 18, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Huveneers, S.; Danen, E.H.J. Adhesion signaling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Tsygankov, A.Y. Cbl-family proteins as regulators of cytoskeleton-dependent phenomena. J. Cell. Physiol. 2013, 228, 2285–2293. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Choi, C.K.; Horwitz, A.R. Integrins in cell migration--the actin connection. J. Cell Sci. 2009, 122, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Shifrin, Y.; Arora, P.D.; Ohta, Y.; Calderwood, D.A.; McCulloch, C.A. The role of FilGAP-filamin A interactions in mechanoprotection. Mol. Biol. Cell 2009, 20, 1269–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, J.-C.; Han, X.; Hsiao, C.-T.; Yates Iii, J.R.; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Fritz, R.D.; Menshykau, D.; Martin, K.; Reimann, A.; Pontelli, V.; Pertz, O. SrGAP2-dependent integration of membrane geometry and Slit-Robo-repulsive cues regulates fibroblast contact inhibition of locomotion. Dev. Cell 2015, 35, 78–92. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.; Reimann, A.; Fritz, R.D.; Ryu, H.; Jeon, N.L.; Pertz, O. Spatio-temporal co-ordination of RhoA, Rac1 and Cdc42 activation during prototypical edge protrusion and retraction dynamics. Sci. Rep. 2016, 6, 21901. [Google Scholar] [CrossRef] [Green Version]
- Fritz, R.D.; Letzelter, M.; Reimann, A.; Martin, K.; Fusco, L.; Ritsma, L.; Ponsioen, B.; Fluri, E.; Schulte-Merker, S.; van Rheenen, J.; et al. A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Sci. Signal. 2013, 6, rs12. [Google Scholar] [CrossRef] [PubMed]
- Orlandini, M.; Nucciotti, S.; Galvagni, F.; Bardelli, M.; Rocchigiani, M.; Petraglia, F.; Oliviero, S. Morphogenesis of human endothelial cells is inhibited by DAB2 via Src. FEBS Lett. 2008, 582, 2542–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, A.P.; Kennedy, A.; Gregory, B.; Brennan, F.M. Simplified production and concentration of lentiviral vectors to achieve high transduction in primary human T cells. BMC Biotechnol. 2013, 13, 98. [Google Scholar] [CrossRef] [Green Version]
- Galvagni, F.; Anselmi, F.; Salameh, A.; Orlandini, M.; Rocchigiani, M.; Oliviero, S. Vascular endothelial growth factor receptor-3 activity is modulated by its association with caveolin-1 on endothelial membrane. Biochemistry 2007, 46, 3998–4005. [Google Scholar] [CrossRef] [PubMed]
- Stepensky, D. FRETcalc plugin for calculation of FRET in non-continuous intracellular compartments. Biochem. Biophys. Res. Commun. 2007, 359, 752–758. [Google Scholar] [CrossRef]
- Boscher, C.; Gaonac’h-Lovejoy, V.; Delisle, C.; Gratton, J.-P. Polarization and sprouting of endothelial cells by angiopoietin-1 require PAK2 and paxillin-dependent Cdc42 activation. Mol. Biol. Cell 2019, 30, 2227–2239. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbera, S.; Raucci, L.; Lugano, R.; Tosi, G.M.; Dimberg, A.; Santucci, A.; Galvagni, F.; Orlandini, M. CD93 Signaling via Rho Proteins Drives Cytoskeletal Remodeling in Spreading Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 12417. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212417
Barbera S, Raucci L, Lugano R, Tosi GM, Dimberg A, Santucci A, Galvagni F, Orlandini M. CD93 Signaling via Rho Proteins Drives Cytoskeletal Remodeling in Spreading Endothelial Cells. International Journal of Molecular Sciences. 2021; 22(22):12417. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212417
Chicago/Turabian StyleBarbera, Stefano, Luisa Raucci, Roberta Lugano, Gian Marco Tosi, Anna Dimberg, Annalisa Santucci, Federico Galvagni, and Maurizio Orlandini. 2021. "CD93 Signaling via Rho Proteins Drives Cytoskeletal Remodeling in Spreading Endothelial Cells" International Journal of Molecular Sciences 22, no. 22: 12417. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212417