
Statins and Bempedoic Acid: Different Actions of Cholesterol Inhibitors on Macrophage Activation

 and
and
Abstract
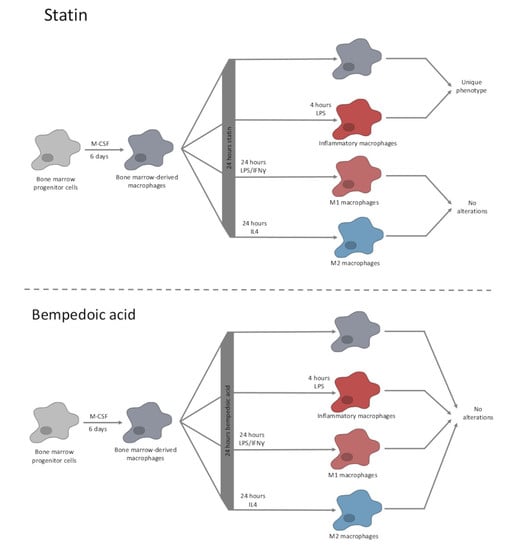
:
1. Introduction
2. Results
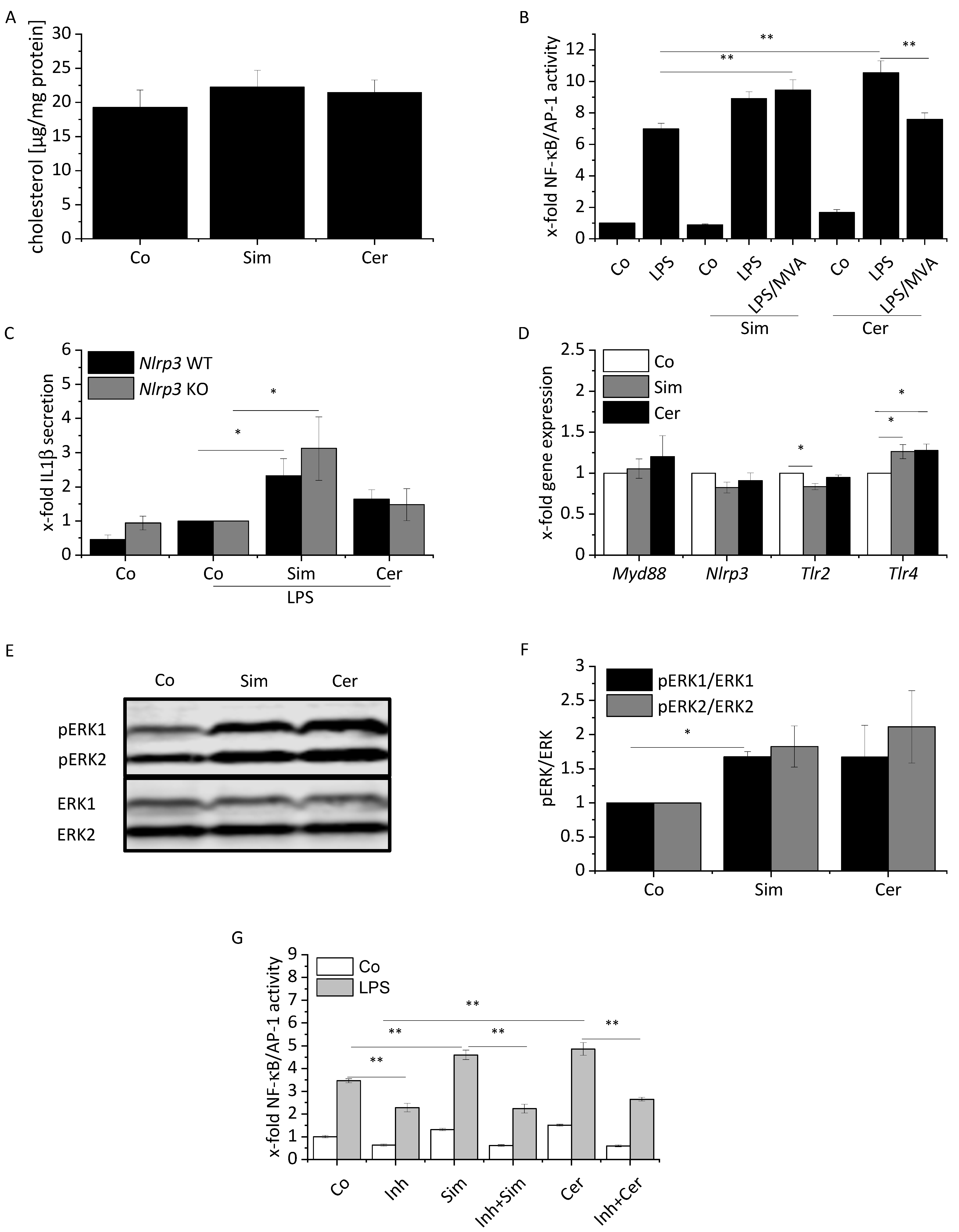
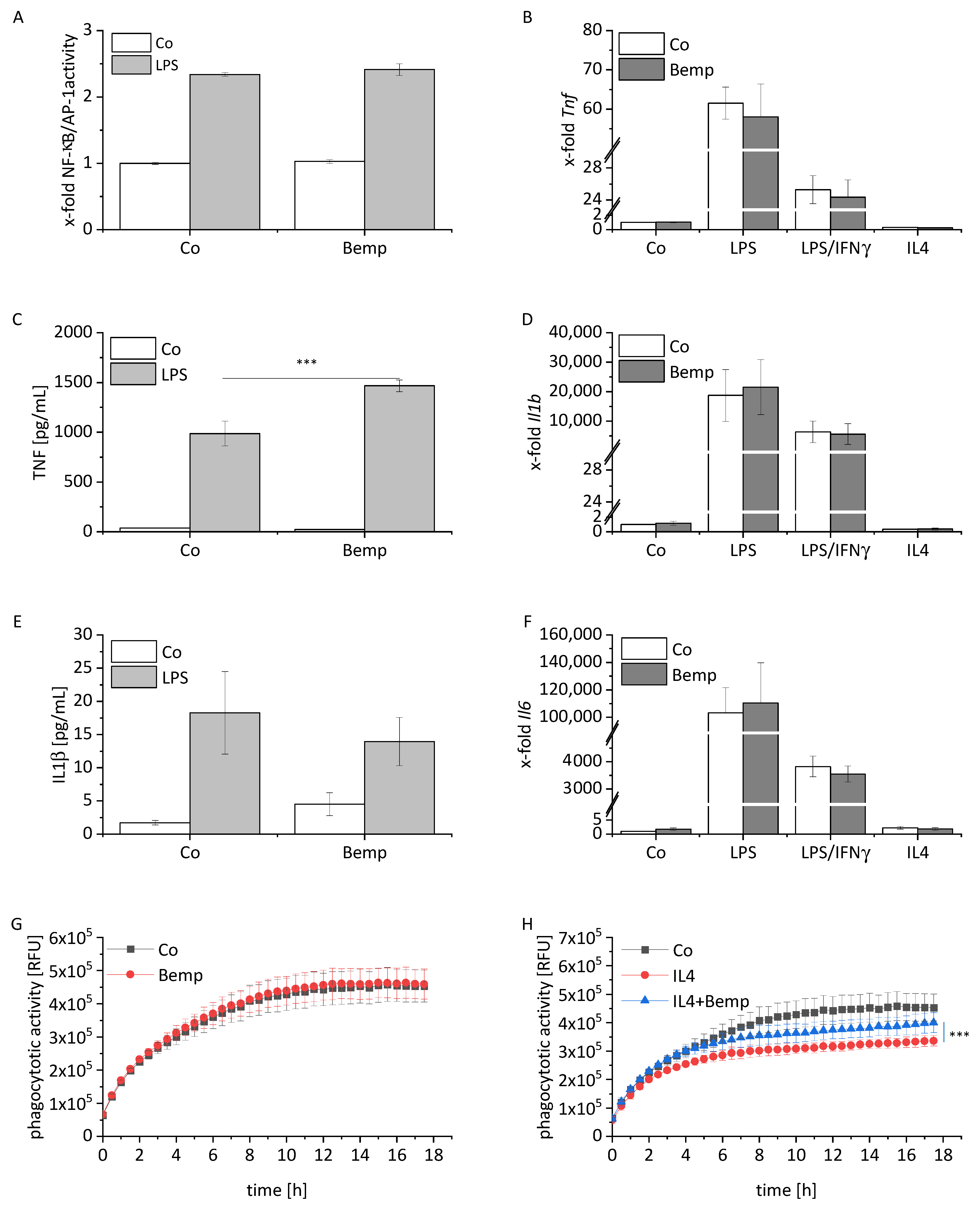
2.1. Modulation of Inflammatory and Anti-Inflammatory Mediator Expression in Statin-Treated Macrophages
2.2. Statins Modulate the Phagocytotic Activity of Macrophages
2.3. ERK Activation Contributes to Statin-Induced Inflammation
2.4. Bempedoic Acid Treatment Has Minimal Impact on the Phenotype of Macrophages
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Mice
4.4. Endotoxin Quantification
4.5. Cytotoxcity Measurements
4.6. Gene Expression in Human Macrophages
4.7. RNA Isolation, Reverse Transcription, and Quantitative PCR (RT–qPCR)
4.8. Western Blot
4.9. Cytokine Quantification
4.10. NF-κB/AP-1 Reporter Cells
4.11. Griess Assay
4.12. Cholesterol Quantification
4.13. Phagocytotic Activity
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Endo, A.; Kuroda, M.; Tanzawa, K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by Ml-236a and Ml-236b fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976, 72, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.; Deplazes, E.; Cranfield, C.G.; Garcia, A. The role of structure and biophysical properties in the pleiotropic effects of statins. Int. J. Mol. Sci. 2020, 21, 8745. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 5 June 2021).
- ClinCalc DrugStats Database. The Top 300 of 2019. Available online: https://clincalc.com/DrugStats/Top300Drugs.aspx (accessed on 5 June 2021).
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Igel, M.; Sudhop, T.; von Bergmann, K. Pharmacology of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins), including rosuvastatin and pitavastatin. J. Clin. Pharmacol. 2002, 42, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [Green Version]
- Palinski, W. New evidence for beneficial effects of statins unrelated to lipid lowering. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, A.; Shamburek, R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis 2009, 203, 325–330. [Google Scholar] [CrossRef] [PubMed]
- McAuley, D.F.; Laffey, J.G.; O’Kane, C.M.; Perkins, G.D.; Mullan, B.; Trinder, T.J.; Johnston, P.; Hopkins, P.A.; Johnston, A.J.; McDowell, C.; et al. Simvastatin in the acute respiratory distress syndrome. N. Engl. J. Med. 2014, 371, 1695–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grudzinska, F.S.; Dosanjh, D.P.S.; Parekh, D.; Dancer, R.C.A.; Patel, J.; Nightingale, P.; Walton, G.M.; Sapey, E.; Thickett, D.R. Statin therapy in patients with community-acquired pneumonia. Clin. Med. 2017, 17, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Nassaji, M.; Ghorbani, R.; Afshar, R.K. The Effect of Statins Use on the Risk and Outcome of Acute Bacterial Infections in Adult Patients. J. Clin. Diagn. Res. 2015, 9, OC09-12. [Google Scholar] [CrossRef] [PubMed]
- Bjorkhem-Bergman, L.; Bergman, P.; Andersson, J.; Lindh, J.D. Statin treatment and mortality in bacterial infections--a systematic review and meta-analysis. PLoS ONE 2010, 5, e10702. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Madhavan, M.V.; Poterucha, T.J.; DeFilippis, E.M.; Hennessey, J.A.; Redfors, B.; Eckhardt, C.; Bikdeli, B.; Platt, J.; Nalbandian, A.; et al. Association between antecedent statin use and decreased mortality in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 1325. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, M.W.; Nilsson, J. Simvastatin stimulates macrophage interleukin-1beta secretion through an isoprenylation-dependent mechanism. Vascul. Pharmacol. 2007, 46, 91–96. [Google Scholar] [CrossRef]
- Kiener, P.A.; Davis, P.M.; Murray, J.L.; Youssef, S.; Rankin, B.M.; Kowala, M. Stimulation of inflammatory responses in vitro and in vivo by lipophilic HMG-CoA reductase inhibitors. Int. Immunopharmacol. 2001, 1, 105–118. [Google Scholar] [CrossRef]
- Das, H.; Kumar, A.; Lin, Z.; Patino, W.D.; Hwang, P.M.; Feinberg, M.W.; Majumder, P.K.; Jain, M.K. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 6653–6658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bont, N.; Netea, M.G.; Rovers, C.; Smilde, T.; Demacker, P.N.; van der Meer, J.W.; Stalenhoef, A.F. LPS-induced cytokine production and expression of LPS-receptors by peripheral blood mononuclear cells of patients with familial hypercholesterolemia and the effect of HMG-CoA reductase inhibitors. Atherosclerosis 1998, 139, 147–152. [Google Scholar] [CrossRef]
- Benati, D.; Ferro, M.; Savino, M.T.; Ulivieri, C.; Schiavo, E.; Nuccitelli, A.; Pasini, F.L.; Baldari, C.T. Opposite effects of simvastatin on the bactericidal and inflammatory response of macrophages to opsonized S. aureus. J. Leukoc. Biol. 2010, 87, 433–442. [Google Scholar] [CrossRef]
- Churchward, M.A.; Todd, K.G. Statin treatment affects cytokine release and phagocytic activity in primary cultured microglia through two separable mechanisms. Mol. Brain 2014, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Healy, A.; Berus, J.M.; Christensen, J.L.; Lee, C.; Mantsounga, C.; Dong, W.; Watts, J.P., Jr.; Assali, M.; Ceneri, N.; Nilson, R.; et al. Statins disrupt macrophage Rac1 regulation leading to increased atherosclerotic plaque calcification. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 714–732. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.H.M.; Chakrabarty, J.K.; Udden, S.M.N.; Zaki, M.H.; Chowdhury, S.M. Inflammatory proteomic network analysis of statin-treated and lipopolysaccharide-activated macrophages. Sci. Rep. 2018, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovland, A.; Retterstol, K.; Mollnes, T.E.; Halvorsen, B.; Aukrust, P.; Lappegard, K.T. Anti-inflammatory effects of non-statin low-density lipoprotein cholesterol-lowering drugs: An unused potential? Scand. Cardiovasc. J. 2020, 54, 274–279. [Google Scholar] [CrossRef]
- Leitinger, N.; Schulman, I.G. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1120–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppstädter, J.; Hachenthal, N.; Valbuena-Perez, J.V.; Lampe, S.; Astanina, K.; Kunze, M.M.; Bruscoli, S.; Riccardi, C.; Schmid, T.; Diesel, B.; et al. Induction of Glucocorticoid-induced Leucine Zipper (GILZ) Contributes to Anti-inflammatory Effects of the Natural Product Curcumin in Macrophages. J. Biol. Chem. 2016, 291, 22949–22960. [Google Scholar] [CrossRef] [Green Version]
- Dahlem, C.; Siow, W.X.; Lopatniuk, M.; Tse, W.K.F.; Kessler, S.M.; Kirsch, S.H.; Hoppstädter, J.; Vollmar, A.M.; Müller, R.; Luzhetskyy, A.; et al. Thioholgamide A, a new anti-proliferative anti-tumor agent, modulates macrophage polarization and metabolism. Cancers 2020, 12, 1288. [Google Scholar] [CrossRef] [PubMed]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allerg. Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef]
- Tuomisto, T.T.; Lumivuori, H.; Kansanen, E.; Häkkinen, S.; Turunen, M.P.; van Thienen, J.V.; Horrevoets, A.J.; Levonen, A.; Ylä-Herttuala, S. Simvastatin has an anti-inflammatory effect on macrophages via upregulation of an atheroprotective transcription factor, Kruppel-like factor 2. Cardiovasc. Res. 2019, 78, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.H.; Lin, Y.C.; Tsao, S.T.; Lin, Y.C.; Yang, A.J.; Huang, C.T.; Huang, K.C.; Lin, W.W. HMG-CoA reductase inhibitors activate caspase-1 in human monocytes depending on ATP release and P2X7 activation. J. Leukoc. Biol. 2013, 93, 289–299. [Google Scholar] [CrossRef]
- Chen, A.O.; Chen, Z.W.; Zhou, Y.; Wu, Y.; Xia, Y.; Lu, D.B.; Fan, M.K.; Li, S.; Chen, J.X.; Sun, A.J.; et al. Rosuvastatin protects against coronary microembolization-induced cardiac injury via inhibiting NLRP3 inflammasome activation. Cell Death Dis. 2021, 12, 78. [Google Scholar] [CrossRef]
- Sweet, D.R.; Vasudevan, N.T.; Fan, L.; Booth, C.E.; Keerthy, K.S.; Liao, X.; Vinayachandran, V.; Takami, Y.; Tugal, D.; Sharma, N.; et al. Myeloid Kruppel-like factor 2 is a critical regulator of metabolic inflammation. Nat. Commun. 2020, 11, 5872. [Google Scholar] [CrossRef]
- Filippov, S.; Pinkosky, S.L.; Lister, R.J.; Pawloski, C.; Hanselman, J.C.; Cramer, C.T.; Srivastava, R.A.K.; Hurley, T.R.; Bradshaw, C.D.; Spahr, M.A.; et al. ETC-1002 regulates immune response, leukocyte homing, and adipose tissue inflammation via LKB1-dependent activation of macrophage AMPK. J. Lipid Res. 2013, 54, 2095–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.; Virani, S.S. CLEAR Serenity Trial: More Clarity for the Future of Bempedoic Acid in Patients Unable to Take Statins? J. Am. Heart Assoc. 2019, 8, e012352. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.; Banach, M.; Chianetta, R.; Luzzu, L.M.; Stoian, A.P.; Diaconu, C.C.; Citarrella, R.; Montalto, G.; Rizzo, M. An overview of statin-induced myopathy and perspectives for the future. Expert Opin. Drug Saf. 2020, 19, 601–615. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Valbuena Perez, J.V.; Linnenberger, R.; Dahlem, C.; Legroux, T.M.; Hecksteden, A.; Tse, W.K.F.; Flamini, S.; Andreas, A.; Herrmann, J.; et al. The glucocorticoid-induced leucine zipper mediates statin-induced muscle damage. FASEB J. 2020, 34, 4684–4701. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.C.; Chen, C.W.; Chen, J.C.; Lin, W.W. HMG-CoA reductase inhibitors inhibit inducible nitric oxide synthase gene expression in macrophages. J. Biomed. Sci. 2003, 10, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 1980, 21, 505–517. [Google Scholar] [CrossRef]
- Schrijvers, D.M.; De Meyer, G.R.Y.; Herman, A.G.; Martinet, W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc. Res. 2007, 73, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Kapellos, T.S.; Taylor, L.; Lee, H.; Cowley, S.A.; James, W.S.; Iqbal, A.J.; Greaves, D.R. A novel real time imaging platform to quantify macrophage phagocytosis. Biochem. Pharmacol. 2016, 116, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Coronel, E.; Ortega, E. Macrophage Polarization Modulates FcgammaR- and CD13-Mediated Phagocytosis and Reactive Oxygen Species Production, Independently of Receptor Membrane Expression. Front. Immunol. 2017, 8, 303. [Google Scholar] [CrossRef] [Green Version]
- Keidar, S.; Aviram, M.; Maor, I.; Oiknine, J.; Brook, J.G. Pravastatin inhibits cellular cholesterol synthesis and increases low density lipoprotein receptor activity in macrophages: In vitro and in vivo studies. Br. J. Clin. Pharmacol. 1994, 38, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Hoppstädter, J.; Dembek, A.; Horing, M.; Schymik, H.S.; Dahlem, C.; Sultan, A.; Wirth, N.; Al-Fityan, S.; Diesel, B.; Gasparoni, G.; et al. Dysregulation of cholesterol homeostasis in human lung cancer tissue and tumour-associated macrophages. EBioMedicine 2021, 72, 103578. [Google Scholar] [CrossRef]
- Rimpelova, S.; Kolar, M.; Strnad, H.; Ruml, T.; Vitek, L.; Gbelcova, H. Comparison of Transcriptomic Profiles of MiaPaCa-2 Pancreatic Cancer Cells Treated with Different Statins. Molecules 2021, 26, 3528. [Google Scholar] [CrossRef]
- Henriksbo, B.D.; Lau, T.C.; Cavallari, J.F.; Denou, E.; Chi, W.; Lally, J.S.; Crane, J.D.; Duggan, B.M.; Foley, K.P.; Fullerton, M.D.; et al. Fluvastatin causes NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes 2014, 63, 3742–3747. [Google Scholar] [CrossRef] [Green Version]
- Hoppstädter, J.; Kessler, S.M.; Bruscoli, S.; Huwer, H.; Riccardi, C.; Kiemer, A.K. Glucocorticoid-induced leucine zipper: A critical factor in macrophage endotoxin tolerance. J. Immunol. 2015, 194, 6057–6067. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.; Addy, K.; Campbell, S.; Wilkinson, P. Primary prevention of cardiovascular disease: Updated review of contemporary guidance and literature. JRSM Cardiovasc. Dis. 2020, 9, 2048004020949326. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H.; Buerke, M.; Schlitt, A.; Muller-Werdan, U.; Werdan, K. Anti-inflammatory effect of statins in an atherosclerosis-related coculture model. FASEB J. 2011, 25, 638.3. [Google Scholar] [CrossRef]
- Matsumoto, M.; Einhaus, D.; Gold, E.S.; Aderem, A. Simvastatin augments lipopolysaccharide-induced proinflammatory responses in macrophages by differential regulation of the c-Fos and c-Jun transcription factors. J. Immunol. 2004, 172, 7377–7384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giroux, L.M.; Davignon, J.; Naruszewicz, M. Simvastatin inhibits the oxidation of low-density lipoproteins by activated human monocyte-derived macrophages. Biochim. Biophys. Acta 1993, 1165, 335–338. [Google Scholar] [CrossRef]
- Kuijk, L.M.; Mandey, S.H.; Schellens, I.; Waterham, H.R.; Rijkers, G.T.; Coffer, P.J.; Frenkel, J. Statin synergizes with LPS to induce IL-1beta release by THP-1 cells through activation of caspase-1. Mol. Immunol. 2008, 45, 2158–2165. [Google Scholar] [CrossRef]
- Hohensinner, P.J.; Baumgartner, J.; Ebenbauer, B.; Thaler, B.; Fischer, M.B.; Huber, K.; Speidl, W.S.; Wojta, J. Statin treatment reduces matrix degradation capacity of proinflammatory polarized macrophages. Vascul. Pharmacol. 2018, 110, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Alabdullah, M.; Grossmann, J.; Spieler, F.; Abdosh, R.; Lutz, V.; Kalies, K.; Knopp, K.; Rieckmann, M.; Koch, S.; et al. The differential statin effect on cytokine production of monocytes or macrophages is mediated by differential geranylgeranylation-dependent Rac1 activation. Cell Death Dis. 2019, 10, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Methe, H.; Kim, J.O.; Kofler, S.; Nabauer, M.; Weis, M. Statins decrease toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, F.; Burger, F.; Pelli, G.; Poku, N.K.; Berlier, C.; Steffens, S.; Mach, F. Statins inhibit C-reactive protein-induced chemokine secretion, ICAM-1 upregulation and chemotaxis in adherent human monocytes. Rheumatology 2009, 48, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.R.; Cauvi, D.M.; Rivera, I.; Hawisher, D.; De Maio, A. Changes in macrophage function modulated by the lipid environment. Innate Immun. 2016, 22, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redondo-Morata, L.; Sanford, R.L.; Andersen, O.S.; Scheuring, S. Effect of statins on the nanomechanical properties of supported lipid bilayers. Biophys. J. 2016, 111, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, D.X.; Griffin, G.; Lichtman, A.H. Mechanisms for the anti-inflammatory effects of statins. Curr. Opin. Lipidol. 2011, 22, 165–170. [Google Scholar] [CrossRef]
- Akula, M.K.; Ibrahim, M.X.; Ivarsson, E.G.; Khan, O.M.; Kumar, I.T.; Erlandsson, M.; Karlsson, C.; Xu, X.; Brisslert, M.; Brakebusch, C.; et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat. Commun. 2019, 10, 3975. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.K.; Park, E.J.; Kim, E.K.; Jin, J.; Kim, J.S.; Shin, I.J.; Kim, B.Y.; Lee, H.; Kim, D.E. Atorvastatin and simvastatin, but not pravastatin, up-regulate LPS-induced MMP-9 expression in macrophages by regulating phosphorylation of ERK and CREB. Cell. Physiol. Biochem. 2012, 30, 499–511. [Google Scholar] [CrossRef]
- Loike, J.D.; Shabtai, D.Y.; Neuhut, R.; Malitzky, S.; Lu, E.; Husemann, J.; Goldberg, I.J.; Silverstein, S.C. Statin inhibition of Fc receptor-mediated phagocytosis by macrophages is modulated by cell activation and cholesterol. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2051–2056. [Google Scholar] [CrossRef] [Green Version]
- Djaldetti, M.; Salman, H.; Bergman, M.; Bessler, H. Effect of pravastatin, simvastatin and atorvastatin on the phagocytic activity of mouse peritoneal macrophages. Exp. Mol. Pathol. 2006, 80, 160–164. [Google Scholar] [CrossRef]
- Tanaka, N.; Abe-Dohmae, S.; Iwamoto, N.; Fitzgerald, M.L.; Yokoyama, S. HMG-CoA reductase inhibitors enhance phagocytosis by upregulating ATP-binding cassette transporter A7. Atherosclerosis 2011, 217, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordle, A.; Koenigsknecht-Talboo, J.; Wilkinson, B.; Limpert, A.; Landreth, G. Mechanisms of statin-mediated inhibition of small G-protein function. J. Biol. Chem. 2005, 280, 34202–34209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by Rho GTPases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Cramer, C.T.; Goetz, B.; Hopson, K.L.M.; Fici, G.J.; Ackermann, R.M.; Brown, S.C.; Bisgaier, C.L.; Rajeswaran, W.G.; Oniciu, D.C.; Pape, M.E. Effects of a novel dual lipid synthesis inhibitor and its potential utility in treating dyslipidemia and metabolic syndrome. J. Lipid Res. 2004, 45, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Laufs, U.; Banach, M.; Mancini, G.B.J.; Gaudet, D.; Bloedon, L.T.; Sterling, L.R.; Kelly, S.; Stroes, E.S.G. Efficacy and safety of bempedoic acid in patients with hypercholesterolemia and statin intolerance. J. Am. Heart Assoc. 2019, 8, e011662. [Google Scholar] [CrossRef] [Green Version]
- Bays, H.E.; Banach, M.; Catapano, A.L.; Duell, P.B.; Gotto, A.M., Jr.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Bloedon, L.T.; et al. Bempedoic acid safety analysis: Pooled data from four phase 3 clinical trials. J. Clin. Lipidol. 2020, 14, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Stahl, A. SLC27 fatty acid transport proteins. Mol. Asp. Med. 2013, 34, 516–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton, E.M.; Cerny, R.L.; Watkins, P.A.; DiRusso, C.C.; Black, P.N. Human fatty acid transport protein 2a/very long chain acyl-CoA synthetase 1 (FATP2a/Acsvl1) has a preference in mediating the channeling of exogenous n-3 fatty acids into phosphatidylinositol. J. Biol. Chem. 2011, 286, 30670–30679. [Google Scholar] [CrossRef] [Green Version]
- Ballantyne, C.M.; Bays, H.; Catapano, A.L.; Goldberg, A.; Ray, K.K.; Saseen, J.J. Role of Bempedoic Acid in Clinical Practice. Cardiovasc. Drugs Ther. 2021, 35, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Amore, B.M.; Cramer, C.T.; MacDougall, D.E.; Sasiela, W.J.; Emery, M.G. Absence of effect of steady state bempedoic acid on cardiac repolarization: Results of a thorough QT/QTc study in healthy volunteers. Clin. Transl. Sci. 2021, 00, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, A.; Monteiro, L.B.; Bakker, N.V.; Sanin, D.E.; Rana, N.; Corrado, M.; Cameron, A.M.; Hassler, F.; Matsushita, M.; Caputa, G.; et al. Triacylglycerol synthesis enhances macrophage inflammatory function. Nat. Commun. 2020, 11, 4107. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.; He, L.; Zimmerman, M.; Yang, G.Z.; Koster, S.; Ouimet, M.; Wang, H.; Moore, K.J.; Dartois, V.; Schilling, J.D.; et al. Inhibition of fatty acid oxidation promotes macrophage control of mycobacterium tuberculosis. Mbio 2020, 11, e01139-20. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.B.; Zmijewski, J.W.; Deshane, J.S.; Tadie, J.M.; Chaplin, D.D.; Takashima, S.; Abraham, E. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J. 2011, 25, 4358–4368. [Google Scholar] [CrossRef] [Green Version]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Liberale, L.; Carbone, F.; Montecucco, F.; Sahebkar, A. Statins reduce vascular inflammation in atherogenesis: A review of underlying molecular mechanisms. Int. J. Biochem. Cell Biol. 2020, 122, 105735. [Google Scholar] [CrossRef]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Stanley, E.R. Murine bone marrow-derived macrophages. Methods Mol. Biol. 1997, 75, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Czepukojc, B.; Abuhaliema, A.; Barghash, A.; Tierling, S.; Nass, N.; Simon, Y.; Korbel, C.; Cadenas, C.; van Hul, N.; Sachinidis, A.; et al. IGF2 mRNA binding protein 2 transgenic mice are more prone to develop a ductular reaction and to progress toward cirrhosis. Front. Med. 2019, 6, 179. [Google Scholar] [CrossRef]
- Kiemer, A.K.; Müller, C.M.; Vollmar, A.M. Inhibition of LPS-induced nitric oxide and TNF-alpha production by alpha-lipoic acid in rat Kupffer cells and in RAW 264.7 murine macrophages. Immunol. Cell Biol. 2002, 80, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Hoppstädter, J.; Diesel, B.; Linnenberger, R.; Hachenthal, N.; Flamini, S.; Minet, M.; Leidinger, P.; Backes, C.; Grässer, F.; Meese, E.; et al. Amplified Host Defense by Toll-Like Receptor-Mediated Downregulation of the Glucocorticoid-Induced Leucine Zipper (GILZ) in Macrophages. Front. Immunol. 2019, 9, 3111. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Forward Primer Sequence 5′-3′ | Reverse Primer Sequence 5′-3′ |
|---|---|---|---|
| Acly | NM_134037.3 | ATGCCCCAAGGAAAGAGTGC | CTCGGGAACACACGTAGTCA |
| Arg1 | NM_007482.3 | ACAAGACAGGGCTCCTTTCAG | GGCTTATGGTTACCCTCCCG |
| Hmgcr | NM_008255.2 | ATCCAGGAGCGAACCAAGAGAG | CAGAAGCCCCAAGCACAAAC |
| Il10 | NM_010548.2 | GCCCAGAAATCAAGGAGCAT | GAAATCGATGACAGCGCCT |
| Il6 | NM_031168.2 | AAGAAATGATGGATGCTACCAAACTG | GTACTCCAGAAGACCAGAGGAAATT |
| Klf2 | NM_008452.2 | CCTTGCACATGAAGCGACAC | ACTTGTCCGGCTCTGTCCTA |
| Myd88 | NM_010851.3 | TAAGTTGTGTGTGTCCGACCG | CATGCGGCGACACCTTTTCT |
| Nos2 | NM_010927.3 | CTTCCTGGACATTACGACCC | TACTCTGAGGGCTGACACAA |
| Ppia | NM_008907.1 | GCGTCTCCTTCGAGCTGTTT | CACCCTGGCACATGAATCCT |
| Slc27a2 | NM_011978.2 | AGCGGAGAGACCTCCTGATGAT | CAGAAGCCCCAACAAGCACAAAC |
| Tlr2 | NM_011905.3 | CACTGCCCGTAGATGAAGTC | TACCTCCGACAGTTCCAAGA |
| Tlr4 | NM_021297.3 | TCCCTGCATAGAGGTAGTTCC | TCAAGGGGTTGAAGCTCAGA |
| Tnf | NM_013693.2 | CCATTCCTGAGTTCTGCAAAGG | AGGTAGGAAGGCCTGAGATCTTATC |
| Tgfb1 | NM_011577.1 | ACCCTGCCCCTATATTTGGA | CGGGTTGTGTTGGTTGTAGAG |
| Tsc22d3 | NM_010286.4 | GCTGCTTGAGAAGAACTCCCA | GAACTTTTCCAGTTGCTCGGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linnenberger, R.; Hoppstädter, J.; Wrublewsky, S.; Ampofo, E.; Kiemer, A.K. Statins and Bempedoic Acid: Different Actions of Cholesterol Inhibitors on Macrophage Activation. Int. J. Mol. Sci. 2021, 22, 12480. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212480
Linnenberger R, Hoppstädter J, Wrublewsky S, Ampofo E, Kiemer AK. Statins and Bempedoic Acid: Different Actions of Cholesterol Inhibitors on Macrophage Activation. International Journal of Molecular Sciences. 2021; 22(22):12480. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212480
Chicago/Turabian StyleLinnenberger, Rebecca, Jessica Hoppstädter, Selina Wrublewsky, Emmanuel Ampofo, and Alexandra K. Kiemer. 2021. "Statins and Bempedoic Acid: Different Actions of Cholesterol Inhibitors on Macrophage Activation" International Journal of Molecular Sciences 22, no. 22: 12480. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212480