1. Introduction
Chronic diseases that lead to organ fibrosis are associated with significant mortality and morbidity, accounting for up to 45% of deaths in developed countries [
1]. The prevalence of fibrotic diseases is steadily increasing and is an important public health problem.
Fibrotic diseases can affect all organs, such as the liver, kidneys, lungs, and heart. Pathological fibrosis is characterized by the exaggerated deposition of components of the extracellular matrix (ECM), such as collagen. Such ECM accumulation destroys the normal architecture of the organ and leads to organ dysfunction and failure with alteration of the specialized functions of each organ, i.e., for the liver, the function of detoxification, for the lung, the function of gas exchange, and for the kidney, the function of filtration. In addition, fibrosis can promote the development of cancer. Transplantation to replace the fibrotic organ often represents the best therapeutic option.
The process of fibrosis occurs in response to an injury or tissue damage following a persistent or overly strong inflammatory response. Fibrosis, which is initially reversible, can evolve to an irreversible state [
2]. The liver can be injured by viruses (hepatitis B, C, D, E), fungal toxins, parasites, auto-antibodies, a high-fat diet, and excessive alcohol consumption, which is a frequent etiology of liver fibrosis [
3] and even the predominant cause in certain countries. When the insults are repeated, the liver develops chronic hepatitis with fibrosis followed by advanced fibrosis or cirrhosis, which is a predisposing state for hepatocellular carcinoma (HCC).
In the lung, idiopathic pulmonary fibrosis (IPF) represents the most common of a group of diseases that includes hypersensitivity fibrosis and rheumatoid lung. This chronic progressive fibrosing interstitial lung disease of unknown origin is rare, affecting three million people worldwide. However, IPF is associated with early death. Indeed, IPF leads to advanced respiratory failure and also represents an independent risk factor for lung cancer [
4]. Lung transplantation should be considered as an option for young patients with advanced disease.
Renal fibrosis is the final common pathway of numerous progressive kidney diseases. The incidence of chronic kidney disease, leading to end-stage renal disease, has significantly increased, affecting 10% of the worldwide population, with high mortality [
5]. In addition, patients with chronic kidney disease have an increased risk of developing kidney cancer (up to 10 times that of the general population), with frequent bilateral and/or multifocal damage [
6].
Regardless of the organ that develops fibrosis, it is important to understand the mechanisms involved in its emergence to develop therapies to prevent it. In particular, chronic inflammation leads to liver or renal fibrosis, and controlling it may make it possible to limit its progression and the onset of organ failure or cancer. Cytokines and chemokines play a central role in both the orientation of the immune response and the maintenance of inflammation [
7,
8]. In addition to these immune molecules, other proteins, such as HLA-G, a class Ib HLA molecule well known for its immunomodulating properties, have been investigated [
9]. We previously demonstrated that HLA-G is expressed by mast cells that are associated with the area of hepatitis C virus-induced liver fibrosis [
10,
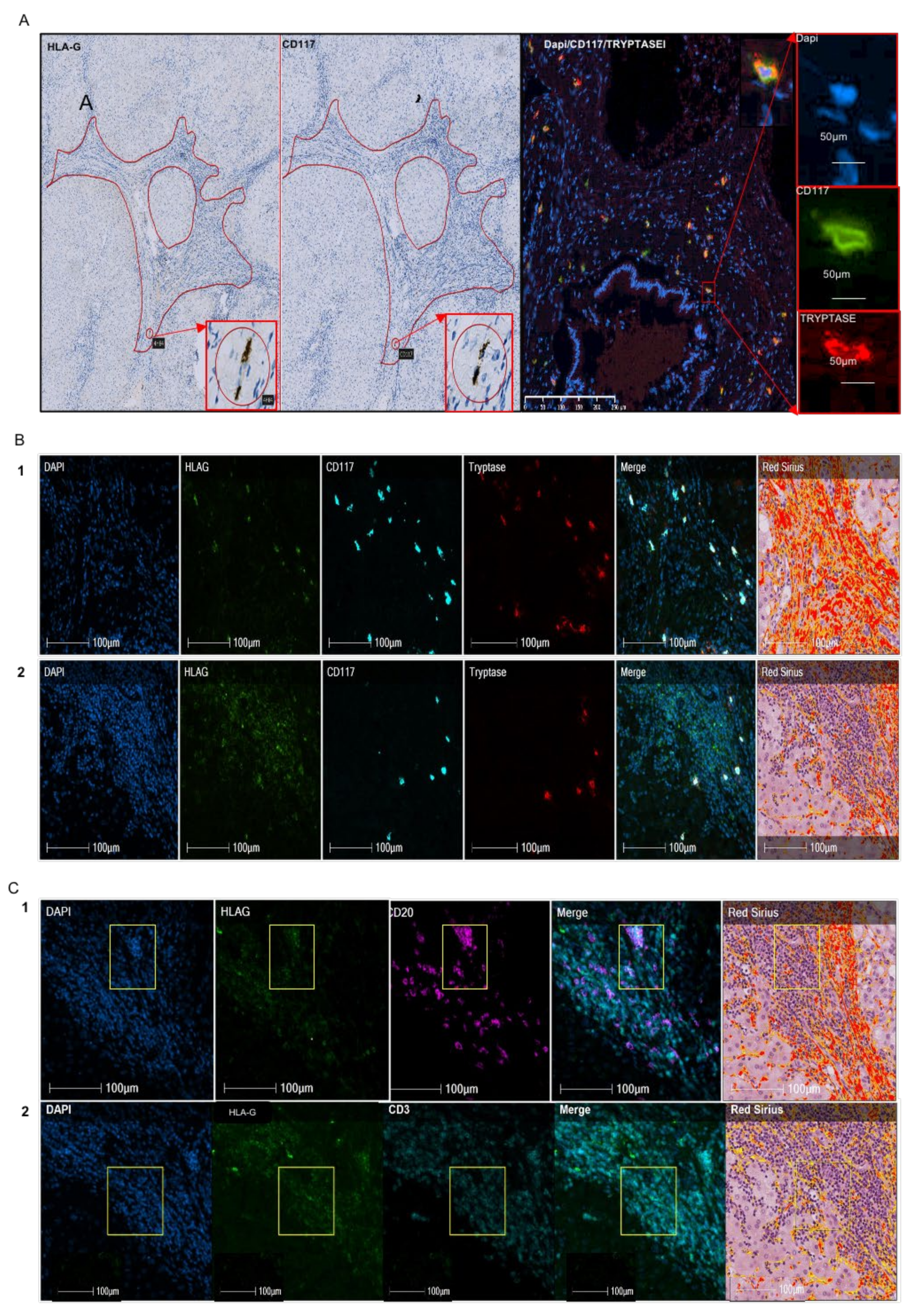
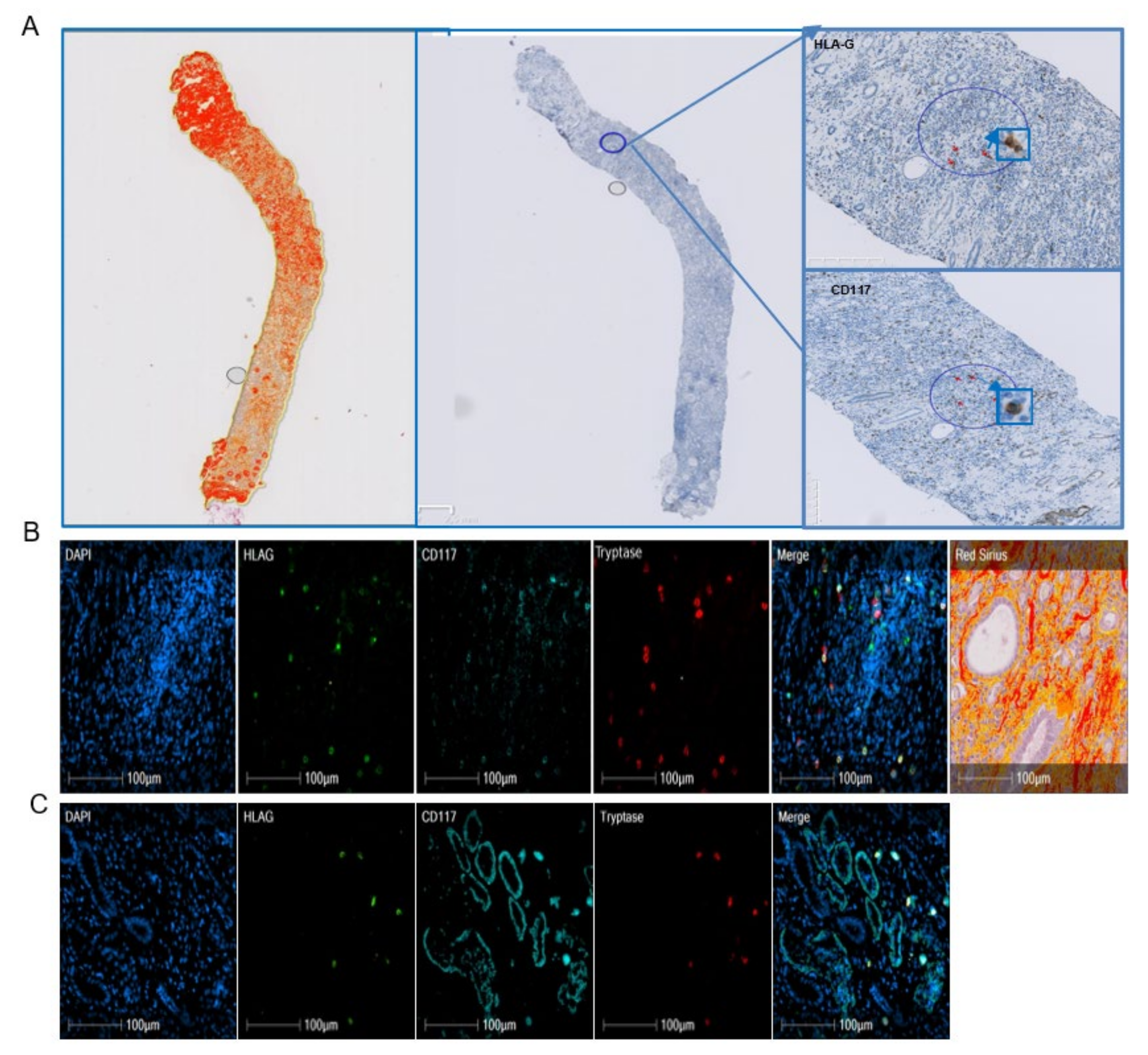
11]. In the present study, we investigated whether HLA-G expression in mast cells is specific to viral etiology, the liver, or the process of fibrosis, irrespective of the organ. We characterized HLA-G expression in mast cells and immune cells on paraffin blocks of cohorts of 41 patients with alcohol-induced cirrhosis, 10 with IPF, and 10 with renal fibrosis. Precise identification of HLA-G-expressing cells was performed using quadruple immunofluorescence on paraffin sections and software that separately analyzes the fluorescence and merges it on three cases of liver alcohol-induced fibrosis and two of IPF.
3. Discussion
The expression of HLA-G proteins was first demonstrated in cytotrophoblasts at the fetal–maternal interface [
12]. Under basal conditions, its expression is largely restricted to specific tissues, such as the cornea [
13], thymus [
14], and β islets of the pancreas [
15]. However, certain types of cells are also able to express it, such as bronchial epithelial cells [
16], mesenchymal cells [
17], cells of monocytic lineage [
18,
19,
20], and erythroid and endothelial precursors [
21], in peculiar conditions.
We previously demonstrated that mast cells can express HLA-G in the basal state, with increased expression in certain cytokine-rich environments, in particular, fibrotic liver tissue. We investigated whether HLA-G can be expressed by mast cells associated with liver fibrosis from another etiology or fibrosis in another organ by studying 41 cases of alcohol-induced liver cirrhosis, 10 of IPF, and 10 of renal fibrosis.
Infections, toxic and metabolic injuries, and idiopathic inflammatory diseases can promote the development of fibrosis because chronic injury induces an apoptosis of parenchymal cells which release profibrogenic and inflammatory cytokines such as TGF-β. The collagen-producing cells differentiate from the resident mesenchymal cells in response to the injury. Epithelial to mesenchymal transition is a phenomenon of cell transdifferentiation that is observed for cholangiocytes in liver, pneumocytes in lung, and tubular epithelial cells in kidney [
22]. Apoptotic cells induce an increase in the concentration of TGF-β in all organs. However, specific features of fibrogenesis may be distinguished in the different organs. In the liver, apoptosis concerns hepatocytes, whereas it affects epithelial cells in lung and kidney [
22]. Thus, resident fibroblasts in kidney and lung activate into a myofibroblast expressing a-SMA, collagen1, whereas liver myofibroblast retain their neural-specific markers [
23].
In the liver, the hepatic stellate cells contribute more than 80% of all collagen-producing cells. In lungs, the damage of pneumocytes is associated to the apoptosis of endothelial cells. The role of inflammation in IPF is controversial. Typical IPF does not show an influx of inflammatory cells, but some authors suggest a role of inflammation in the differentiation of pulmonary fibroblasts into ECM-producing myofibroblasts [
24]. Repeated alveolar epithelial lesions of unknown etiology and alveolar epithelial apoptosis are involved in IPF [
25]. In the kidney and liver, myelomonocytic cells are recruited from the bone marrow and represent respectively 14 to 15% and 8 to 12% of the myofibroblasts. No reversibility of fibrosis is observed in the lung in contrast to the liver and kidney in the absence of injury and if the point of no return has not been reached. Indeed, inflammatory processes are limited in IPF, in particular at the early phase of the disease, whereas repeated alveolar epithelial lesions of unknown etiology and alveolar epithelial apoptosis can promote the proliferation and activation of pulmonary fibroblasts or myofibroblasts [
25].
As for liver fibrosis, a failed wound-healing process of the kidney tissue after chronic, sustained injury leads to the production and secretion of proinflammatory cytokines, as well as TGF-β, which plays a key role in the fibrotic process. For example, in liver fibrosis, TGF-β, which is expressed as a minute amount in quiescent HSC, is quickly produced by this type of cells after liver injury. In addition to the HSC, other sources of TGF-β have been described as platelets, macrophages, hepatocytes, and also mast cells [
26]. TGF-β1 is stored in the matrix in its latent form, and once activated, it promotes the transition from fibroblast to myofibroblast, which is fundamental for the fibrosis process. In addition, it inhibits ECM degradation by suppressing metalloproteases and promoting a natural inhibitor TIMP. Thus, it induces the production of ECM through SMAD3-dependent or non-SMAD-associated mechanisms [
27]. Indeed, a mutual interaction exists between mast cells and TGF-β. TGF-β is a potent attractant for mast cells; indeed, the pathologic processes mediated by TGF-β are often associated with mast cell accumulation [
28]. In addition, mast cells are one of the primary sources of IL-17 that drive TGF-β-dependent fibrosis [
29]. TGF-β has been also reported to promote or suppress mast cells functions. Indeed, TGF-β inhibits the expression of the high-affinity IgE receptor Fc1RI, which activates mast cells [
30]. On the other hand, it inhibits mast cell proliferation, degranulation, and the production of several effector molecules such as histamine and TNF-β [
31]. Given the increase in MC in fibrosis, the effect of TGF-β on MC functions can be important in the regulation of inflammatory responses that maintain the fibrosis process.
As in hepatitis C virus-induced liver fibrosis, we found half of the HLA-G
+ cells in alcohol-induced cirrhosis to be mast cells (
Table 2) and only 34% of mast cells expressed HLA-G, with high individual variability shown by the standard deviation (
Table 1). In addition, we observed a distinct repartition according to the region of the liver, in which 63–92% of the HLA-G
+ cells in fibrotic regions were mast cells, whereas only 3–23% were mast cells in cellular nodes (
Table 3). Similarly, a different pattern is observed for mast cells, since 92% of the mast cells in fibrotic regions expressed HLA-G, whereas only 23% expressed HLA-G in cellular nodes (data not shown). Thus, the expression of HLA-G is not restricted to the viral etiology of liver cirrhosis. Indeed, we obtained a similar result for lung. In IPF, 63% of HLA-G
+ cells in fibrotic regions were mast cells, whereas only 7% of those in nodes were mast cells (
Table 6). The cases of renal fibrosis were particular. As a result of the large number of tubules, it was not possible to properly count the cells in the fibrotic regions because mast cells had infiltrated the tubules. Only qualitative microscopic observation could be performed, showing a number of HLA-G
+ cells to be mast cells, without being able to differentiate tubules from fibrotic regions (
Figure 3). Thus, HLA-G appears to be expressed by mast cells in fibrotic disease through cell surface and intra-cytoplasmic molecules, irrespective of the organ. We have previously demonstrated that human mast cells in culture were able to produce soluble HLA-G forms in the conditioned medium at basal state and that secretion increased after stimulation with cytokines, including IL-10 [
10].
The higher percentage of mast cells (more than half) expressing HLA-G in the liver and lung may be explained by the inflammatory components of fibrosis. Indeed, as innate immune cells, the number of mast cells increases in inflammatory conditions, and they can also release proinflammatory mediators [
32].
The number of mast cells is elevated in fibrotic diseases. Indeed, mast cell density is higher in the lungs of patients with IPF than those with other lung pathologies [
33] and normal lung. Similarly, human renal diseases are accompanied by an increase in the number of mast cells in the renal cortex, especially in the region of fibrosis [
34], as mast cells are rarely observed in healthy kidneys.
Previous publications stated that mast cells are absent from or only sparsely found in normal human liver, lungs, and kidneys [
35]. The progress of the knowledge on mast cells has shown that mast cells as innate immune cells can be observed in all the tissues, but they are more abundant at sites exposed to the environment. Moreover, they display a large repertoire of receptors allowing them to respond to stimuli and to interact with other cells [
36]. Renal mast cells functionally resemble those in the lung. Contradictory data have been reported for the role of mast cells in fibrosis. A number of authors have proposed that mast cells are involved in fibrosis because they play a role in acute and chronic inflammation, which initiates it. In addition, mast cells are able to secrete histamine, heparin, and IL-4, which enhance the proliferation of fibroblasts. However, others [
37,
38], including us [
39], have shown that mast cells play an antifibrotic role: for example, in animal models, such as mast cell-deficient Ws/Ws mice and rats. Okazaki et al. showed that induced fibrosis was more severe in mast cell-deficient rats than in wild-type rats [
38].
Moreover, mast cells have been shown to be polarized in cancer, similarly to macrophages [
40]. Anti-inflammatory mast cells express cytokines, such as IL-10, and their number is inversely associated with the severity of inflammation, whereas proinflammatory mast cells correspond to a proinflammatory setting. It is likely that anti-inflammatory mast cells express HLA-G, in particular, because (i) an association has been shown in several models between IL-10 levels and HLA-G expression and (ii) HLA-G has an anti-inflammatory action. HLA-G-expressing mast cells may be present at an early stage of the disease, during the inflammatory phase, to counteract inflammation, which is the first reaction to the lesion. In the literature, it was reported that Il-10, by reducing inflammatory response, may inhibit the proliferation and collagen synthesis of the myofibroblasts [
41]. Indeed, IL-10 may play a protective role in alcoholic liver disease [
42]. In contrast, higher serum levels of IL-10 were found in patients with IPF than normal subjects, and the highest level of IL-10 in the bronchoalveolar lavage was demonstrated in patients with IPF compared with sarcoidosis or hypersensitivity pneumonitis [
43]. We could explain that by the less important inflammatory component. In renal fibrosis, it was demonstrated in a mouse model that a lack of IL-10 aggravated kidney inflammation and fibrosis [
44]. In humans, treatment with local IL-10 immunotherapy associated with TGF-β antagonist improves chronic kidney disease [
45].
Another relevant result is the observation of HLA-G
+ cells in cell nodes, near the fibrotic regions in rare cases of alcoholic cirrhosis. These cells are morphologically characterized by a cluster of easily recognizable cells of small to medium size, suggesting a lymphoid aggregate. Their morphological characteristics are suggestive of follicles, which are structures formed mainly by B lymphocytes. Quadruple immunofluorescence in the nodes confirmed this hypothesis, as 68% of HLA-G
+ cells co-expressed CD20, which is a specific marker of B lymphocytes (
Table 3). Similar structures were also observed in IPF, with a similar result, showing 76% of HLA-G
+ cells in nodes to be B lymphocytes (
Table 6). Lymphoid neogenesis has been reported in fibrosis. Thus, under certain pathological conditions, such as persistent inflammation, the cellular aggregates may develop into a highly organized structure resembling secondary lymphoid tissue, i.e., tertiary lymphoid organs or ectopic lymphoid follicles [
46]. Such lymphoid follicles contain T-cell-rich areas and distinct B-cell follicles with germinal centers [
47]. The mechanism by which infiltrating B cells organize the ectopic follicle and germinal center is controlled by lymphotoxin-α
1β
2 and lymphoid chemokines, such as CC-chemokine ligand 19 (CCL19), CCL21, CXC-chemokine ligand 12 (CXCL12), and CXCL13, which regulate lymphocyte homing. In addition to lymphotoxin and chemokines, antigenic stimulation is also required to induce and maintain follicle formation. Such follicles were not observed in our cohort of hepatitis C virus-induced liver fibrosis and were only found in one of three cases of alcohol-induced liver fibrosis, suggesting a distinct stage of the disease. Indeed, the function of ectopic lymphoid organs and their correlation with inflammation and fibrosis is not yet clear. A number of studies have shown a novel and surprising role for B cells in regulating fibroblasts in fibrosis, in which their profibrotic effect is analogous to that of TGF-β and also enhanced by B-cell activating factor (BAFF) [
48].
The relevance of the cellular source of HLA-G in fibrosis is not only descriptive but also functional. HLA-G has an inhibitory effect on the function of all types of lymphocytes [
49] and dendritic cells [
20,
50] through its specific receptors, such as ILT2 and ILT4. The presence of HLA-G on these immune cells, in addition to being a marker of inflammation, is also a sign of an appropriate immune reaction by also decreasing inflammation. Indeed, HLA-G is known to play a protective role against exaggerated inflammatory reactions, as previously shown in septic shock [
51]. In addition, we previously studied the reciprocal interaction between mast cells and hepatic stellate cells and showed that it leads to the attraction of mast cells and a significant decrease in collagen production by HSC cells through HLA-G production [
39]. In addition, the expression of HLA-G by B cells in ectopic follicles may also contribute to counteract the profibrotic effect of B lymphocytes on myofibroblasts by inhibiting B cells via an autocrine mechanism.


 ,
, 



{kind=link}
{kind=link}
{kind=link}