
Placental Endocrine Activity: Adaptation and Disruption of Maternal Glucose Metabolism in Pregnancy and the Influence of Fetal Sex


 , and
, and 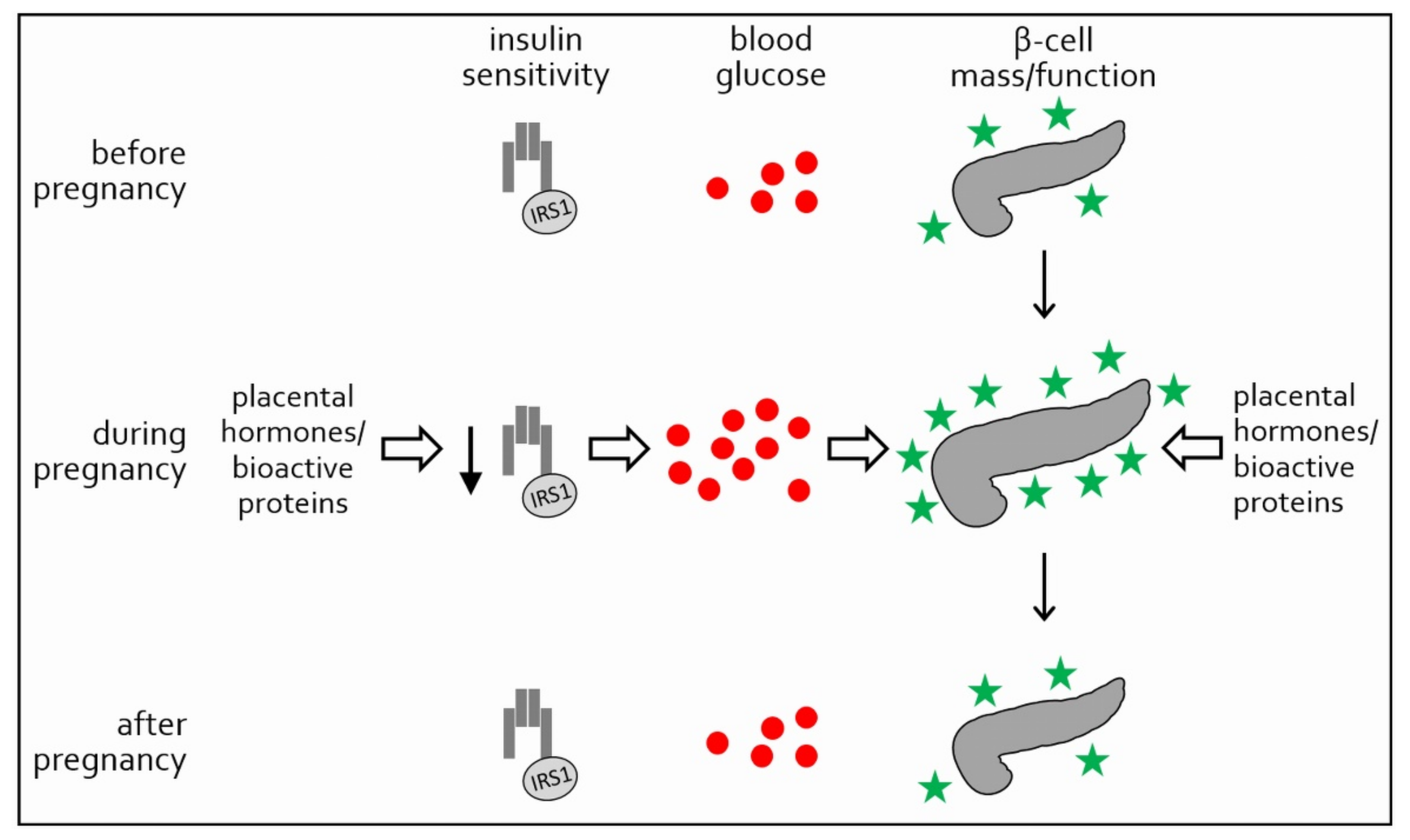{kind=link}
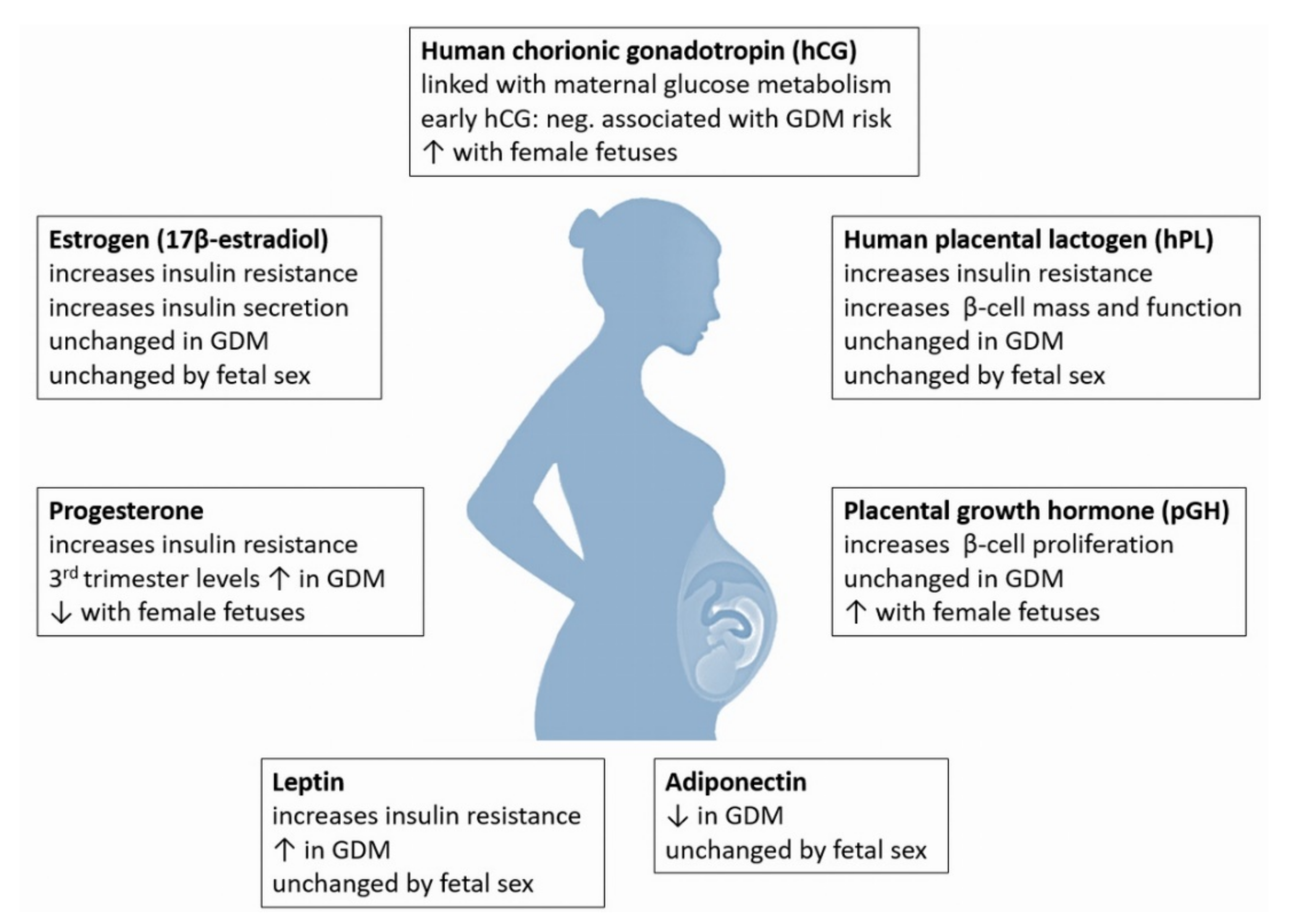{kind=link}
Abstract
:1. Modulation of Glucose Metabolism in Normal Pregnancy
1.1. Establishment of Insulin Resistance in Pregnancy
1.2. Balancing of Insulin Resistance by Expansion of β-Cell Function
1.3. Outlook
2. Methodology
3. Gestational Diabetes, Pathophysiology and Screening
3.1. Pathophysiology of GDM: Insulin Resistance and β-Cell Failure
3.2. GDM Screening and Risk Factors
4. Adaptation and Disruption of Maternal Glucose Metabolism Depends on Fetal Sex
5. The Placenta: Endocrine Organ of the Fetus
6. Placental Hormones: Role in Glucose Metabolism and Sex Dimorphism
6.1. Early hCG Is Negatively Associated with the Risk for GDM Diagnosis
6.2. Placental Lactogen Increases Maternal β-Cell Mass and Function
6.3. Placental Growth Hormone Is Not Altered in GDM but Can Be Associated with Macrosomia
6.4. Adiponectin/Leptin Ratio Is Inversely Associated with GDM
6.5. Maternal Estrogen and Adipose Estrogen Receptor Expression Are Altered in GDM
6.6. Progesterone Is Inversely Associated with Glucose and Insulin in Early Pregnancy
7. Hormonal and Molecular Causes Underlying Fetal and Placental Sexual Dimorphisms
8. Conclusions
AuthorContributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poon, L.C.; McIntyre, H.D.; Hyett, J.A.; da Fonseca, E.B.; Hod, M. FIGO Pregnancy and NCD Committee. The first-trimester of pregnancy—A window of opportunity for prediction and prevention of pregnancy complications and future life. Diabetes Res. Clin. Pract. 2018, 145, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouzon, S.H.; Lassance, L. Endocrine and metabolic adaptations to pregnancy, impact of obesity. Horm. Mol. Biol. Clin. Investig. 2015, 24, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.; Smith, L.; Bowe, J. Placental peptides regulating islet adaptation to pregnancy, Clinical potential in gestational diabetes mellitus. Curr. Opin. Pharmacol. 2018, 43, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadden, D.R.; McLaughlin, C. Normal and abnormal maternal metabolism during pregnancy. Semin. Fetal. Neonatal. Med. 2009, 14, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Lain, K.Y.; Catalano, P.M. Metabolic changes in pregnancy. Clin. Obstet. Gynecol. 2007, 50, 938–948. [Google Scholar] [CrossRef]
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butte, N.F. Carbohydrate and lipid metabolism in pregnancy, Normal compared with gestational diabetes mellitus. Am. J. Clin. Nutr. 2000, 71, 1256S–1261S. [Google Scholar] [CrossRef]
- Jones, C.W. Gestational diabetes and its impact on the neonate. Neonatal. Netw. 2001, 20, 17–23. [Google Scholar] [CrossRef]
- Baeyens, L.; Hindi, S.; Sorenson, R.L.; German, M.S. Beta-Cell adaptation in pregnancy. Diabetes Obes. Metab. 2016, 18, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Westgren, M.; Carlsson, C.; Lindholm, T.; Thysell, H.; Ingemarsson, I. Continuous maternal glucose measurements and fetal glucose and insulin levels after administration of terbutaline in term labor. Acta Obstet. Gynecol. Scand. Suppl. 1982, 108, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Economides, D.L.; Nicolaides, K.H.; Campbell, S. Relation between maternal-to-fetal blood glucose gradient and uterine and umbilical Doppler blood flow measurements. Br. J. Obstet. Gynaecol. 1990, 97, 543–544. [Google Scholar] [CrossRef]
- Parrettini, S.; Caroli, A.; Torlone, E. Nutrition and Metabolic Adaptations in Physiological and Complicated Pregnancy, Focus on Obesity and Gestational Diabetes. Front. Endocrinol. 2020, 11, 611929. [Google Scholar] [CrossRef] [PubMed]
- Rieck, S.; Kaestner, K.H. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol. Metab. 2010, 21, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, T.A.; Xiang, A.H. Gestational diabetes mellitus. J. Clin. Investig. 2005, 115, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hou, W.; Meng, X.; Zhao, W.; Pan, J.; Tang, J.; Huang, Y.; Tao, M.; Liu, F. Heterogeneity of insulin resistance and beta cell dysfunction in gestational diabetes mellitus, A prospective cohort study of perinatal outcomes. J. Transl. Med. 2018, 16, 289. [Google Scholar] [CrossRef]
- Catalano, P.M. Trying to understand gestational diabetes. Diabetes Med. 2014, 31, 273–281. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Mamerto, T.P.; Chung, G.; Villavieja, A.; Gaus, N.L.; Morgan, E.; Pineda-Cortel, M.R.B. Gestational Diabetes Mellitus, A Harbinger of the Vicious Cycle of Diabetes. Int. J. Mol. Sci. 2020, 21, 5003. [Google Scholar] [CrossRef]
- Cornejo, M.; Fuentes, G.; Valero, P.; Vega, S.; Grismaldo, A.; Toledo, F.; Pardo, F.; Moore-Carrasco, R.; Subiabre, M.; Casanello, P.; et al. Gestational diabesity and foetoplacental vascular dysfunction. Acta Physiol. 2021, 232, e13671. [Google Scholar] [CrossRef]
- McIntyre, H.D.; Catalano, P.; Zhang, C.; Desoye, G.; Mathiesen, E.R.; Damm, P. Gestational diabetes mellitus. Nat. Rev. Dis. Primers 2019, 5, 47. [Google Scholar] [CrossRef]
- Egan, A.M.; Dunne, F.P. Optimal management of gestational diabetes. Br. Med. Bull. 2019, 131, 97–108. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Singh, R.; Pradeep, Y.; Kapoor, D.; Rani, A.K.; Pradhan, S.; Bhatia, E.; Yadav, S.B. Evaluation of the prevalence of gestational diabetes mellitus in North Indians using the International Association of Diabetes and Pregnancy Study groups (IADPSG) criteria. J. Postgrad. Med. 2015, 61, 155–158. [Google Scholar]
- International Association of Diabetes and Pregnancy Study Groups Consensus Panel; Metzger, B.E.; Gabbe, S.G.; Persson, B.; Buchanan, T.A.; Catalano, P.A.; Damm, P.; Dyer, A.R.; Leiva, A. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 2010, 33, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Egan, A.M.; Dow, M.L.; Vella, A. A Review of the Pathophysiology and Management of Diabetes in Pregnancy. Mayo Clin. Proc. 2020, 95, 2734–2746. [Google Scholar] [CrossRef]
- Verburg, P.E.; Tucker, G.; Scheil, W.; Erwich, J.J.; Dekker, G.A.; Roberts, C.T. Sexual Dimorphism in Adverse Pregnancy Outcomes—A Retrospective Australian Population Study 1981–2011. PLoS ONE 2016, 11, e0158807. [Google Scholar] [CrossRef]
- Retnakaran, R.; Kramer, C.K.; Ye, C.; Kew, S.; Hanley, A.J.; Connelly, P.W.; Sermer, M.; Zinman, B. Fetal sex and maternal risk of gestational diabetes mellitus, the impact of having a boy. Diabetes Care 2015, 38, 844–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retnakaran, R.; Shah, B.R. Fetal Sex and the Natural History of Maternal Risk of Diabetes During and After Pregnancy. J. Clin. Endocrinol. Metab. 2015, 100, 2574–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannubilo, S.R.; Pasculli, A.; Ballatori, C.; Biagini, A.; Ciavattini, A. Fetal Sex, Need for Insulin, and Perinatal Outcomes in Gestational Diabetes Mellitus, An Observational Cohort Study. Clin. Ther. 2018, 40, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Retnakaran, R.; Shah, B.R. Sex of the baby and future maternal risk of Type 2 diabetes in women who had gestational diabetes. Diabet. Med. 2016, 33, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Jaskolka, D.; Retnakaran, R.; Zinman, B.; Kramer, C.K. Sex of the baby and risk of gestational diabetes mellitus in the mother, A systematic review and meta-analysis. Diabetologia 2015, 58, 2469–2475. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.M.; Segurado, R.; Mahony, R.M.; Foley, M.E.; McAuliffe, F.M. The Effects of Fetal Gender on Maternal and Fetal Insulin Resistance. PLoS ONE 2015, 10, e0137215. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Geng, L.; Zhang, Y.; Lu, H.; Shen, Y.; Chen, R.; Fang, P.; Tao, M.; Wang, C.; Jia, W. Fetal sex influences maternal fasting plasma glucose levels and basal beta-cell function in pregnant women with normal glucose tolerance. Acta Diabetol. 2017, 54, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Yasuhi, I.; Koga, M.; Sugimi, S.; Umezaki, Y.; Fukuoka, M.; Suga, S.; Fukuda, M.; Kusuda, N. Fetal sex and maternal insulin resistance during mid-pregnancy, A retrospective cohort study. BMC Pregnancy Childbirth 2020, 20, 560. [Google Scholar] [CrossRef]
- Xiao, L.; Zhao, J.P.; Nuyt, A.M.; Fraser, W.D.; Luo, Z.C. Female fetus is associated with greater maternal insulin resistance in pregnancy. Diabet. Med. 2014, 31, 1696–16701. [Google Scholar] [CrossRef] [PubMed]
- Rafferty, A.R.; Geraghty, A.A.; Kennelly, M.A.; O’Brien, E.C.; Reji, R.M.; Mehegan, J.; Segurado, R.; Smith, T.; Maguire, O.; Cronin, M.; et al. Limited Impact of Fetal Sex and Maternal Body Mass Index on Fetal and Maternal Insulin Resistance and Lipid Metabolism, Findings from the PEARs Study. Reprod. Sci. 2020, 27, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, S.A.; Wooding, F.B. An immunogold, cryoultrastructural study of sites of synthesis and storage of chorionic gonadotropin and placental lactogen in human syncytiotrophoblast. Cell Tissue Res. 1990, 261, 375–382. [Google Scholar] [CrossRef]
- Hay, D.L.; Lopata, A. Chorionic gonadotropin secretion by human embryos in vitro. J. Clin. Endocrinol. Metab. 1988, 67, 1322–1324. [Google Scholar] [CrossRef]
- Tal, R.; Taylor, H.S. Endocrinology of Pregnancy; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Cole, L.A. Biological functions of hCG and hCG-related molecules. Reprod. Biol. Endocrinol. 2010, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Benirschke, K.; Burton, G.J.; Baergen, R.N. Early Development of the Human Placenta Pathology of the Human Placenta; Springer: Berlin/Heidelberg, Germany, 2012; pp. 41–53. [Google Scholar]
- Benirschke, K.; Burton, G.J.; Baergen, R.N. Nonvillous Parts and Trophoblast Invasion. Pathology of the Human Placenta; Springer: Berlin/Heidelberg, Germany, 2012; pp. 157–240. [Google Scholar]
- He, N.; van Iperen, L.; de Jong, D.; Szuhai, K.; Helmerhorst, F.M.; van der Westerlaken, L.A.; Chuva de Sousa Lopes, S.M. Human Extravillous Trophoblasts Penetrate Decidual Veins and Lymphatics before Remodeling Spiral Arteries during Early Pregnancy. PLoS ONE 2017, 12, e0169849. [Google Scholar] [CrossRef]
- Moser, G.; Weiss, G.; Sundl, M.; Gauster, M.; Siwetz, M.; Lang-Olip, I.; Huppertz, B. Extravillous trophoblasts invade more than uterine arteries, Evidence for the invasion of uterine veins. Histochem. Cell Biol. 2017, 147, 353–366. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Stenman, U.H.; Tiitinen, A.; Alfthan, H.; Valmu, L. The classification, functions and clinical use of different isoforms of HCG. Hum. Reprod. Update 2006, 12, 769–784. [Google Scholar] [CrossRef]
- De Medeiros, S.F.; Norman, R.J. Human choriogonadotrophin protein core and sugar branches heterogeneity, Basic and clinical insights. Hum. Reprod. Update 2009, 15, 69–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, L.A. hCG, the wonder of today’s science. Reprod. Biol. Endocrinol. 2012, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Guo, F.; Maraka, S.; Zhang, Y.; Zhang, C.; Korevaar, T.I.M.; Fan, J. Associations between Human Chorionic Gonadotropin, Maternal Free Thyroxine, and Gestational Diabetes Mellitus. Thyroid 2021, 31, 1282–1288. [Google Scholar] [CrossRef]
- Visconti, F.; Quaresima, P.; Chiefari, E.; Caroleo, P.; Arcidiacono, B.; Puccio, L.; Mirabelli, M.; Foti, D.P.; Di Carlo, C.; Vero, R.; et al. First Trimester Combined Test (FTCT) as a Predictor of Gestational Diabetes Mellitus. Int. J. Environ. Res. Public Health 2019, 16, 3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, K.; Cowans, N.J. The association between gestational diabetes mellitus and first trimester aneuploidy screening markers. Ann. Clin. Biochem. 2013, 50, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Donovan, B.M.; Nidey, N.L.; Jasper, E.A.; Robinson, J.G.; Bao, W.; Saftlas, A.F.; Ryckman, K.K. First trimester prenatal screening biomarkers and gestational diabetes mellitus, A systematic review and meta-analysis. PLoS ONE 2018, 13, e0201319. [Google Scholar] [CrossRef] [PubMed]
- Valent, A.M.; Choi, H.; Kolahi, K.S.; Thornburg, K.L. Hyperglycemia and gestational diabetes suppress placental glycolysis and mitochondrial function and alter lipid processing. FASEB J. 2021, 35, e21423. [Google Scholar] [CrossRef]
- Hong, Y.; Xie, Q.X.; Chen, C.Y.; Yang, C.; Li, Y.Z.; Chen, D.M.; Xie, M.Q. Insulin resistance in first-trimester pregnant women with pre-pregnant glucose tolerance and history of recurrent spontaneous abortion. J. Biol. Regul. Homeost. Agents 2013, 27, 225–231. [Google Scholar]
- Hsieh, T.T.; Chen, K.C.; Hsu, J.J.; Chiu, T.H.; Hsieh, C.C.; Wang, H.S. Effects of glucose on placental hormones in the human term placenta in vitro. J. Formos. Med. Assoc. 1997, 96, 309–313. [Google Scholar]
- Barnea, E.R.; Neubrun, D.; Shurtz-Swirski, R. Effect of insulin on human chorionic gonadotrophin secretion by placental explants. Hum. Reprod. 1993, 8, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Mandl, M.; Haas, J.; Bischof, P.; Nohammer, G.; Desoye, G. Serum-dependent effects of IGF-I and insulin on proliferation and invasion of human first trimester trophoblast cell models. Histochem. Cell Biol. 2002, 117, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Deville, J.L.; Gaspard, U.; Reuter, A.M.; Franchimont, P.; Lambotte, R. Maternal serum levels of chorionic gonadotropin and its alpha and beta free subunits as a function of the fetal sex: Preliminary study. Comptes Rendus Seances Soc. Biol. Fil. 1980, 174, 365–369. [Google Scholar]
- Yaron, Y.; Lehavi, O.; Orr-Urtreger, A.; Gull, I.; Lessing, J.B.; Amit, A.; Ben-Yosef, D. Maternal serum HCG is higher in the presence of a female fetus as early as week 3 post-fertilization. Hum. Reprod. 2002, 17, 485–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Li, R.; Wang, Y.; Li, M.; Wang, L.; Zhen, X.; Liu, P. Increased maternal serum hCG concentrations in the presence of a female fetus as early as 2 weeks after IVF-ET. J. Gynecol. Obstet. Hum. Reprod. 2021, 50, 102053. [Google Scholar] [CrossRef]
- Larsen, S.O.; Wojdemann, K.R.; Shalmi, A.C.; Sundberg, K.; Christiansen, M.; Tabor, A. Gender impact on first trimester markers in Down syndrome screening. Prenat. Diagn. 2002, 22, 1207–1208. [Google Scholar] [CrossRef]
- Spencer, K. The influence of fetal sex in screening for Down syndrome in the second trimester using AFP and free beta-hCG. Prenat. Diagn. 2000, 20, 648–651. [Google Scholar] [CrossRef]
- Cowans, N.J.; Stamatopoulou, A.; Maiz, N.; Spencer, K.; Nicolaides, K.H. The impact of fetal gender on first trimester nuchal translucency and maternal serum free beta-hCG and PAPP-A MoM in normal and trisomy 21 pregnancies. Prenat. Diagn. 2009, 29, 578–581. [Google Scholar] [CrossRef]
- Adibi, J.J.; Lee, M.K.; Saha, S.; Boscardin, W.J.; Apfel, A.; Currier, R.J. Fetal sex differences in human chorionic gonadotropin fluctuate by maternal race, age, weight and by gestational age. J. Dev. Orig. Health Dis. 2015, 6, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Gol, M.; Altunyurt, S.; Cimrin, D.; Guclu, S.; Bagci, M.; Demir, N. Different maternal serum hCG levels in pregnant women with female and male fetuses, Does fetal hypophyseal-adrenal-gonadal axis play a role? J. Perinat. Med. 2004, 32, 342–345. [Google Scholar] [CrossRef]
- Steier, J.A.; Myking, O.L.; Ulstein, M. Human chorionic gonadotropin in cord blood and peripheral maternal blood in singleton and twin pregnancies at delivery. Acta Obstet. Gynecol. Scand. 1989, 68, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.H.; Fitzpatrick, S.L.; Barrera-Saldana, H.A.; Resendez-Perez, D.; Saunders, G.F. The human placental lactogen genes, Structure, function, evolution and transcriptional regulation. Endocr. Rev. 1991, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Handwerger, S.; Freemark, M. The roles of placental growth hormone and placental lactogen in the regulation of human fetal growth and development. J. Pediatr. Endocrinol. Metab. 2000, 13, 343–356. [Google Scholar] [CrossRef]
- Ryan, E.A.; Enns, L. Role of gestational hormones in the induction of insulin resistance. J. Clin. Endocrinol. Metab. 1988, 67, 341–347. [Google Scholar] [CrossRef]
- Le, T.N.; Elsea, S.H.; Romero, R.; Chaiworapongsa, T.; Francis, G.L. Prolactin receptor gene polymorphisms are associated with gestational diabetes. Genet. Test. Mol. Biomark. 2013, 17, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Brelje, T.C.; Scharp, D.W.; Lacy, P.E.; Ogren, L.; Talamantes, F.; Robertson, M.; Friesen, H.G.; Sorenson, R.L. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets, Implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 1993, 132, 879–887. [Google Scholar] [CrossRef]
- Ngala, R.A.; Fondjo, L.A.; Gmagna, P.; Ghartey, F.N.; Awe, M.A. Placental peptides metabolism and maternal factors as predictors of risk of gestational diabetes in pregnant women: A case-control study. PLoS ONE 2017, 12, e0181613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retnakaran, R.; Ye, C.; Kramer, C.K.; Connelly, P.W.; Hanley, A.J.; Sermer, M.; Zinman, B. Evaluation of Circulating Determinants of Beta-Cell Function in Women with and Without Gestational Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 2683–2691. [Google Scholar] [CrossRef]
- Houghton, D.J.; Shackleton, P.; Obiekwe, B.C.; Chard, T. Relationship of maternal and fetal levels of human placental lactogen to the weight and sex of the fetus. Placenta 1984, 5, 455–458. [Google Scholar] [CrossRef]
- Lacroix, M.C.; Guibourdenche, J.; Frendo, J.L.; Muller, F.; Evain-Brion, D. Human placental growth hormone—A review. Placenta 2002, 23, S87–S94. [Google Scholar] [CrossRef]
- Frankenne, F.; Closset, J.; Gomez, F.; Scippo, M.L.; Smal, J.; Hennen, G. The physiology of growth hormones (GHs) in pregnant women and partial characterization of the placental GH variant. J. Clin. Endocrinol. Metab. 1988, 66, 1171–1180. [Google Scholar] [CrossRef]
- Mirlesse, V.; Frankenne, F.; Alsat, E.; Poncelet, M.; Hennen, G.; Evain-Brion, D. Placental growth hormone levels in normal pregnancy and in pregnancies with intrauterine growth retardation. Pediatr. Res. 1993, 34, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Alsat, E.; Guibourdenche, J.; Luton, D.; Frankenne, F.; Evain-Brion, D. Human placental growth hormone. Am. J. Obstet. Gynecol. 1997, 177, 1526–1534. [Google Scholar] [CrossRef]
- Patel, N.; Alsat, E.; Igout, A.; Baron, F.; Hennen, G.; Porquet, D.; Evain-Brion, D. Glucose inhibits human placental GH secretion, in vitro. J. Clin. Endocrinol. Metab. 1995, 80, 1743–1746. [Google Scholar]
- Wang, S.; Wu, J.; Wang, N.; Zeng, L.; Wu, Y. The role of growth hormone receptor in beta cell function. Growth Horm. IGF Res. 2017, 36, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Moldrup, A.; Petersen, E.D.; Nielsen, J.H. Effects of sex and pregnancy hormones on growth hormone and prolactin receptor gene expression in insulin-producing cells. Endocrinology 1993, 133, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Vickers, M.H.; Taylor, R.S.; Fraser, M.; McCowan, L.M.E.; Baker, P.N.; Perry, J.K. Maternal serum placental growth hormone, insulin-like growth factors and their binding proteins at 20 weeks’ gestation in pregnancies complicated by gestational diabetes mellitus. Hormones 2017, 16, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Chellakooty, M.; Skibsted, L.; Skouby, S.O.; Andersson, A.M.; Petersen, J.H.; Main, K.M.; Skakkebaek, N.E.; Juul, A. Longitudinal study of serum placental GH in 455 normal pregnancies, Correlation to gestational age, fetal gender, and weight. J. Clin. Endocrinol. Metab. 2002, 87, 2734–2739. [Google Scholar] [CrossRef]
- Fruhbeck, G.; Catalan, V.; Rodriguez, A.; Ramirez, B.; Becerril, S.; Salvador, J.; Portincasa, P.; Colina, I.; Gomez-Ambrosi, J. Involvement of the leptin-adiponectin axis in inflammation and oxidative stress in the metabolic syndrome. Sci. Rep. 2017, 7, 6619. [Google Scholar] [CrossRef]
- Masuzaki, H.; Ogawa, Y.; Sagawa, N.; Hosoda, K.; Matsumoto, T.; Mise, H.; Nishimura, H.; Yoshimasa, Y.; Tanaka, I.; Mori, T.; et al. Nonadipose tissue production of leptin, Leptin as a novel placenta-derived hormone in humans. Nat. Med. 1997, 3, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tan, B.; Karteris, E.; Zervou, S.; Digby, J.; Hillhouse, E.W.; Vatish, M.; Randeva, H.S. Secretion of adiponectin by human placenta, Differential modulation of adiponectin and its receptors by cytokines. Diabetologia 2006, 49, 1292–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabe, K.; Lehrke, M.; Parhofer, K.G.; Broedl, U.C. Adipokines and insulin resistance. Mol. Med. 2008, 14, 741–751. [Google Scholar] [CrossRef]
- Grasman, J. Reconstruction of the drive underlying food intake and its control by leptin and dieting. PLoS ONE 2013, 8, e74997. [Google Scholar] [CrossRef] [PubMed]
- Hauguel-de Mouzon, S.; Lepercq, J.; Catalano, P. The known and unknown of leptin in pregnancy. Am. J. Obstet Gynecol. 2006, 194, 1537–1545. [Google Scholar] [CrossRef]
- Senaris, R.; Garcia-Caballero, T.; Casabiell, X.; Gallego, R.; Castro, R.; Considine, R.V.; Dieguez, C.; Casanueva, F.F. Synthesis of leptin in human placenta. Endocrinology 1997, 138, 4501–4504. [Google Scholar] [CrossRef]
- Linnemann, K.; Malek, A.; Sager, R.; Blum, W.F.; Schneider, H.; Fusch, C. Leptin production and release in the dually in vitro perfused human placenta. J. Clin. Endocrinol. Metab. 2000, 85, 4298–4301. [Google Scholar]
- Chardonnens, D.; Cameo, P.; Aubert, M.L.; Pralong, F.P.; Islami, D.; Campana, A.; Gaillard, R.C.; Bischof, P. Modulation of human cytotrophoblastic leptin secretion by interleukin-1alpha and 17beta-oestradiol and its effect on HCG secretion. Mol. Hum. Reprod. 1999, 5, 1077–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, L.; Saget, S.; Lu, C.; Hay, W.W., Jr.; Karsenty, G.; Shao, J. Adiponectin Promotes Maternal Beta-Cell Expansion Through Placental Lactogen Expression. Diabetes 2021, 70, 132–142. [Google Scholar] [CrossRef]
- Williams, M.A.; Qiu, C.; Muy-Rivera, M.; Vadachkoria, S.; Song, T.; Luthy, D.A. Plasma adiponectin concentrations in early pregnancy and subsequent risk of gestational diabetes mellitus. J. Clin. Endocrinol. Metab. 2004, 89, 2306–2311. [Google Scholar] [CrossRef] [Green Version]
- Kharroubi, I.; Rasschaert, J.; Eizirik, D.L.; Cnop, M. Expression of adiponectin receptors in pancreatic beta cells. Biochem. Biophys. Res. Commun. 2003, 312, 1118–1122. [Google Scholar] [CrossRef]
- Staiger, K.; Stefan, N.; Staiger, H.; Brendel, M.D.; Brandhorst, D.; Bretzel, R.G.; Machicao, F.; Kellerer, M.; Stumvoll, M.; Fritsche, A.; et al. Adiponectin is functionally active in human islets but does not affect insulin secretory function or beta-cell lipoapoptosis. J. Clin. Endocrinol. Metab. 2005, 90, 6707–6713. [Google Scholar] [CrossRef] [Green Version]
- McLachlan, K.A.; O’Neal, D.; Jenkins, A.; Alford, F.P. Do adiponectin, TNFalpha, leptin and CRP relate to insulin resistance in pregnancy? Studies in women with and without gestational diabetes, during and after pregnancy. Diabetes Metab. Res. Rev. 2006, 22, 131–138. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, H.D.; Chang, A.M.; Callaway, L.K.; Cowley, D.M.; Dyer, A.R.; Radaelli, T.; Farrell, K.A.; Huston-Presley, L.; Amini, S.B.; Kirwan, J.P.; et al. Hormonal and metabolic factors associated with variations in insulin sensitivity in human pregnancy. Diabetes Care 2010, 33, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Shang, M.; Dong, X.; Hou, L. Correlation of adipokines and markers of oxidative stress in women with gestational diabetes mellitus and their newborns. J. Obstet. Gynaecol. Res. 2018, 44, 637–646. [Google Scholar] [CrossRef]
- Qiu, C.; Williams, M.A.; Vadachkoria, S.; Frederick, I.O.; Luthy, D.A. Increased maternal plasma leptin in early pregnancy and risk of gestational diabetes mellitus. Obstet. Gynecol. 2004, 103, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Fernandez-Galaz, C.; Fernandez-Agullo, T.; Arribas, C.; Andres, A.; Ros, M.; Carrascosa, J.M. Leptin impairs insulin signaling in rat adipocytes. Diabetes 2004, 53, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroqui, L.; Gonzalez, A.; Neco, P.; Caballero-Garrido, E.; Vieira, E.; Ripoll, C.; Nadal, A.; Quesada, I. Role of leptin in the pancreatic beta-cell, Effects and signaling pathways. J. Mol. Endocrinol. 2012, 49, R9–R17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Atawi, F.; Warsy, A.; Babay, Z.; Addar, M. Fetal sex and leptin concentrations in pregnant females. Ann. Saudi Med. 2005, 25, 124–128. [Google Scholar] [CrossRef]
- Fazeli Daryasari, S.R.; Tehranian, N.; Kazemnejad, A.; Razavinia, F.; Tork Tatari, F.; Pahlavan, F. Adiponectin levels in maternal serum and umbilical cord blood at birth by mode of delivery, Relationship to anthropometric measurements and fetal sex. BMC Pregnancy Childbirth 2019, 19, 344. [Google Scholar] [CrossRef]
- Berkane, N.; Liere, P.; Oudinet, J.P.; Hertig, A.; Lefevre, G.; Pluchino, N.; Schumacher, M.; Chabbert-Buffet, N. From Pregnancy to Preeclampsia, A Key Role for Estrogens. Endocr. Rev. 2017, 38, 123–144. [Google Scholar] [CrossRef]
- Husen, B.; Adamski, J.; Bruns, A.; Deluca, D.; Fuhrmann, K.; Moller, G.; Schwabe, I.; Einspanier, A. Characterization of 17beta-hydroxysteroid dehydrogenase type 7 in reproductive tissues of the marmoset monkey. Biol. Reprod. 2003, 68, 2092–2099. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F. Role of Sex Steroids in beta Cell Function, Growth, and Survival. Trends Endocrinol. Metab. 2016, 27, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.; Cho, E.H.; Baek, K.H.; Lee, K.J. Prediction of Gestational Diabetes Mellitus by Unconjugated Estriol Levels in Maternal Serum. Int. J. Med. Sci. 2017, 14, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiblova, P.; Dostalova, I.; Bartlova, M.; Lacinova, Z.; Ticha, I.; Krejci, V.; Springer, D.; Kleibl, Z.; Haluzik, M. Expression of adipokines and estrogen receptors in adipose tissue and placenta of patients with gestational diabetes mellitus. Mol. Cell. Endocrinol. 2010, 314, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knabl, J.; Hiden, U.; Huttenbrenner, R.; Riedel, C.; Hutter, S.; Kirn, V.; Gunthner-Biller, M.; Desoye, G.; Kainer, F.; Jeschke, U. GDM Alters Expression of Placental Estrogen Receptor alpha in a Cell Type and Gender-Specific Manner. Reprod. Sci. 2015, 22, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Toriola, A.T.; Vaarasmaki, M.; Lehtinen, M.; Zeleniuch-Jacquotte, A.; Lundin, E.; Rodgers, K.G.; Lakso, H.A.; Chen, T.; Schock, H.; Hallmans, G.; et al. Determinants of maternal sex steroids during the first half of pregnancy. Obstet. Gynecol. 2011, 118, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Jarvela, I.Y.; Zackova, T.; Laitinen, P.; Ryynanen, M.; Tekay, A. Effect of parity and fetal sex on placental and luteal hormones during early first trimester. Prenat. Diagn. 2012, 32, 160–167. [Google Scholar] [CrossRef]
- Lutterodt, M.; Byskov, A.G.; Skouby, S.O.; Tabor, A.; Yding Andersen, C. Anti-Mullerian hormone in pregnant women in relation to other hormones, fetal sex and in circulation of second trimester fetuses. Reprod. Biomed. Online 2009, 18, 694–699. [Google Scholar] [CrossRef]
- Vejrazkova, D.; Vcelak, J.; Vankova, M.; Lukasova, P.; Bradnova, O.; Halkova, T.; Kancheva, R.; Bendlova, B. Steroids and insulin resistance in pregnancy. J. Steroid Biochem. Mol. Biol. 2014, 139, 122–129. [Google Scholar] [CrossRef]
- Freemark, M. Regulation of maternal metabolism by pituitary and placental hormones, Roles in fetal development and metabolic programming. Horm. Res. 2006, 65, 41–49. [Google Scholar] [CrossRef]
- Costa, M.A. The endocrine function of human placenta: An overview. Reprod. Biomed. Online 2016, 32, 14–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Renzo, G.C.; Giardina, I.; Clerici, G.; Brillo, E.; Gerli, S. Progesterone in normal and pathological pregnancy. Horm. Mol. Biol. Clin. Investig. 2016, 27, 35–48. [Google Scholar] [CrossRef]
- Branisteanu, D.D.; Mathieu, C. Progesterone in gestational diabetes mellitus, Guilty or not guilty? Trends Endocrinol. Metab. 2003, 14, 54–56. [Google Scholar] [CrossRef]
- Tachibana, T.; Kasajima, A.; Aoki, T.; Tabata, T.; McNamara, K.; Yazdani, S.; Satoko, S.; Fujishima, F.; Motoi, F.; Unno, M.; et al. Progesteron receptor expression in insulin producing cells of neuroendocrine neoplasms. J. Steroid Biochem. Mol. Biol. 2020, 201, 105694. [Google Scholar] [CrossRef]
- Li, M.; Song, Y.; Rawal, S.; Hinkle, S.N.; Zhu, Y.; Tekola-Ayele, F.; Ferrara, A.; Tsai, M.Y.; Zhang, C. Plasma Prolactin and Progesterone Levels and the Risk of Gestational Diabetes, A Prospective and Longitudinal Study in a Multiracial Cohort. Front. Endocrinol. 2020, 11, 83. [Google Scholar] [CrossRef]
- Zhang, Z.; Kang, X.; Guo, Y.; Zhang, J.; Xie, J.; Shao, S.; Xiang, Y.; Chen, G.; Yu, X. Association of circulating galectin-3 with gestational diabetes mellitus, progesterone, and insulin resistance. J. Diabetes 2021, 13, 54–62. [Google Scholar] [CrossRef]
- Rebarber, A.; Istwan, N.B.; Russo-Stieglitz, K.; Cleary-Goldman, J.; Rhea, D.J.; Stanziano, G.J.; Saltzman, D.H. Increased incidence of gestational diabetes in women receiving prophylactic 17alpha-hydroxyprogesterone caproate for prevention of recurrent preterm delivery. Diabetes Care 2007, 30, 2277–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuu, J.; Hellerstein, S.; Lipworth, L.; Wide, L.; Xu, B.; Yu, G.P.; Kuper, H.; Lagiou, P.; Hankinson, S.E.; Ekbom, A.; et al. Correlates of pregnancy oestrogen, progesterone and sex hormone-binding globulin in the USA and China. Eur. J. Cancer Prev. 2002, 11, 283–293. [Google Scholar] [CrossRef]
- Adamcova, K.; Kolatorova, L.; Skodova, T.; Simkova, M.; Parizek, A.; Starka, L.; Duskova, M. Steroid hormone levels in the peripartum period—Differences caused by fetal sex and delivery type. Physiol. Res. 2018, 67, S489–S497. [Google Scholar] [CrossRef]
- Mittwoch, U. Blastocysts prepare for the race to be male. Hum. Reprod. 1993, 8, 1550–1555. [Google Scholar] [CrossRef]
- Bukowski, R.; Smith, G.C.; Malone, F.D.; Ball, R.H.; Nyberg, D.A.; Comstock, C.H.; Hankins, G.D.; Berkowitz, R.L.; Gross, S.J.; Dugoff, L.; et al. Human sexual size dimorphism in early pregnancy. Am. J. Epidemiol. 2007, 165, 1216–1218. [Google Scholar] [CrossRef]
- Eriksson, J.G.; Kajantie, E.; Osmond, C.; Thornburg, K.; Barker, D.J. Boys live dangerously in the womb. Am. J. Hum. Biol. 2010, 22, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Berta, P.; Hawkins, J.R.; Sinclair, A.H.; Taylor, A.; Griffiths, B.L.; Goodfellow, P.N.; Fellous, M. Genetic evidence equating SRY and the testis-determining factor. Nature 1990, 348, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.H.; Berta, P.; Palmer, M.S.; Hawkins, J.R.; Griffiths, B.L.; Smith, M.J.; Foster, J.W.; Frischauf, A.M.; Lovell-Badge, R.; Goodfellow, P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Siiteri, P.K.; Wilson, J.D. Testosterone formation and metabolism during male sexual differentiation in the human embryo. J. Clin. Endocrinol. Metab. 1974, 38, 113–125. [Google Scholar] [CrossRef]
- Kunzig, H.J.; Meyer, U.; Schmitz-Roeckerath, B.; Broer, K.H. Influence of fetal sex on the concentration of amniotic fluid testosterone, Antenatal sex determination? Arch. Gynakol. 1977, 223, 75–84. [Google Scholar] [CrossRef]
- Erdmann, K.; Schaal, N.K.; Meinlschmidt, G.; Tegethoff, M.; Frohlich, S.; Kozlowski, P.; Rivet, N.; Jamey, C.; Reix, N.; Kintz, P.; et al. Sex specific relationships between infants’ mental rotation ability and amiotic sex hormones. Neurosci. Lett. 2019, 707, 134298. [Google Scholar] [CrossRef]
- Simmons, D.; France, J.T.; Keelan, J.A.; Song, L.; Knox, B.S. Sex differences in umbilical cord serum levels of inhibin, testosterone, oestradiol, dehydroepiandrosterone sulphate, and sex hormone-binding globulin in human term neonates. Biol. Neonate 1994, 65, 287–294. [Google Scholar] [CrossRef]
- Steier, J.A.; Ulstein, M.; Myking, O.L. Human chorionic gonadotropin and testosterone in normal and preeclamptic pregnancies in relation to fetal sex. Obstet. Gynecol. 2002, 100, 552–556. [Google Scholar] [PubMed]
- Hickey, M.; Hart, R.; Keelan, J.A. The relationship between umbilical cord estrogens and perinatal characteristics. Cancer Epidemiol. Biomark. Prev. 2014, 23, 946–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inkster, A.M.; Fernandez-Boyano, I.; Robinson, W.P. Sex Differences Are Here to Stay, Relevance to Prenatal Care. J. Clin. Med. 2021, 10, 3000. [Google Scholar] [CrossRef]
- Petropoulos, S.; Edsgard, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Plaza Reyes, A.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, T.L.; Sun, T.; Koeppel, A.F.; Lee, B.; Wang, E.T.; Farber, C.R.; Rich, S.S.; Sundheimer, L.W.; Buttle, R.A.; Chen, Y.I.; et al. Sex differences in the late first trimester human placenta transcriptome. Biol. Sex Differ. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cvitic, S.; Longtine, M.S.; Hackl, H.; Wagner, K.; Nelson, M.D.; Desoye, G.; Hiden, U. The human placental sexome differs between trophoblast epithelium and villous vessel endothelium. PLoS ONE 2013, 8, e79233. [Google Scholar] [CrossRef] [PubMed]
- Clifton, V.L.; Murphy, V.E. Maternal asthma as a model for examining fetal sex-specific effects on maternal physiology and placental mechanisms that regulate human fetal growth. Placenta 2004, 25, S45–S52. [Google Scholar] [CrossRef]
- Powell, T.L.; Barner, K.; Madi, L.; Armstrong, M.; Manke, J.; Uhlson, C.; Jansson, T.; Ferchaud-Roucher, V. Sex-specific responses in placental fatty acid oxidation, esterification and transfer capacity to maternal obesity. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2021, 1866, 158861. [Google Scholar] [CrossRef]
- Sedlmeier, E.M.; Meyer, D.M.; Stecher, L.; Sailer, M.; Daniel, H.; Hauner, H.; Bader, B.L. Fetal sex modulates placental microRNA expression, potential microRNA-mRNA interactions, and levels of amino acid transporter expression and substrates, INFAT study subpopulation analysis of n-3 LCPUFA intervention during pregnancy and associations with offspring body composition. BMC Mol. Cell Biol. 2021, 22, 15. [Google Scholar]
- Cvitic, S.; Strutz, J.; Appel, H.M.; Weiss, E.; Brandl, W.T.; Thuringer, A.; Bernhart, E.M.; Lassance, L.; Wadsack, C.; Schliefsteiner, C.; et al. Sexual dimorphism of miRNA signatures in feto-placental endothelial cells is associated with altered barrier function and actin organization. Clin. Sci. 2020, 134, 39–51. [Google Scholar] [CrossRef]
- Guo, S.; Huang, S.; Jiang, X.; Hu, H.; Han, D.; Moreno, C.S.; Fairbrother, G.L.; Hughes, D.A.; Stoneking, M.; Khaitovich, P. Variation of microRNA expression in the human placenta driven by population identity and sex of the newborn. BMC Genom. 2021, 22, 286. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stern, C.; Schwarz, S.; Moser, G.; Cvitic, S.; Jantscher-Krenn, E.; Gauster, M.; Hiden, U. Placental Endocrine Activity: Adaptation and Disruption of Maternal Glucose Metabolism in Pregnancy and the Influence of Fetal Sex. Int. J. Mol. Sci. 2021, 22, 12722. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312722
Stern C, Schwarz S, Moser G, Cvitic S, Jantscher-Krenn E, Gauster M, Hiden U. Placental Endocrine Activity: Adaptation and Disruption of Maternal Glucose Metabolism in Pregnancy and the Influence of Fetal Sex. International Journal of Molecular Sciences. 2021; 22(23):12722. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312722
Chicago/Turabian StyleStern, Christina, Sarah Schwarz, Gerit Moser, Silvija Cvitic, Evelyn Jantscher-Krenn, Martin Gauster, and Ursula Hiden. 2021. "Placental Endocrine Activity: Adaptation and Disruption of Maternal Glucose Metabolism in Pregnancy and the Influence of Fetal Sex" International Journal of Molecular Sciences 22, no. 23: 12722. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312722