
The Role of EREG/EGFR Pathway in Tumor Progression

 , ,
, ,
Abstract
:1. Introduction
Erythroblastic Leukemia Viral Oncogene Homolog Signaling
2. Critical Role of EREG in Early Tumor Development
2.1. The Normal Biological Functions of EREG Signaling
2.2. EREG Promotes Early Cancer Development
3. The Expression Levels of EREG during Cancer Progression
3.1. Elevated Expressions of EREG in Various Cancer Types
3.2. The Outcomes of EREG Expression and KRAS Mutation
3.3. Alternative Signaling Pathways
4. Actions of EREG in the TME
4.1. EREG Promotes Tumorigenicity
4.2. EREG Mediates Tumor Metastasis
4.3. EREG Expression Correlates with Cancer Stem Cell Characteristics
4.4. EREG Mediates Drug Resistance
5. Potential Therapeutic Targeting of EREG/EGFR and Alternative Pathways
5.1. Targeting the EREG/EGFR Pathway
5.2. Targeting the Hypermethylation of EREG
5.3. Targeting the Alternative Activation Signaling Pathway
5.4. Targeting TAM in the TME
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, W. ErBb Family Proteins in Cholangiocarcinoma and Clinical Implications. J. Clin. Med. 2020, 9, 2255. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Ledonne, A.; Mercuri, N.B. On the Modulatory Roles of Neuregulins/ErbB Signaling on Synaptic Plasticity. Int. J. Mol. Sci. 2019, 21, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdiel-Acer, M.; Maia, A.; Hristova, Z.; Borgoni, S.; Vetter, M.; Burmester, S.; Becki, C.; Michels, B.; Abnaof, K.; Binenbaum, I.; et al. Stromal NRG1 in luminal breast cancer defines pro-fibrotic and migratory cancer-associated fibroblasts. Oncogene 2021, 40, 2651–2666. [Google Scholar] [CrossRef]
- Zhao, W.J.; Ou, G.Y.; Lin, W.W. Integrative Analysis of Neuregulin Family Members-Related Tumor Microenvironment for Predicting the Prognosis in Gliomas. Front. Immunol. 2021, 12, 682415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Karthaus, W.R.; Lee, Y.S.; Gao, V.R.; Wu, C.; Russo, J.W.; Liu, M.; Mota, J.M.; Abida, W.; Linton, E.; et al. Tumor Microenvironment-Derived NRG1 Promotes Antiandrogen Resistance in Prostate Cancer. Cancer Cell 2020, 38, 279–296. [Google Scholar] [CrossRef]
- Falls, D.L. Neuregulins: Functions, forms, and signaling strategies. Exp. Cell Res. 2003, 284, 14–30. [Google Scholar] [CrossRef]
- Ou, G.Y.; Lin, W.W.; Zhao, W.J. Neuregulins in Neurodegenerative Diseases. Front. Aging Neurosci. 2021, 13, 662474. [Google Scholar] [CrossRef]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Han, Y.; Xia, W.; Zhang, L.; Xu, S.; Ju, H.; Zhang, X.; Ren, G.; Liu, L.; et al. EREG-driven oncogenesis of Head and Neck Squamous Cell Carcinoma exhibits higher sensitivity to Erlotinib therapy. Theranostics 2020, 10, 10589–10605. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Wolf, E. The epidermal growth factor receptor ligands at a glance. J. Cell Physiol. 2009, 218, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef]
- Hinkle, C.L.; Sunnarborg, S.W.; Loiselle, D.; Parker, C.E.; Stevenson, M.; Russell, W.E.; Lee, D.C. Selective roles for tumor necrosis factor alpha-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: The juxtamembrane stalk determines cleavage efficiency. J. Biol. Chem. 2004, 279, 24179–24188. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.L.; Coffey, R.J.; Dempsey, P.J. The proamphiregulin cytoplasmic domain is required for basolateral sorting, but is not essential for constitutive or stimulus-induced processing in polarized Madin-Darby canine kidney cells. J. Biol. Chem. 2001, 276, 29538–29549. [Google Scholar] [CrossRef] [Green Version]
- Higashiyama, S.; Iwabuki, H.; Morimoto, C.; Hieda, M.; Inoue, H.; Matsushita, N. Membrane-anchored growth factors, the epidermal growth factor family: Beyond receptor ligands. Cancer Sci. 2008, 99, 214–220. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Komurasaki, T.; Uchida, D.; Takayama, Y.; Isobe, T.; Okuyama, T.; Hanada, K. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J. Biol. Chem. 1995, 270, 7495–7500. [Google Scholar] [PubMed] [Green Version]
- Ornskov, D.; Nexo, E.; Sorensen, B.S. Insulin induces a transcriptional activation of epiregulin, HB-EGF and amphiregulin, by a PI3K-dependent mechanism: Identification of a specific insulin-responsive promoter element. Biochem. Biophys. Res. Commun. 2007, 354, 885–891. [Google Scholar] [CrossRef]
- Li, X.; Massa, P.E.; Hanidu, A.; Peet, G.W.; Aro, P.; Savitt, A.; Mische, S.; Li, J.; Marcu, K.B. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J. Biol. Chem. 2002, 277, 45129–45140. [Google Scholar] [CrossRef] [Green Version]
- Orso, F.; Penna, E.; Cimino, D.; Astanina, E.; Maione, F.; Valdembri, D.; Giraudo, E.; Serini, G.; Sismondi, P.; De Bortoli, M.; et al. AP-2alpha and AP-2gamma regulate tumor progression via specific genetic programs. FASEB J. 2008, 22, 2702–2714. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Komurasaki, T.; Toyoda, H.; Uchida, D.; Morimoto, S. Epiregulin binds to epidermal growth factor receptor and ErbB-4 and induces tyrosine phosphorylation of epidermal growth factor receptor, ErbB-2, ErbB-3 and ErbB-4. Oncogene 1997, 15, 2841–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.S.; Cheng, X.; Pawlowski, J.E.; Wallace, A.R.; Ferrer, P.; Molloy, C.J. Epiregulin is a potent vascular smooth muscle cell-derived mitogen induced by angiotensin II, endothelin-1, and thrombin. Proc. Natl. Acad. Sci. USA 1999, 96, 1633–1638. [Google Scholar] [CrossRef] [Green Version]
- Shirasawa, S.; Sugiyama, S.; Baba, I.; Inokuchi, J.; Sekine, S.; Ogino, K.; Kawamura, Y.; Dohi, T.; Fujimoto, M.; Sasazuki, T. Dermatitis due to epiregulin deficiency and a critical role of epiregulin in immune-related responses of keratinocyte and macrophage. Proc. Natl. Acad. Sci. USA 2004, 101, 13921–13926. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, S.; Nakabayashi, K.; Baba, I.; Sasazuki, T.; Shirasawa, S. Role of epiregulin in peptidoglycan-induced proinflammatory cytokine production by antigen presenting cells. Biochem. Biophys. Res. Commun. 2005, 337, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Rink, C.; Biswas, S.; Sen, C.K. Characterization of the acute temporal changes in excisional murine cutaneous wound inflammation by screening of the wound-edge transcriptome. Physiol. Genom. 2008, 34, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Komurasaki, T.; Ikeda, Y.; Yoshimoto, M.; Morimoto, S. Molecular cloning of mouse epiregulin, a novel epidermal growth factor-related protein, expressed in the early stage of development. FEBS Lett. 1995, 377, 403–407. [Google Scholar] [PubMed] [Green Version]
- Toyoda, H.; Komurasaki, T.; Uchida, D.; Morimoto, S. Distribution of mRNA for human epiregulin, a differentially expressed member of the epidermal growth factor family. Biochem. J. 1997, 326 Pt 1, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of Angiogenesis to Inflammation and Cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunaga, N.; Kaira, K.; Imai, H.; Shimizu, K.; Nakano, T.; Shames, D.S.; Girard, L.; Soh, J.; Sato, M.; Iwasaki, Y.; et al. Oncogenic KRAS-induced epiregulin overexpression contributes to aggressive phenotype and is a promising therapeutic target in non-small-cell lung cancer. Oncogene 2013, 32, 4034–4042. [Google Scholar] [CrossRef] [Green Version]
- Lindvall, C.; Hou, M.; Komurasaki, T.; Zheng, C.; Henriksson, M.; Sedivy, J.M.; Bjorkholm, M.; Teh, B.T.; Nordenskjold, M.; Xu, D. Molecular characterization of human telomerase reverse transcriptase-immortalized human fibroblasts by gene expression profiling: Activation of the epiregulin gene. Cancer Res. 2003, 63, 1743–1747. [Google Scholar]
- Vermeer, P.D.; Panko, L.; Karp, P.; Lee, J.H.; Zabner, J. Differentiation of human airway epithelia is dependent on erbB2. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L175–L180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, E.K.; Foley, J.S.; Cheng, J.; Patel, A.S.; Drazen, J.M.; Tschumperlin, D.J. Bronchial epithelial compression regulates epidermal growth factor receptor family ligand expression in an autocrine manner. Am. J. Respir. Cell Mol. Biol. 2005, 32, 373–380. [Google Scholar] [CrossRef]
- Weissferdt, A.; Lin, H.; Woods, D.; Tang, X.; Fujimoto, J.; Wistuba, I.I.; Moran, C.A. HER family receptor and ligand status in thymic carcinoma. Lung Cancer 2012, 77, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, A.; Shezen, E.; Raanan, C.; Slilat, Y.; Ben-Arie, A.; Prus, D.; Schreiber, L. Epiregulin as a marker for the initial steps of ovarian cancer development. Int. J. Oncol. 2011, 39, 1165–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jing, Y.; Ding, L.; Zhang, X.; Song, Y.; Chen, S.; Zhao, X.; Huang, X.; Pu, Y.; Wang, Z.; et al. Epiregulin reprograms cancer-associated fibroblasts and facilitates oral squamous cell carcinoma invasion via JAK2-STAT3 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 274. [Google Scholar] [CrossRef] [Green Version]
- Farooqui, M.; Bohrer, L.R.; Brady, N.J.; Chuntova, P.; Kemp, S.E.; Wardwell, C.T.; Nelson, A.C.; Schwertfeger, K.L. Epiregulin contributes to breast tumorigenesis through regulating matrix metalloproteinase 1 and promoting cell survival. Mol. Cancer 2015, 14, 138. [Google Scholar] [CrossRef] [Green Version]
- Abba, M.C.; Canzoneri, R.; Gurruchaga, A.; Lee, J.; Tatineni, P.; Kil, H.; Lacunza, E.; Aldaz, C.M. LINC00885 a Novel Oncogenic Long Non-Coding RNA Associated with Early Stage Breast Cancer Progression. Int. J. Mol. Sci. 2020, 21, 7407. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tsuang, B.J.; Chiang, C.J.; Ku, K.C.; Tseng, J.S.; Yang, T.Y.; Hsu, K.H.; Chen, K.C.; Yu, S.L.; Lee, W.C.; et al. The Relationship Between Air Pollution and Lung Cancer in Nonsmokers in Taiwan. J. Thorac. Oncol. 2019, 14, 784–792. [Google Scholar] [CrossRef]
- Patel, R.D.; Kim, D.J.; Peters, J.M.; Perdew, G.H. The aryl hydrocarbon receptor directly regulates expression of the potent mitogen epiregulin. Toxicol. Sci. 2006, 89, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, A.K.; Velmurugan, K.; Xiong, K.N.; Alexander, C.M.; Xiong, J.; Brooks, R. Epiregulin is required for lung tumor promotion in a murine two-stage carcinogenesis model. Mol. Carcinog. 2017, 56, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.L.; Chen, K.Y.; Lee, K.Y.; Feng, P.H.; Wu, S.M. Nicotinic-nAChR signaling mediates drug resistance in lung cancer. J. Cancer 2020, 11, 1125–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.Y.; Tseng, C.H.; Feng, P.H.; Sun, W.L.; Ho, S.C.; Lin, C.W.; Van Hiep, N.; Luo, C.S.; Tseng, Y.H.; Chen, T.T.; et al. 3-Nitrobenzanthrone promotes malignant transformation in human lung epithelial cells through the epiregulin-signaling pathway. Cell Biol. Toxicol. 2021. [Google Scholar] [CrossRef]
- Singh, B.; Bogatcheva, G.; Washington, M.K.; Coffey, R.J. Transformation of polarized epithelial cells by apical mistrafficking of epiregulin. Proc. Natl. Acad. Sci. USA 2013, 110, 8960–8965. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Pearsall, R.S.; Das, S.; Dey, S.K.; Godfrey, V.L.; Threadgill, D.W. Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol. Cell Biol. 2004, 24, 8907–8916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufert, C.; Becker, C.; Tureci, O.; Waldner, M.J.; Backert, I.; Floh, K.; Atreya, I.; Leppkes, M.; Jefremow, A.; Vieth, M.; et al. Tumor fibroblast-derived epiregulin promotes growth of colitis-associated neoplasms through ERK. J. Clin. Investig. 2013, 123, 1428–1443. [Google Scholar] [CrossRef] [Green Version]
- Gregorieff, A.; Liu, Y.; Inanlou, M.R.; Khomchuk, Y.; Wrana, J.L. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015, 526, 715–718. [Google Scholar] [CrossRef]
- Szymaniak, A.D.; Mi, R.; McCarthy, S.E.; Gower, A.C.; Reynolds, T.L.; Mingueneau, M.; Kukuruzinska, M.; Varelas, X. The Hippo pathway effector YAP is an essential regulator of ductal progenitor patterning in the mouse submandibular gland. Elife 2017, 6, e23499. [Google Scholar] [CrossRef]
- Riese, D.J., 2nd; Cullum, R.L. Epiregulin: Roles in normal physiology and cancer. Semin. Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Thogersen, V.B.; Sorensen, B.S.; Poulsen, S.S.; Orntoft, T.F.; Wolf, H.; Nexo, E. A subclass of HER1 ligands are prognostic markers for survival in bladder cancer patients. Cancer Res. 2001, 61, 6227–6233. [Google Scholar]
- Nicholson, B.E.; Frierson, H.F.; Conaway, M.R.; Seraj, J.M.; Harding, M.A.; Hampton, G.M.; Theodorescu, D. Profiling the evolution of human metastatic bladder cancer. Cancer Res. 2004, 64, 7813–7821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Colby, J.K.; Rengel, R.C.; Fischer, S.M.; Clinton, S.K.; Klein, R.D. Overexpression of cyclooxygenase-2 (COX-2) in the mouse urinary bladder induces the expression of immune- and cell proliferation-related genes. Mol. Carcinog. 2009, 48, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auf, G.; Jabouille, A.; Delugin, M.; Guerit, S.; Pineau, R.; North, S.; Platonova, N.; Maitre, M.; Favereaux, A.; Vajkoczy, P.; et al. High epiregulin expression in human U87 glioma cells relies on IRE1alpha and promotes autocrine growth through EGF receptor. BMC Cancer 2013, 13, 597. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Hinohara, K.; Wang, L.; Nishimura, T.; Urushido, M.; Yachi, K.; Tsuda, M.; Tanino, M.; Kimura, T.; Nishihara, H.; et al. Epiregulin enhances tumorigenicity by activating the ERK/MAPK pathway in glioblastoma. Neuro-Oncology 2014, 16, 960–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Li, Q.; Wu, G.; Huang, Y. Identification of Glioma Cancer Stem Cell Characteristics Based on Weighted Gene Prognosis Module Co-Expression Network Analysis of Transcriptome Data Stemness Indices. J. Mol. Neurosci. 2020, 70, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, S.; Wu, P.H.; Yakabe, T.; Giaccia, A.J.; Le, Q.T.; Aoyama, H.; Shimizu, S.; Shirato, H.; Onodera, Y.; Nam, J.M. Rab27b contributes to radioresistance and exerts a paracrine effect via epiregulin in glioblastoma. Neurooncol. Adv. 2020, 2, vdaa091. [Google Scholar] [CrossRef]
- Revillion, F.; Lhotellier, V.; Hornez, L.; Bonneterre, J.; Peyrat, J.P. ErbB/HER ligands in human breast cancer, and relationships with their receptors, the bio-pathological features and prognosis. Ann. Oncol. 2008, 19, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massague, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Addison, J.B.; Koontz, C.; Fugett, J.H.; Creighton, C.J.; Chen, D.; Farrugia, M.K.; Padon, R.R.; Voronkova, M.A.; McLaughlin, S.L.; Livengood, R.H.; et al. KAP1 promotes proliferation and metastatic progression of breast cancer cells. Cancer Res. 2015, 75, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Jin, Q.; Chen, C.; Liu, Y.; Ye, X.; Jiang, Y.; Ji, F.; Qian, H.; Gan, D.; Yue, S.; et al. The miR-186-3p/EREG axis orchestrates tamoxifen resistance and aerobic glycolysis in breast cancer cells. Oncogene 2019, 38, 5551–5565. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Munkacsy, G.; Gyorffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Li, X.D.; Miao, S.Y.; Wang, G.L.; Yang, L.; Shu, Y.Q.; Yin, Y.M. Amphiregulin and epiregulin expression in colorectal carcinoma and the correlation with clinicopathological characteristics. Onkologie 2010, 33, 353–358. [Google Scholar] [CrossRef]
- Watanabe, T.; Kobunai, T.; Yamamoto, Y.; Kanazawa, T.; Konishi, T.; Tanaka, T.; Matsuda, K.; Ishihara, S.; Nozawa, K.; Eshima, K.; et al. Prediction of liver metastasis after colorectal cancer using reverse transcription-polymerase chain reaction analysis of 10 genes. Eur. J. Cancer 2010, 46, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Stahler, A.; Heinemann, V.; Giessen-Jung, C.; Crispin, A.; Schalhorn, A.; Stintzing, S.; Fischer von Weikersthal, L.; Vehling-Kaiser, U.; Stauch, M.; Quietzsch, D.; et al. Influence of mRNA expression of epiregulin and amphiregulin on outcome of patients with metastatic colorectal cancer treated with 5-FU/LV plus irinotecan or irinotecan plus oxaliplatin as first-line treatment (FIRE 1-trial). Int. J. Cancer 2016, 138, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stintzing, S.; Ivanova, B.; Ricard, I.; Jung, A.; Kirchner, T.; Tannapfel, A.; Juette, H.; Hegewisch-Becker, S.; Arnold, D.; Reinacher-Schick, A. Amphiregulin (AREG) and Epiregulin (EREG) Gene Expression as Predictor for Overall Survival (OS) in Oxaliplatin/Fluoropyrimidine Plus Bevacizumab Treated mCRC Patients-Analysis of the Phase III AIO KRK-0207 Trial. Front. Oncol. 2018, 8, 474. [Google Scholar] [CrossRef]
- Chen, S.; Yue, T.; Huang, Z.; Zhu, J.; Bu, D.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. Inhibition of hydrogen sulfide synthesis reverses acquired resistance to 5-FU through miR-215-5p-EREG/TYMS axis in colon cancer cells. Cancer Lett. 2019, 466, 49–60. [Google Scholar] [CrossRef]
- Lin, C.Y.; Hsieh, P.L.; Chou, C.L.; Yang, C.C.; Lee, S.W.; Tian, Y.F.; Shiue, Y.L.; Li, W.S. High EREG Expression Is Predictive of Better Outcomes in Rectal Cancer Patients Receiving Neoadjuvant Concurrent Chemoradiotherapy. Oncology 2020, 98, 549–557. [Google Scholar] [CrossRef]
- Westendorp, F.; Karpus, O.N.; Koelink, P.J.; Vermeulen, J.L.M.; Meisner, S.; Koster, J.; Buller, N.; Wildenberg, M.E.; Muncan, V.; van den Brink, G.R. Epithelium-derived Indian Hedgehog restricts stromal expression of ErbB family members that drive colonic tumor cell proliferation. Oncogene 2021, 40, 1628–1643. [Google Scholar] [CrossRef]
- Shigeishi, H.; Higashikawa, K.; Hiraoka, M.; Fujimoto, S.; Mitani, Y.; Ohta, K.; Takechi, M.; Kamata, N. Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol. Rep. 2008, 19, 1557–1564. [Google Scholar]
- Kogashiwa, Y.; Inoue, H.; Kuba, K.; Araki, R.; Yasuda, M.; Nakahira, M.; Sugasawa, M. Prognostic role of epiregulin/amphiregulin expression in recurrent/metastatic head and neck cancer treated with cetuximab. Head Neck 2018, 40, 2424–2431. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Li, S.L.; Gan, Y.H.; Wang, C.Y.; Yu, G.Y. Epiregulin promotes migration and invasion of salivary adenoid cystic carcinoma cell line SACC-83 through activation of ERK and Akt. Oral Oncol. 2009, 45, 156–163. [Google Scholar] [CrossRef]
- Liu, S.; Ye, D.; Xu, D.; Liao, Y.; Zhang, L.; Liu, L.; Yu, W.; Wang, Y.; He, Y.; Hu, J.; et al. Autocrine epiregulin activates EGFR pathway for lung metastasis via EMT in salivary adenoid cystic carcinoma. Oncotarget 2016, 7, 25251–25263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; He, H.W.; Sun, H.X.; Ren, K.H.; Shao, R.G. Dual knockdown of N-ras and epiregulin synergistically suppressed the growth of human hepatoma cells. Biochem. Biophys. Res. Commun. 2009, 387, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Sunaga, N.; Kaira, K. Epiregulin as a therapeutic target in non-small-cell lung cancer. Lung Cancer 2015, 6, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Iwanaga, K.; Choi, K.C.; Wislez, M.; Raso, M.G.; Wei, W.; Wistuba, I.I.; Kurie, J.M. Intratumoral epiregulin is a marker of advanced disease in non-small cell lung cancer patients and confers invasive properties on EGFR-mutant cells. Cancer Prev. Res. 2008, 1, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, W.; Leng, S.; Padilla, M.T.; Saxton, B.; Hutt, J.; Tessema, M.; Kato, K.; Kim, K.C.; Belinsky, S.A.; et al. Muc1 knockout potentiates murine lung carcinogenesis involving an epiregulin-mediated EGFR activation feedback loop. Carcinogenesis 2017, 38, 604–614. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, L.; Ren, Y.; Dai, W.; Chen, T.; Luo, L.; Zeng, J.; Mi, K.; Lang, J.; Cao, B. Epiregulin confers EGFR-TKI resistance via EGFR/ErbB2 heterodimer in non-small cell lung cancer. Oncogene 2021, 40, 2596–2609. [Google Scholar] [CrossRef]
- Zhu, Z.; Kleeff, J.; Friess, H.; Wang, L.; Zimmermann, A.; Yarden, Y.; Buchler, M.W.; Korc, M. Epiregulin is Up-regulated in pancreatic cancer and stimulates pancreatic cancer cell growth. Biochem. Biophys. Res. Commun. 2000, 273, 1019–1024. [Google Scholar] [CrossRef]
- Torring, N.; Jorgensen, P.E.; Sorensen, B.S.; Nexo, E. Increased expression of heparin binding EGF (HB-EGF), amphiregulin, TGF alpha and epiregulin in androgen-independent prostate cancer cell lines. Anticancer Res. 2000, 20, 91–95. [Google Scholar]
- Yun, J.; Song, S.H.; Park, J.; Kim, H.P.; Yoon, Y.K.; Lee, K.H.; Han, S.W.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; et al. Gene silencing of EREG mediated by DNA methylation and histone modification in human gastric cancers. Lab. Investig. 2012, 92, 1033–1044. [Google Scholar] [CrossRef]
- Xia, Q.; Zhou, Y.; Yong, H.; Wang, X.; Zhao, W.; Ding, G.; Zhu, J.; Li, X.; Feng, Z.; Wang, B. Elevated epiregulin expression predicts poor prognosis in gastric cancer. Pathol. Res. Pract. 2019, 215, 873–879. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Sharma, S.V.; Settleman, J. ErbBs in lung cancer. Exp. Cell Res. 2009, 315, 557–571. [Google Scholar] [CrossRef]
- Jacobi, N.; Seeboeck, R.; Hofmann, E.; Eger, A. ErbB Family Signalling: A Paradigm for Oncogene Addiction and Personalized Oncology. Cancers 2017, 9, 33. [Google Scholar] [CrossRef] [Green Version]
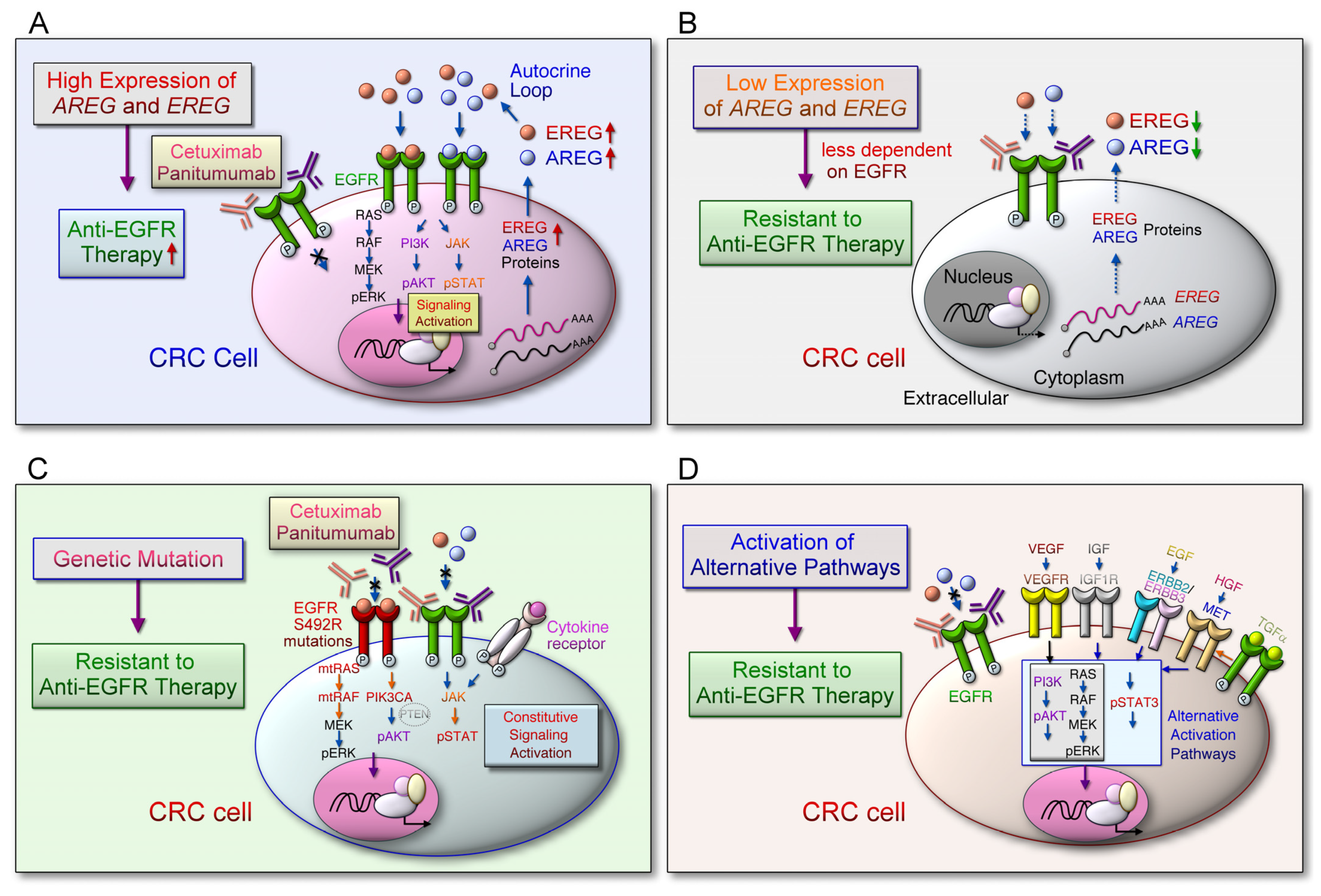
- Li, Q.H.; Wang, Y.Z.; Tu, J.; Liu, C.W.; Yuan, Y.J.; Lin, R.; He, W.L.; Cai, S.R.; He, Y.L.; Ye, J.N. Anti-EGFR therapy in metastatic colorectal cancer: Mechanisms and potential regimens of drug resistance. Gastroenterol. Rep. 2020, 8, 179–191. [Google Scholar] [CrossRef]
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.S.; Seong, Y.S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Shen, W.X.; Hu, X.F.; Zheng, D.Y.; Wu, X.Y.; Huang, Y.F.; Chen, J.Z.; Mao, C.; Tang, J.L. EGFR gene copy number as a predictive biomarker for the treatment of metastatic colorectal cancer with anti-EGFR monoclonal antibodies: A meta-analysis. J. Hematol. Oncol. 2012, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 2021, 12, 217–237. [Google Scholar] [CrossRef]
- Seligmann, J.F.; Elliott, F.; Richman, S.D.; Jacobs, B.; Hemmings, G.; Brown, S.; Barrett, J.H.; Tejpar, S.; Quirke, P.; Seymour, M.T. Combined Epiregulin and Amphiregulin Expression Levels as a Predictive Biomarker for Panitumumab Therapy Benefit or Lack of Benefit in Patients With RAS Wild-Type Advanced Colorectal Cancer. JAMA Oncol. 2016, 2, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.B.; Dutta, D.; Watson, D.; Maddala, T.; Munneke, B.M.; Shak, S.; Rowinsky, E.K.; Xu, L.A.; Harbison, C.T.; Clark, E.A.; et al. Tumour gene expression predicts response to cetuximab in patients with KRAS wild-type metastatic colorectal cancer. Br. J. Cancer 2011, 104, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K.; et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007, 25, 3230–3237. [Google Scholar] [CrossRef]
- Vale, C.L.; Tierney, J.F.; Fisher, D.; Adams, R.A.; Kaplan, R.; Maughan, T.S.; Parmar, M.K.; Meade, A.M. Does anti-EGFR therapy improve outcome in advanced colorectal cancer? A systematic review and meta-analysis. Cancer Treat. Rev. 2012, 38, 618–625. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Seymour, M.T.; Brown, S.R.; Middleton, G.; Maughan, T.; Richman, S.; Gwyther, S.; Lowe, C.; Seligmann, J.F.; Wadsley, J.; Maisey, N.; et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): A prospectively stratified randomised trial. Lancet Oncol. 2013, 14, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Bormann, F.; Stinzing, S.; Tierling, S.; Morkel, M.; Markelova, M.R.; Walter, J.; Weichert, W.; Rossner, F.; Kuhn, N.; Perner, J.; et al. Epigenetic regulation of Amphiregulin and Epiregulin in colorectal cancer. Int. J. Cancer 2019, 144, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Jing, C.; Jin, Y.H.; You, Z.; Qiong, Q.; Jun, Z. Prognostic value of amphiregulin and epiregulin mRNA expression in metastatic colorectal cancer patients. Oncotarget 2016, 7, 55890–55899. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, B.; De Roock, W.; Piessevaux, H.; Van Oirbeek, R.; Biesmans, B.; De Schutter, J.; Fieuws, S.; Vandesompele, J.; Peeters, M.; Van Laethem, J.L.; et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 2009, 27, 5068–5074. [Google Scholar] [CrossRef]
- Pentheroudakis, G.; Kotoula, V.; De Roock, W.; Kouvatseas, G.; Papakostas, P.; Makatsoris, T.; Papamichael, D.; Xanthakis, I.; Sgouros, J.; Televantou, D.; et al. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: Interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer 2013, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Jonker, D.J.; Karapetis, C.S.; Harbison, C.; O’Callaghan, C.J.; Tu, D.; Simes, R.J.; Malone, D.P.; Langer, C.; Tebbutt, N.; Price, T.J.; et al. Epiregulin gene expression as a biomarker of benefit from cetuximab in the treatment of advanced colorectal cancer. Br. J. Cancer 2014, 110, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seligmann, J.F.; Hatch, A.J.; Richman, S.D.; Elliott, F.; Jacobs, B.; Brown, S.; Hurwitz, H.; Barrett, J.H.; Quirke, P.; Nixon, A.B.; et al. Association of Tumor HER3 Messenger RNA Expression With Panitumumab Efficacy in Advanced Colorectal Cancer. JAMA Oncol. 2018, 4, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Brule, S.Y.; Jonker, D.J.; Karapetis, C.S.; O’Callaghan, C.J.; Moore, M.J.; Wong, R.; Tebbutt, N.C.; Underhill, C.; Yip, D.; Zalcberg, J.R.; et al. Location of colon cancer (right-sided versus left-sided) as a prognostic factor and a predictor of benefit from cetuximab in NCIC CO.17. Eur. J. Cancer 2015, 51, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; McGuffey, E.J.; Morris, J.S.; Manyam, G.; Baladandayuthapani, V.; Wei, W.; Morris, V.K.; Overman, M.J.; Maru, D.M.; Jiang, Z.Q.; et al. Association of CpG island methylator phenotype and EREG/AREG methylation and expression in colorectal cancer. Br. J. Cancer 2016, 114, 1352–1361. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Yang, Z.Y.; Hu, X.F.; Chen, Q.; Tang, J.L. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2012, 23, 1518–1525. [Google Scholar] [CrossRef]
- Esposito, C.; Rachiglio, A.M.; La Porta, M.L.; Sacco, A.; Roma, C.; Iannaccone, A.; Tatangelo, F.; Forgione, L.; Pasquale, R.; Barbaro, A.; et al. The S492R EGFR ectodomain mutation is never detected in KRAS wild-type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol. Ther. 2013, 14, 1143–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, F.; Lampis, A.; Orsenigo, M.; Di Bartolomeo, M.; Gevorgyan, A.; Losa, M.; Frattini, M.; Riva, C.; Andreola, S.; Bajetta, E.; et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann. Oncol. 2009, 20, 84–90. [Google Scholar] [CrossRef]
- Ma, X.T.; Wang, S.; Ye, Y.J.; Du, R.Y.; Cui, Z.R.; Somsouk, M. Constitutive activation of Stat3 signaling pathway in human colorectal carcinoma. World J. Gastroenterol. 2004, 10, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.M.; Fottner, C.; Liu, S.B.; Jung, M.C.; Engelhardt, D.; Baretton, G.B. Overexpression of the insulin-like growth factor I receptor in human colon carcinomas. Cancer 2002, 95, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Richman, S.D.; Southward, K.; Chambers, P.; Cross, D.; Barrett, J.; Hemmings, G.; Taylor, M.; Wood, H.; Hutchins, G.; Foster, J.M.; et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: Analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J. Pathol. 2016, 238, 562–570. [Google Scholar] [CrossRef]
- Bianco, R.; Rosa, R.; Damiano, V.; Daniele, G.; Gelardi, T.; Garofalo, S.; Tarallo, V.; De Falco, S.; Melisi, D.; Benelli, R.; et al. Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin. Cancer Res. 2008, 14, 5069–5080. [Google Scholar] [CrossRef] [Green Version]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.M.; Seligmann, J.F.; Elliott, F.; Shires, M.; Richman, S.D.; Brown, S.; Zhang, L.; Singh, S.; Pugh, J.; Xu, X.M.; et al. Artificial Intelligence-Assisted Amphiregulin and Epiregulin IHC Predicts Panitumumab Benefit in RAS Wild-Type Metastatic Colorectal Cancer. Clin. Cancer Res. 2021, 27, 3422–3431. [Google Scholar] [CrossRef]
- Job, S.; Reynies, A.; Heller, B.; Weiss, A.; Guerin, E.; Macabre, C.; Ledrappier, S.; Bour, C.; Wasylyk, C.; Etienne-Selloum, N.; et al. Preferential Response of Basal-Like Head and Neck Squamous Cell Carcinoma Cell Lines to EGFR-Targeted Therapy Depending on EREG-Driven Oncogenic Addiction. Cancers 2019, 11, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, M.; Anai, M.; Kodama, T.; Shibasaki, Y. Epiregulin-blocking antibody inhibits epiregulin-dependent EGFR signaling. Biochem. Biophys. Res. Commun. 2017, 489, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Yamada-Okabe, H.; Suzuki, M.; Natori, O.; Kato, A.; Matsubara, K.; Jau Chen, Y.; Yamazaki, M.; Funahashi, S.; Yoshida, K.; et al. LGR5-positive colon cancer stem cells interconvert with drug-resistant LGR5-negative cells and are capable of tumor reconstitution. Stem Cells 2012, 30, 2631–2644. [Google Scholar] [CrossRef]
- Ryuge, S.; Sato, Y.; Jiang, S.X.; Wang, G.; Kobayashi, M.; Nagashio, R.; Katono, K.; Iyoda, A.; Satoh, Y.; Masuda, N. The clinicopathological significance of Lgr5 expression in lung adenocarcinoma. Lung Cancer 2013, 82, 143–148. [Google Scholar] [CrossRef]
- Wu, Y.L.; Saijo, N.; Thongprasert, S.; Yang, J.C.; Han, B.; Margono, B.; Chewaskulyong, B.; Sunpaweravong, P.; Ohe, Y.; Ichinose, Y.; et al. Efficacy according to blind independent central review: Post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EGFR mutation-positive advanced NSCLC. Lung Cancer 2017, 104, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal. Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Regales, L.; Gong, Y.; Shen, R.; de Stanchina, E.; Vivanco, I.; Goel, A.; Koutcher, J.A.; Spassova, M.; Ouerfelli, O.; Mellinghoff, I.K.; et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J. Clin. Investig. 2009, 119, 3000–3010. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Sandmann, T.; Frierson, H. Jr.; Fu, L.; Fuentes, E.; Walter, K.; Okrah, K.; Rumpel, C.; Moskaluk, C.; Lu, S.; et al. Integrated genomic analysis of colorectal cancer progression reveals activation of EGFR through demethylation of the EREG promoter. Oncogene 2016, 35, 6403–6415. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.C.; Thiam, T.K.; Lu, Y.J.; Yeh, C.Y.; Tsai, W.S.; You, J.F.; Hung, H.Y.; Tsai, C.N.; Hsu, A.; Chen, H.C.; et al. Mutations of KRAS/NRAS/BRAF predict cetuximab resistance in metastatic colorectal cancer patients. Oncotarget 2016, 7, 22257–22270. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal. Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep. Med. 2021, 2, 100186. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]
- Lee, S.K.; Jeong, W.J.; Cho, Y.H.; Cha, P.H.; Yoon, J.S.; Ro, E.J.; Choi, S.; Oh, J.M.; Heo, Y.; Kim, H.; et al. beta-Catenin-RAS interaction serves as a molecular switch for RAS degradation via GSK3beta. EMBO Rep. 2018, 19, e46060. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Yar Saglam, A.S.; Alp, E.; Elmazoglu, Z.; Menevse, S. Treatment with cucurbitacin B alone and in combination with gefitinib induces cell cycle inhibition and apoptosis via EGFR and JAK/STAT pathway in human colorectal cancer cell lines. Hum. Exp. Toxicol. 2016, 35, 526–543. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Pisanti, S.; Napolitano, F.; Di Somma, S.; Martinelli, R.; Portella, G. Tumor-Associated Macrophage Status in Cancer Treatment. Cancers 2020, 12, 1987. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal. Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Jia, Y.; Li, X.; Jiang, T.; Zhao, S.; Zhao, C.; Zhang, L.; Liu, X.; Shi, J.; Qiao, M.; Luo, J.; et al. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int. J. Cancer 2019, 145, 1432–1444. [Google Scholar] [CrossRef]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wei, T.; Meng, H.; Luo, P.; Zhang, J. Role of the dynamic tumor microenvironment in controversies regarding immune checkpoint inhibitors for the treatment of non-small cell lung cancer (NSCLC) with EGFR mutations. Mol. Cancer 2019, 18, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhang, Y.; Wang, Y.; Ye, P.; Li, J.; Li, H.; Ding, Q.; Xia, J. Amphiregulin Confers Regulatory T Cell Suppressive Function and Tumor Invasion via the EGFR/GSK-3beta/Foxp3 Axis. J. Biol. Chem. 2016, 291, 21085–21095. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | EREG Expression | Co-Existing Genetic Background | Pathways | Tumorigenesis and Clinical Outcomes | Ref |
|---|---|---|---|---|---|
| Bladder | Up | AR, TGF-α, and HB-EGF | - | Short OS | [51] |
| Up | uPA, MMP14, and TIMP-2 | - | Lung Metastasis | [52] | |
| Up | COX2 | ERK | Immune/Stress Response and Cell Cycle/Proliferation | [53] | |
| Brain | Up | HB-EGF | EREG/EGFR autocrine loop, JNK, and under the control of IRE1α | Tumor cell growth, Migration | [54] |
| Up | AEBP1 | EGFR-ERK and abrogated by gefitinib | Tumor cell growth, Colony and Sphere formation, Invasion, Tumor formation, Short OS | [55] | |
| Up | 51 differentially expressed gene (DEG) such as BUB1 | Chromosome segregation and cell cycle functions in DEG | Higher EREG-mRNA stemness scores increased with glioma grade and had worse OS | [56] | |
| Up | Rab27b | Rab27b mediates paracrine EREG/EGFR by ionizingradiation | Tumor cell growth, Radioresistance | [57] | |
| Breast | Up | GRO1, MMP1/2, SPARC, IL13Rα2, VCAM1, ID1, and COX2 | Lung metastasis signature (LMS) | LMS extracellular proteins mediate breast cancer metastasis to lung | [59] |
| Up | COX2, MMP1, and MMP2 | - | Tumor growth, Angiogenesis, and Metastasis | [60] | |
| Up | BTC, TGFα, HB-EGF, and NRG2 | ERBB/HER ligands | Tumor aggressiveness | [58] | |
| Up | MMP1 | Partially regulated by fibroblast growth factor receptor | Tumor growth, Tumor formation of early stage breast cancer | [38] | |
| Up | Active SUMOlyated KAP1, COX2, MMP1/2, and CD44 | KAP1 regulates multiple KRAB-ZNF | Tumor growth and Metastasis | [61] | |
| Up | Keratin 14 (K14) | Dependent upon K14 expression | Distant Metastasis; Metastatic niche remodeling (Tnc, AdamTs1, Jag1) and Metastasis survival (AdamTs1, Birc5) | [62] | |
| Up | GLUT3, HK2, and PDK1 | miR-186-3p/EREG axis, EGFR/AKT/ERK | Tamoxifen resistance and Aerobic glycolysis (Warburg effect) | [63] | |
| Up | LINC00885 | TP53, EREG/EGFR/FOXM1 | Tumor cell proliferation, Migration, Invasion, and 3D growth | [39] | |
| Cervix | Up | - | TGF-β | Replicative immortality | [64] |
| Up | Epithelial cells mainly express | Frequent aggregated form occasionally in serous and mucinous tumors | The potential autocrine and paracrine effect on EGFR | [36] | |
| Colon and Rectum | Up | AREG | - | Tumor invasion and Distant metastases | [65] |
| Up | 10-gene signature including AREG, COX-2, and LCK | - | Prediction of Liver metastasis | [66] | |
| Up | Igl-V1, Ndg1, Lgals2, and Aldh1a3 | ERK | TAF-derived EREG mediated intestinal epithelial cell proliferation and tumor development | [47] | |
| Up | EGFR, HER2 | WNT | Distal carcinomas were more often chromosome instable and patients with metastases responded to anti-EGFR therapy | [67] | |
| Up | Aberrant EGFR or mutant RAS- and PIK3CA expression | The prognostic effect of high EREG expression | Longer OS and DFS in mCRC patients for 5-FU/LV plus irinotecan or irinotecan plus oxaliplatin (FIRE 1-trial) | [68] | |
| Down | Low AREG and EREG level are associated with mutant BRAF | - | Short OS in mCRC patients for Oxaliplatin/fluoropyrimidine plus bevacizumab treatment (Phase III AIO KRK-0207 trial) | [69] | |
| Up | Wild-type RAS and BRAF | - | AREG and EREG expression as predictor for longer OS in the AIO KRK-0207 trial study | [69] | |
| Up | TYMS | miR-215-5p-EREG/TYMS axis, Decreasing H2S synthesis reduced EREG and TYMS expression | Inhibiting H2S synthesis can reverse acquired resistance to 5-Fluorouracil (5-FU) | [70] | |
| Up | - | - | Better outcomes of DSS, LRFS, and MeFS for neoadjuvant concurrent chemoradiotherapy | [71] | |
| Up | EREG, BTC, and NRG1 in gp38+ fibroblasts | Epithelium-derived Indian Hedgehog (Ihh) restricts stromal expression of ERBB ligands | Colonic adenomagenesis in APC and Ihh deficiency mice, Tumor cell proliferation and increased Lgr5+ stem cells | [72] | |
| Head and Neck | Up | AREG | - | Longer PFS and OS in recurrent/ metastatic patients with Cetuximab and chemotherapy | [74] |
| Up | C-Myc | EREG-EGFR-C-Myc, EREG mediates constitutive activation of ERGR/ERK | Tumor cell growth, Tumor formation, Sensitivity to Erlotinib, Shorter OS | [11] | |
| Up | HER2-4 | - | OSCC Cell proliferation, Short OS | [73] | |
| Up | COX-2 | AKT/ERK | SACC Migration, Invasion | [75] | |
| Up | Snail/Slug stability | EREG/EGFR, AKT/ERK and STAT3 | SACC EMT, Blockade of lung metastasis by Erlotinib, Short OS | [76] | |
| Up | α-SMA | IL-6, JAK2/STAT3 | NF-CAF transformation, CAF EMT, OSCC Tumor growth, Shorter OS. | [37] | |
| Liver | Up | Knockdown of N-RAS | Dual knockdown reduced ERK, AKT, and Rb | Dual knockdown of N-RAS and EREG induced cell cycle arrest and inhibited cell growth | [77] |
| Up | - | Intestinal microbiota and TLR4 activation | Proliferation and antiapoptosis | [78] | |
| Lung | Up | EGFR-mutant NSCLC cells | EGFR-dependent, ERK and p38MPAK | Invasion, Proliferation, Antiapoptosis, Metastasis and Short OS | [80] |
| Up | KRAS mutation | Mutant KRAS constitutive activation | Anchorage-dependent and -independent cell growth, Antiapoptosis, Short OS and DFS, Pleural involvement, Lymphatic permeation or vascular invasion | [31,79] | |
| Up | Muc1 deficiency in fibroblasts and malignant cells | EGFR/AKT | Cell proliferation and survival, EREG production in lung cancer Muc1-KO model, Short OS in cancer patients | [81] | |
| Up | Intratumoral EREG action derived from macrophages | ERBB2/AKT | EGFR-TKI resistance | [82] | |
| Down | 87% (20/23) of SCLC cells | - | - | [31] | |
| Pancreas | Up | - | - | Tumor cell growth, Correlated with pancreatic ductal adenocarcinoma | [83] |
| Prostate | Up | TGFα, AREG, HB-EGF, HER1/2 | - | Expression in androgen- independent cell | [84] |
| Stomach | Down | Hypermethylated in gastric cancer cell (7/11, 64%) and primary tumors (4/13, 30%) | Aberrant DNA methylation and histone modification | EREG promoter methylation, 5-Aza-CdR and Cetuximab exerted a synergistic antiproliferation | [85] |
| Up | - | - | Tumor size, Lymph node metastases, Distant metastases, Short OS | [86] | |
| Thymus | Up | TGFα, HER1-3, Rare EGFR and HER2 gene amplification | - | ERBB ligand and receptor proteins commonly in primary squamous cell carcinomas | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.-L.; Feng, P.-H.; Lee, K.-Y.; Chen, K.-Y.; Sun, W.-L.; Van Hiep, N.; Luo, C.-S.; Wu, S.-M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312828
Cheng W-L, Feng P-H, Lee K-Y, Chen K-Y, Sun W-L, Van Hiep N, Luo C-S, Wu S-M. The Role of EREG/EGFR Pathway in Tumor Progression. International Journal of Molecular Sciences. 2021; 22(23):12828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312828
Chicago/Turabian StyleCheng, Wan-Li, Po-Hao Feng, Kang-Yun Lee, Kuan-Yuan Chen, Wei-Lun Sun, Nguyen Van Hiep, Ching-Shan Luo, and Sheng-Ming Wu. 2021. "The Role of EREG/EGFR Pathway in Tumor Progression" International Journal of Molecular Sciences 22, no. 23: 12828. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312828