
Evaluating the Effect of Lenvatinib on Sorafenib-Resistant Hepatocellular Carcinoma Cells

 , ,
, ,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
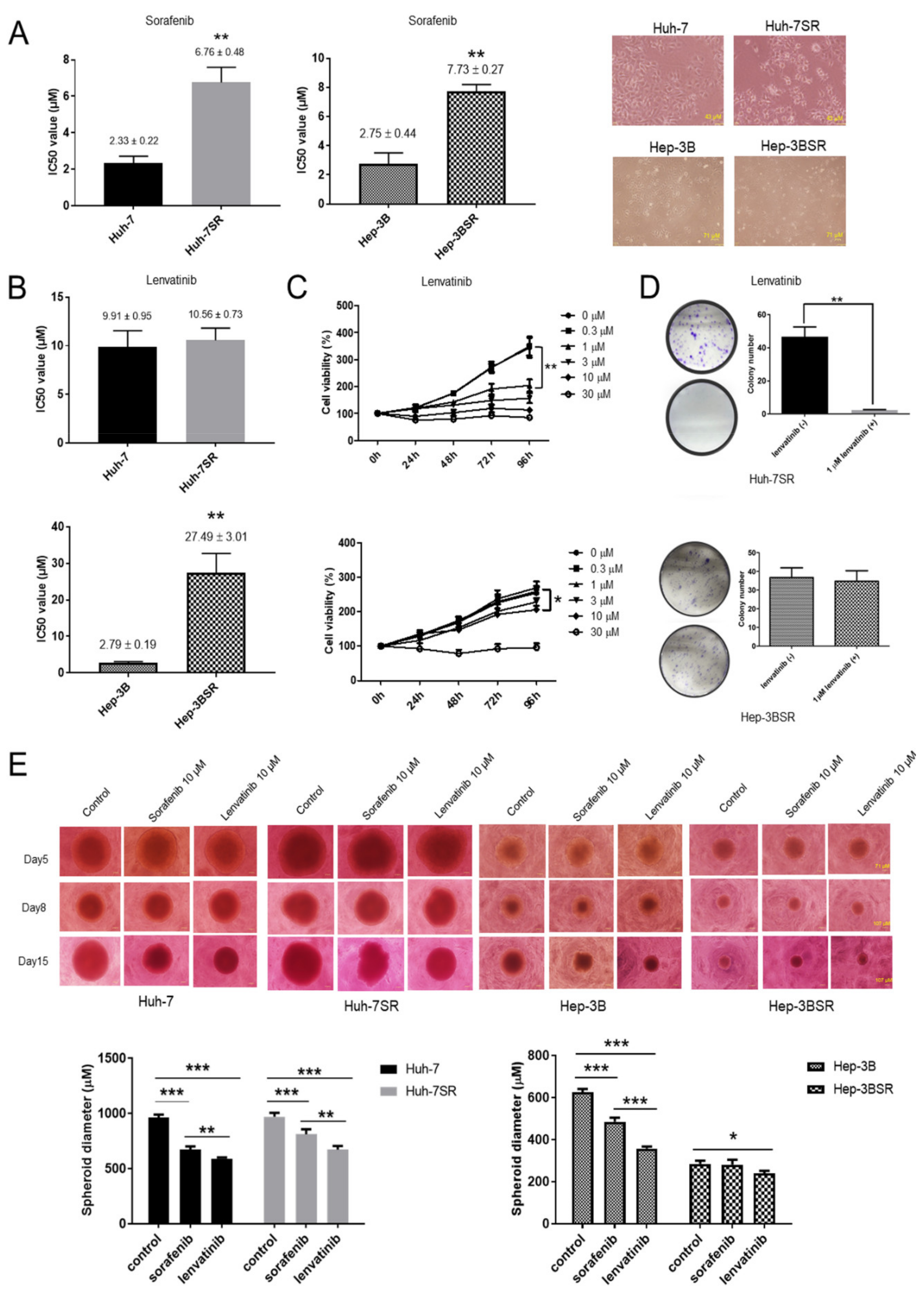
2.1. Lenvatinib Inhibits Sorafenib-Resistant HCC Cell Proliferation
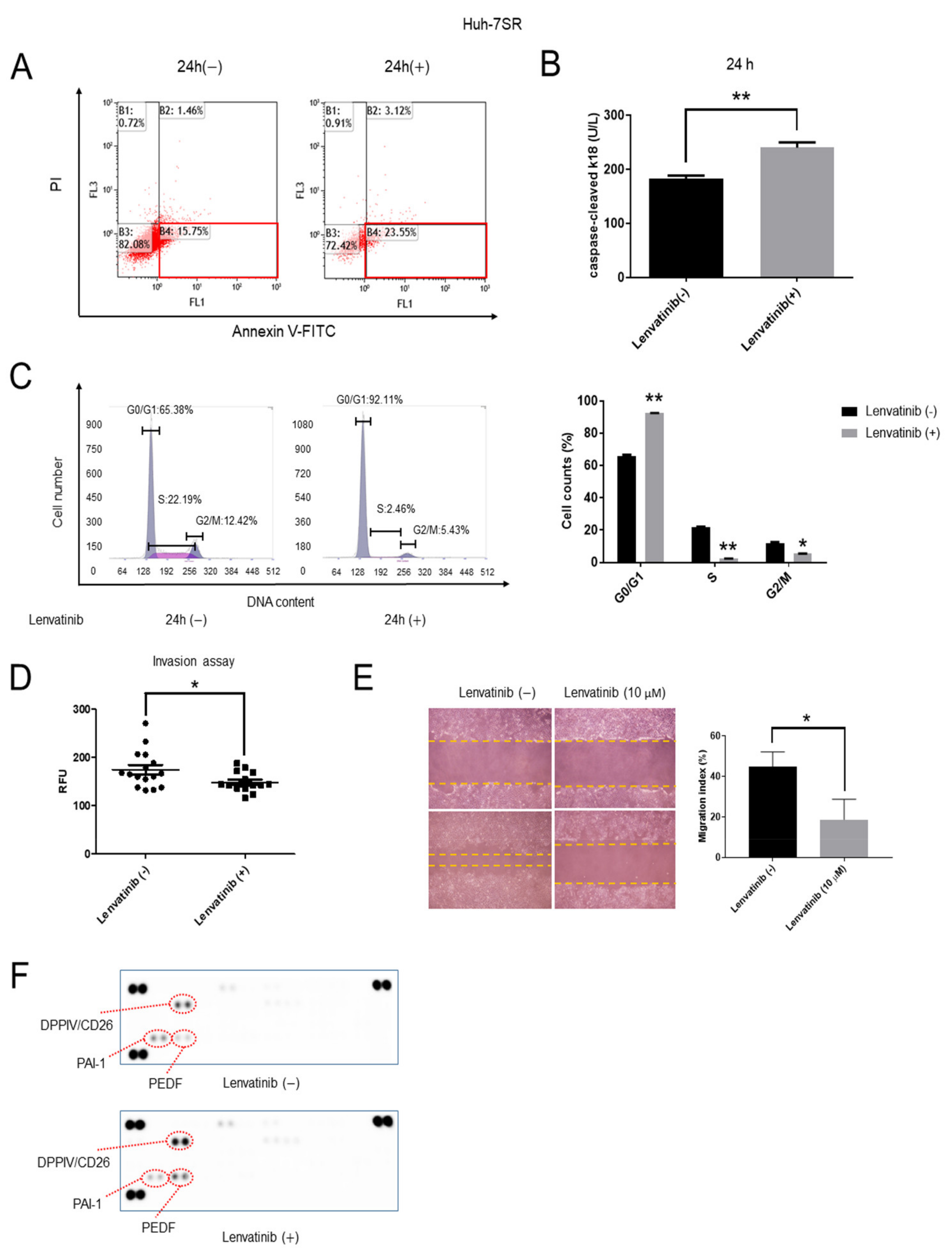
2.2. Lenvatinib Induces Apoptosis and Cell Cycle G1 Phase Arrest, Decreases Invasion and Migration Ability, and Regulates the Expression of Angiogenesis-Related Proteins in Huh-7SR Cells
2.3. Lenvatinib Induces Cell Cycle G1 Phase Arrest, Decreases Migration Ability, and Regulates the Expression of Angiogenesis-Related Proteins in Hep-3BSR Cells
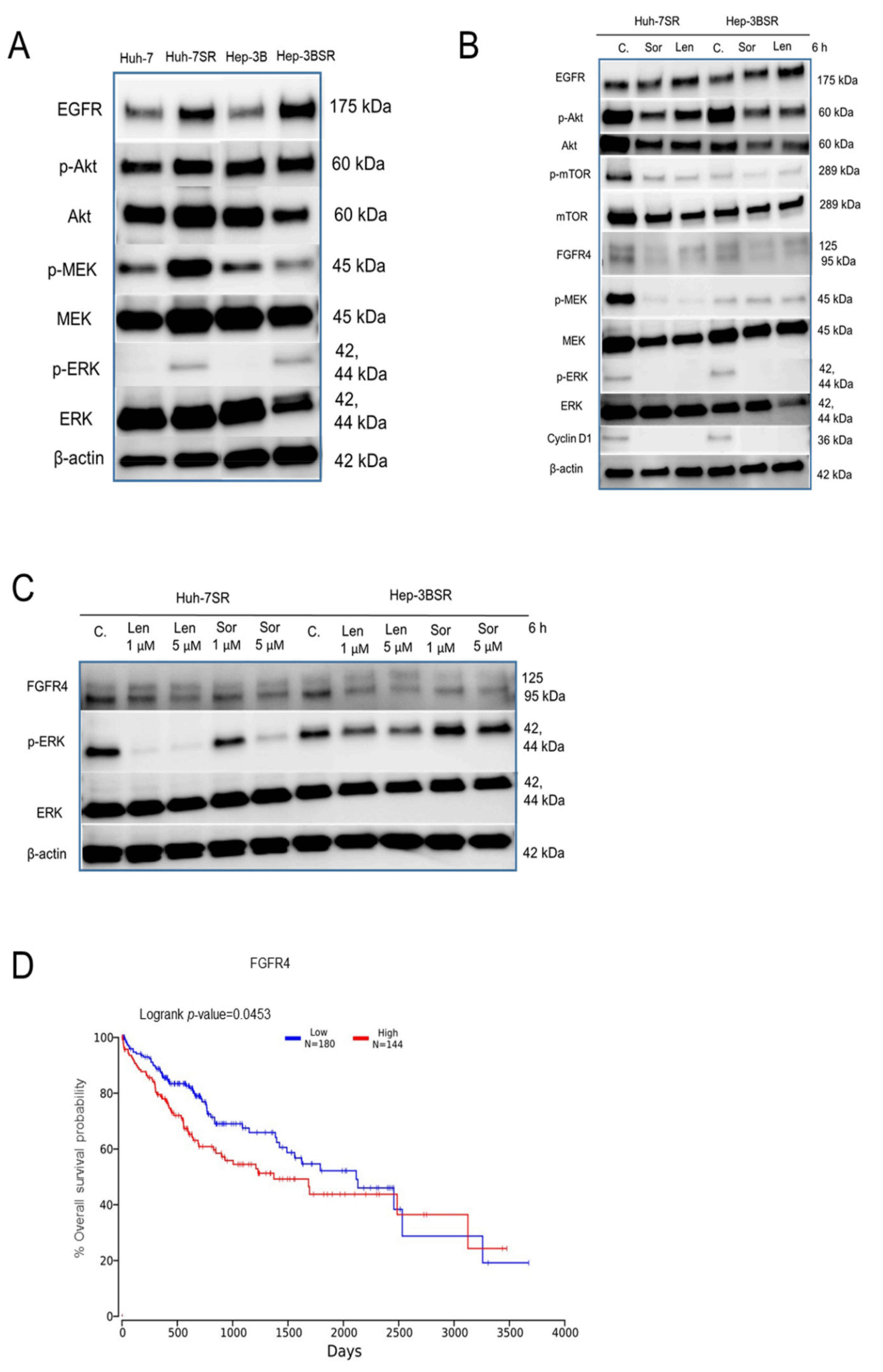
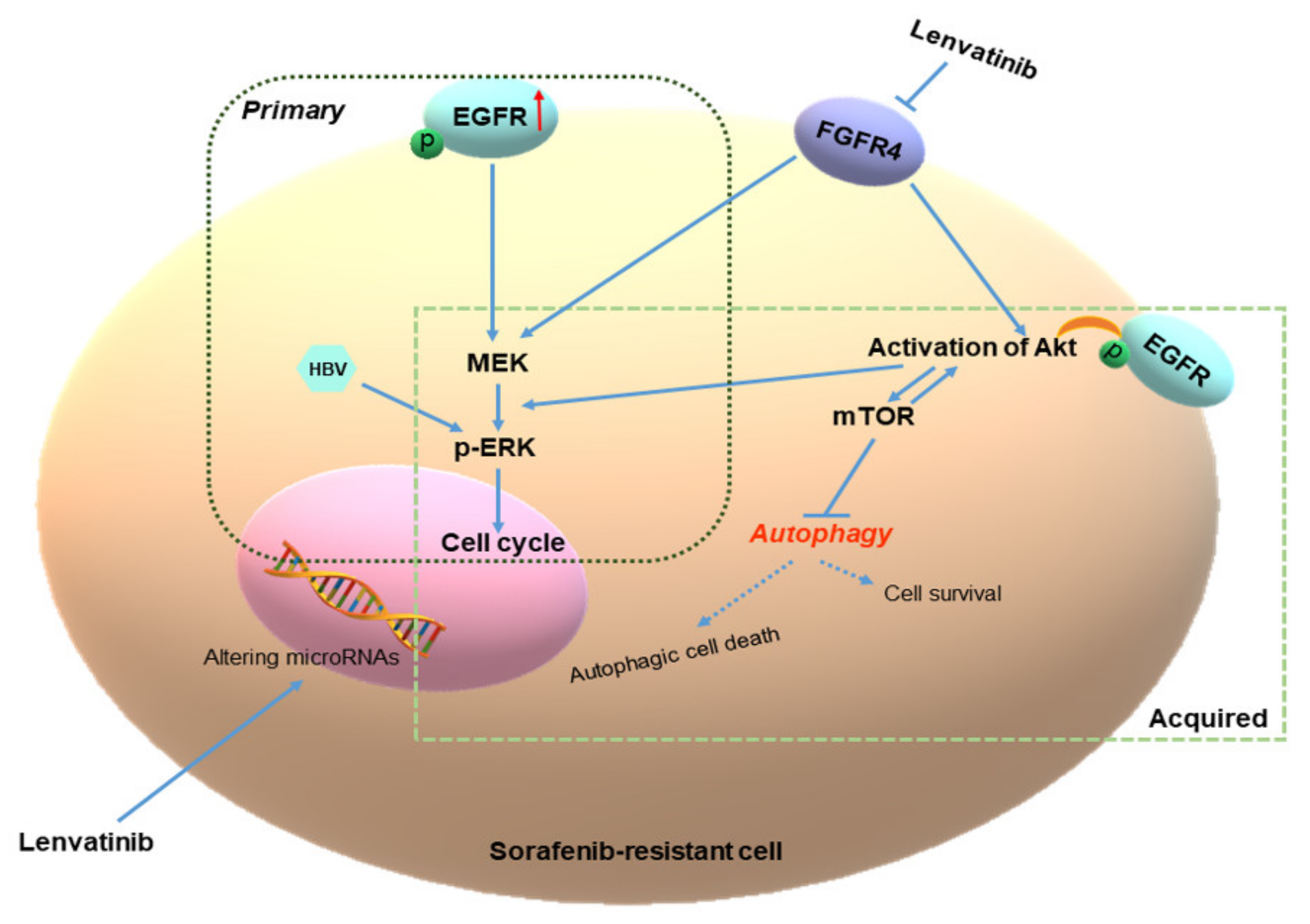
2.4. Lenvatinib Effects against Sorafenib-Resistance in HCC Cells May Be through the FGFR4-ERK Signaling Pathway
2.5. Different Autophagic Responsiveness between Huh-7SR and Hep-3BSR Cells
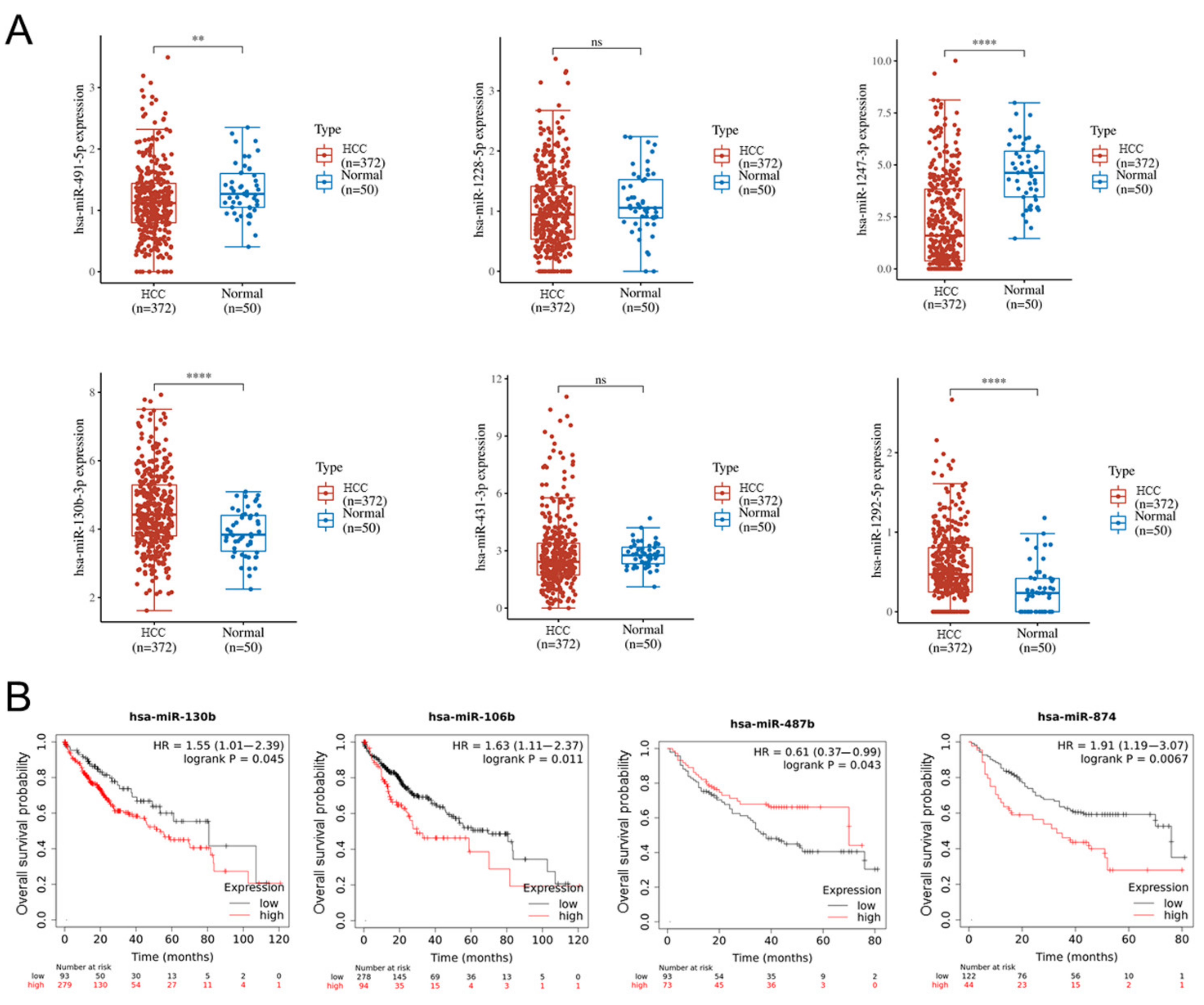
2.6. Lenvatinib Affects microRNA Expression in Sorafenib-Resistant Cells
2.7. Altered miRNA in HCC and Normal Tissues and the Relationship with Overall Survival of HCC Patients Based on TCGA Database Analysis
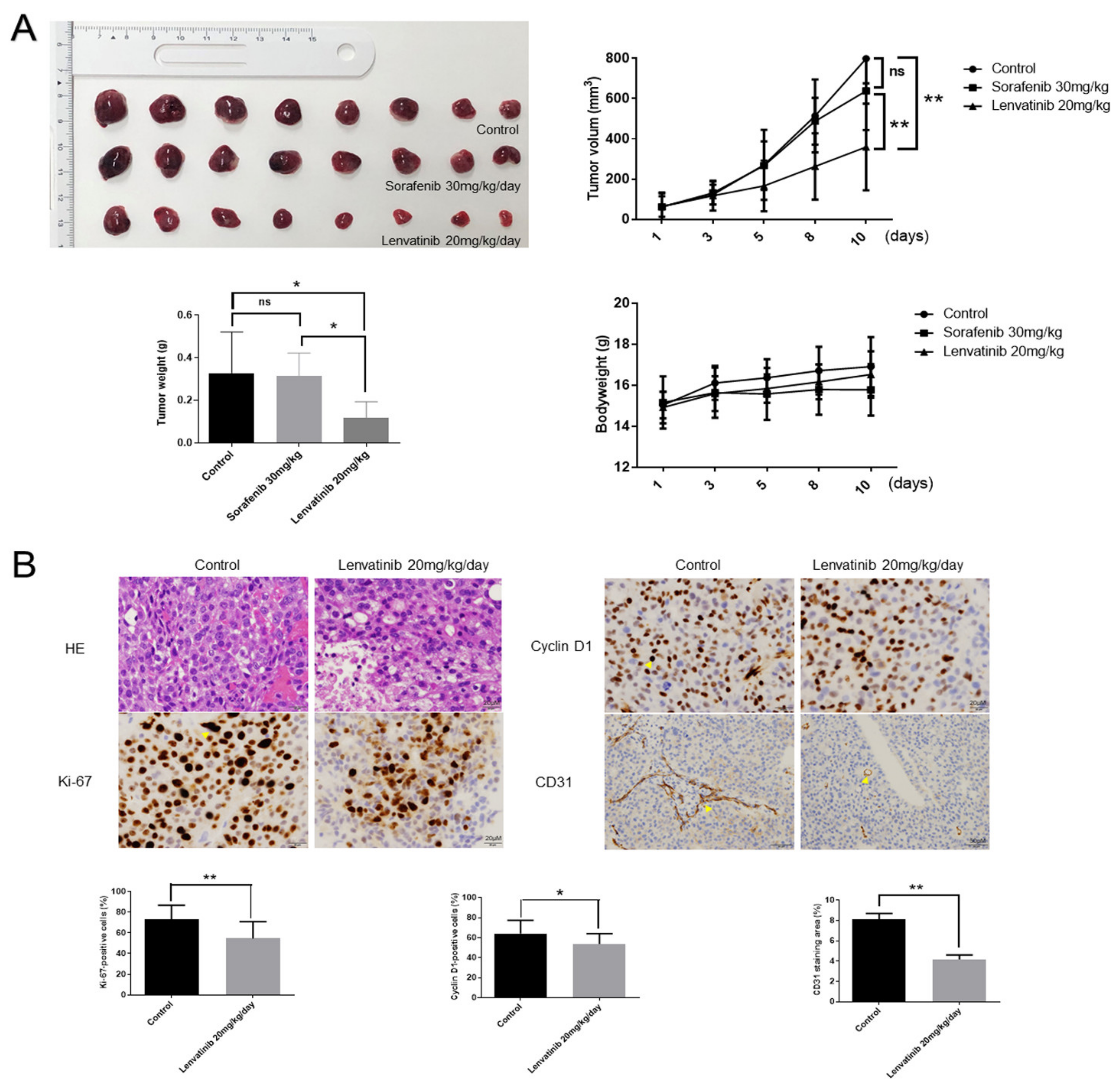
2.8. Lenvatinib Inhibits Huh-7 Sorafenib-Resistant Cell Proliferation In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Cell Culture
4.3. Cell Viability Assay
4.4. Three-Dimensional (3D) Tumor Spheroid Assay (3D Culture)
4.5. Flow Cytometry Analysis of Cell Cycle
4.6. Apoptosis Analysis
4.7. Apoptosis Analysis by Enzyme Linked Immunosorbent Assay (ELISA)
4.8. Antibody Arrays to Analyze Angiogenesis-Related Proteins
4.9. Invasion Assay
4.10. Wound Healing Assay
4.11. miRNA Microarray
4.12. Colony Formation Assay
4.13. Data Set Analysis
4.14. Western Blot
4.15. Xenograft Model Analysis
4.16. Immunohistochemistry
4.17. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lurje, I.; Czigany, Z.; Bednarsch, J.; Roderburg, C.; Isfort, P.; Neumann, U.P.; Lurje, G. Treatment Strategies for Hepatocellular Carcinoma-a Multidisciplinary Approach. Int. J. Mol. Sci. 2019, 20, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Chen, J.; Jin, R.; Zhao, J.; Liu, J.; Ying, H.; Yan, H.; Zhou, S.; Liang, Y.; Huang, D.; Liang, X.; et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015, 367, 1–11. [Google Scholar] [CrossRef]
- Ezzoukhry, Z.; Louandre, C.; Trécherel, E.; Godin, C.; Chauffert, B.; Dupont, S.; Diouf, M.; Barbare, J.C.; Mazière, J.C.; Galmiche, A. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int. J. Cancer 2012, 131, 2961–2969. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Gerolami, R.; Caparello, C.; et al. Outcomes of sequential treatment with sorafenib followed by regorafenib for HCC: Additional analyses from the phase III RESORCE trial. J. Hepatol. 2018, 69, 353–358. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Jindal, A.; Thadi, A.; Shailubhai, K. Hepatocellular Carcinoma: Etiology and Current and Future Drugs. J. Clin. Exp. Hepatol. 2019, 9, 221–232. [Google Scholar] [CrossRef]
- Hao, Z.; Wang, P. Lenvatinib in Management of Solid Tumors. Oncologist 2020, 25, e302–e310. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, A.; Kumada, T.; Kariyama, K.; Takaguchi, K.; Atsukawa, M.; Itobayashi, E.; Tsuji, K.; Tajiri, K.; Hirooka, M.; Shimada, N.; et al. Clinical features of lenvatinib for unresectable hepatocellular carcinoma in real-world conditions: Multicenter analysis. Cancer Med. 2019, 8, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Y.; Wang, C.C.; Liu, Y.W.; Li, W.F.; Chen, Y.H. Clinical impact of lenvatinib in patients with unresectable hepatocellular carcinoma who received sorafenib. PeerJ 2020, 8, e10382. [Google Scholar] [CrossRef]
- Tomonari, T.; Sato, Y.; Tanaka, H.; Tanaka, T.; Fujino, Y.; Mitsui, Y.; Hirao, A.; Taniguchi, T.; Okamoto, K.; Sogabe, M.; et al. Potential use of lenvatinib for patients with unresectable hepatocellular carcinoma including after treatment with sorafenib: Real-world evidence and in vitro assessment via protein phosphorylation array. Oncotarget 2020, 11, 2531–2542. [Google Scholar] [CrossRef]
- Enz, N.; Vliegen, G.; De Meester, I.; Jungraithmayr, W. CD26/DPP4-a potential biomarker and target for cancer therapy. Pharmacol. Ther. 2019, 198, 135–159. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Nakano, D.; Koga, H.; Torimura, T. Effects of a DPP4 Inhibitor on Progression of NASH-related HCC and the p62/Keap1/Nrf2-Pentose Phosphate Pathway in a Mouse Model. Liver Cancer 2019, 8, 359–372. [Google Scholar] [CrossRef]
- Yu, H.; Mei, X.P.; Su, P.F.; Jin, G.Z.; Zhou, H.K. A poor prognosis in human hepatocellular carcinoma is associated with low expression of DPP4. Braz. J. Med. Biol. Res. 2020, 53, e9114. [Google Scholar] [CrossRef]
- Li, C.; Huang, Z.; Zhu, L.; Yu, X.; Gao, T.; Feng, J.; Hong, H.; Yin, H.; Zhou, T.; Qi, W.; et al. The contrary intracellular and extracellular functions of PEDF in HCC development. Cell Death Dis. 2019, 10, 742. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Liang, Z.Y.; Zhou, W.X.; Zhou, L. Expression, clinicopathologic and prognostic significance of plasminogen activator inhibitor 1 in hepatocellular carcinoma. Cancer Biomark. 2020, 27, 285–293. [Google Scholar] [CrossRef]
- Baghy, K.; Tátrai, P.; Regős, E.; Kovalszky, I. Proteoglycans in liver cancer. World J. Gastroenterol. 2016, 22, 379–393. [Google Scholar] [CrossRef]
- Li, Y.; Turpin, C.P.; Wang, S. Role of thrombospondin 1 in liver diseases. Hepatol. Res. 2017, 47, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Poon, R.T.; Chung, K.K.; Cheung, S.T.; Lau, C.P.; Tong, S.W.; Leung, K.L.; Yu, W.C.; Tuszynski, G.P.; Fan, S.T. Clinical significance of thrombospondin 1 expression in hepatocellular carcinoma. Clin. Cancer Res. 2004, 10, 4150–4157. [Google Scholar] [CrossRef] [Green Version]
- Fousek, K.; Horn, L.A.; Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef]
- Matsuki, M.; Hoshi, T.; Yamamoto, Y.; Ikemori-Kawada, M.; Minoshima, Y.; Funahashi, Y.; Matsui, J. Lenvatinib inhibits angiogenesis and tumor fibroblast growth factor signaling pathways in human hepatocellular carcinoma models. Cancer Med. 2018, 7, 2641–2653. [Google Scholar] [CrossRef]
- Schmidt, B.; Wei, L.; DePeralta, D.K.; Hoshida, Y.; Tan, P.S.; Sun, X.; Sventek, J.P.; Lanuti, M.; Tanabe, K.K.; Fuchs, B.C. Molecular subclasses of hepatocellular carcinoma predict sensitivity to fibroblast growth factor receptor inhibition. Int. J. Cancer 2016, 138, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, H.O.; Huang, H.; Tsung, A. Autophagy: Dual Response in the Development of Hepatocellular Carcinoma. Cells. 2019, 8, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, B.; Zhou, H.; Liang, H.; Li, C. Regulation of ERK and AKT pathways by hepatitis B virus X protein via the Notch1 pathway in hepatocellular carcinoma. Int. J. Oncol. 2017, 51, 1449–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stjepanovic, N.; Capdevila, J. Multikinase inhibitors in the treatment of thyroid cancer: Specific role of lenvatinib. Biologics 2014, 8, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohyama, O.; Matsui, J.; Kodama, K.; Hata-Sugi, N.; Kimura, T.; Okamoto, K.; Minoshima, Y.; Iwata, M.; Funahashi, Y. Antitumor activity of lenvatinib (e7080): An angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J. Thyroid Res. 2014, 2014, 638747. [Google Scholar] [CrossRef]
- Kanzaki, H.; Chiba, T.; Ao, J.; Koroki, K.; Kanayama, K.; Maruta, S.; Maeda, T.; Kusakabe, Y.; Kobayashi, K.; Kanogawa, N.; et al. The impact of FGF19/FGFR4 signaling inhibition in antitumor activity of multi-kinase inhibitors in hepatocellular carcinoma. Sci. Rep. 2021, 11, 5303. [Google Scholar] [CrossRef]
- Yamauchi, M.; Ono, A.; Ishikawa, A.; Kodama, K.; Uchikawa, S.; Hatooka, H.; Zhang, P.; Teraoka, Y.; Morio, K.; Fujino, H.; et al. Tumor Fibroblast Growth Factor Receptor 4 Level Predicts the Efficacy of Lenvatinib in Patients With Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2020, 11, e00179. [Google Scholar] [CrossRef]
- Yi, C.; Chen, L.; Lin, Z.; Liu, L.; Shao, W.; Zhang, R.; Lin, J.; Zhang, J.; Zhu, W.; Jia, H.; et al. Lenvatinib Targets FGF Receptor 4 to Enhance Antitumor Immune Response of Anti-Programmed Cell Death-1 in HCC. Hepatology 2021, 74, 2544–2560. [Google Scholar] [CrossRef]
- Shigesawa, T.; Maehara, O.; Suda, G.; Natsuizaka, M.; Kimura, M.; Shimazaki, T.; Yamamoto, K.; Yamada, R.; Kitagataya, T.; Nakamura, A.; et al. Lenvatinib suppresses cancer stem-like cells in HCC by inhibiting FGFR1-3 signaling, but not FGFR4 signaling. Carcinogenesis 2021, 42, 58–69. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, M.A.; Chapresto-Garzón, R.; Cadenas, M.; Navarro-Villarán, E.; Negrete, M.; Gómez-Bravo, M.A.; Victor, V.M.; Padillo, F.J.; Muntané, J. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis. 2020, 11, 339. [Google Scholar] [CrossRef]
- Jin, H.; Shi, Y.; Lv, Y.; Yuan, S.; Ramirez, C.F.A.; Lieftink, C.; Wang, L.; Wang, S.; Wang, C.; Dias, M.H.; et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 2021, 595, 730–734. [Google Scholar] [CrossRef]
- Sun, T.; Liu, H.; Ming, L. Multiple Roles of Autophagy in the Sorafenib Resistance of Hepatocellular Carcinoma. Cell Physiol. Biochem. 2017, 44, 716–727. [Google Scholar] [CrossRef]
- Fischer, T.D.; Wang, J.H.; Vlada, A.; Kim, J.S.; Behrns, K.E. Role of autophagy in differential sensitivity of hepatocarcinoma cells to sorafenib. World J. Hepatol. 2014, 6, 752–758. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Chen, H.L.; Liu, C.Y.; Lin, C.S.; Cheng, A.L.; Chen, P.J.; Chen, K.F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Chen, X.; Li, P.; Wang, K. The Underlying Mechanisms of Noncoding RNAs in the Chemoresistance of Hepatocellular Carcinoma. Mol. Ther. Nucleic Acids 2020, 21, 13–27. [Google Scholar] [CrossRef]
- Oura, K.; Morishita, A.; Masaki, T. Molecular and Functional Roles of MicroRNAs in the Progression of Hepatocellular Carcinoma—A Review. Int. J. Mol. Sci. 2020, 21, 8362. [Google Scholar] [CrossRef]
- Gramantieri, L.; Pollutri, D.; Gagliardi, M.; Giovannini, C.; Quarta, S.; Ferracin, M.; Casadei-Gardini, A.; Callegari, E.; De Carolis, S.; Marinelli, S.; et al. MiR-30e-3p Influences Tumor Phenotype through MDM2/TP53 Axis and Predicts Sorafenib Resistance in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 1720–1734. [Google Scholar] [CrossRef]
- Xu, W.P.; Liu, J.P.; Feng, J.F.; Zhu, C.P.; Yang, Y.; Zhou, W.P.; Ding, J.; Huang, C.K.; Cui, Y.L.; Ding, C.H.; et al. miR-541 potentiates the response of human hepatocellular carcinoma to sorafenib treatment by inhibiting autophagy. Gut 2020, 69, 1309–1321. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, C.; Yang, Z.; Liu, W.; Yuan, Y.; Li, K.; Zhang, Y.; Wang, Y.; Shi, Y.; Qiu, Y.; et al. Dysregulated Sp1/miR-130b-3p/HOXA5 axis contributes to tumor angiogenesis and progression of hepatocellular carcinoma. Theranostics 2020, 10, 5209–5224. [Google Scholar] [CrossRef]
- Xu, Q.; Dou, C.; Liu, X.; Yang, L.; Ni, C.; Wang, J.; Guo, Y.; Yang, W.; Tong, X.; Huang, D. Oviductus ranae protein hydrolysate (ORPH) inhibits the growth, metastasis and glycolysis of HCC by targeting miR-491-5p/PKM2 axis. Biomed. Pharmacother. 2018, 107, 1692–1704. [Google Scholar] [CrossRef]
- Fa, X.; Song, P.; Fu, Y.; Deng, Y.; Liu, K. Long non-coding RNA VPS9D1-AS1 facilitates cell proliferation, migration and stemness in hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 131. [Google Scholar] [CrossRef]
- Jefremow, A.; Wiesmueller, M.; Rouse, R.A.; Dietrich, P.; Kremer, A.E.; Waldner, M.J.; Neurath, M.F.; Siebler, J. Beyond the border: The use of lenvatinib in advanced hepatocellular carcinoma after different treatment lines: A retrospective analysis. J. Physiol. Pharmacol. 2020, 71. [Google Scholar] [CrossRef]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chong, C.C.; Chen, G.G.; Lai, P.B. A Seven-microRNA Expression Signature Predicts Survival in Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0128628. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Cai, Z.; Ma, N.; Xie, W.; Gao, C.; Huang, M.; Bai, Y.; Ni, Y.; Tang, Y. A Novel Ten-Gene Signature Predicting Prognosis in Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2020, 8, 629. [Google Scholar] [CrossRef]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef] [Green Version]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | p-Value | Fold Change (Treated/Untreated) | FDR | Chromosomal Location |
|---|---|---|---|---|
| Upregulated | ||||
| has-miR-718 | 0.000823533 | 2.93 | 0.009177565 | Xq28 |
| hsa-miR-4787-3p | 0.000378539 | 2.82 | 0.005427912 | 3p21.2 |
| hsa-miR-6816-5p | 1.73835 × 10−5 | 2.69 | 0.000977428 | 22q11.21 |
| hsa-miR-6790-3p | 0.000369116 | 2.64 | 0.005427912 | 19p13.3 |
| hsa-miR-1234-3p | 2.9809 × 10−5 | 2.59 | 0.001292429 | 8q24.3 |
| hsa-miR-8063 | 0.000246669 | 2.56 | 0.004141378 | 15q14 |
| hsa-miR-6768-5p | 2.43923 × 10−6 | 2.48 | 0.000450491 | 16p13.3 |
| hsa-miR-6501-3p | 1.236 × 10−5 | 2.47 | 0.000867423 | 21q22.11 |
| hsa-miR-10394-3p | 0.00019664 | 2.40 | 0.003800681 | 19q13.43 |
| hsa-miR-6850-5p | 6.5404 × 10−5 | 2.36 | 0.002074481 | 8q24.3 |
| hsa-miR-12120 | 5.15411 × 10-5 | 2.35 | 0.001850097 | Yq11.221 |
| hsa-miR-4465 | 1.65012 × 10−5 | 2.35 | 0.000971998 | 6q24.1 |
| hsa-miR-491-5p | 1.25706 × 10−6 | 2.22 | 0.000388745 | 9p21.3 |
| hsa-miR-371b-5p | 0.000874032 | 2.20 | 0.009484016 | 19q13.42 |
| hsa-miR-3652 | 1.24497 × 10−5 | 2.15 | 0.000867423 | 12q23.3 |
| hsa-miR-921 | 0.000571511 | 2.04 | 0.007188459 | 1q24.1 |
| hsa-miR-1237-5p | 2.91345 × 10−6 | 2.03 | 0.000450491 | 11q13.1 |
| hsa-miR-1469 | 0.000239265 | 1.95 | 0.004141378 | 15q26.2 |
| hsa-miR-1181 | 0.000576271 | 1.92 | 0.007188459 | 19p13.2 |
| hsa-miR-6869-5p | 7.72727 × 10−5 | 1.88 | 0.00227243 | 20p13 |
| hsa-miR-3665 | 5.65874 × 10−7 | 1.85 | 0.000349993 | 13q22.3 |
| hsa-miR-1290 | 0.000146648 | 1.85 | 0.003194727 | 1p36.13 |
| hsa-miR-1273c | 3.34339 × 10−5 | 1.84 | 0.001292429 | 6q25.2 |
| hsa-miR-4535 | 0.000456305 | 1.81 | 0.00613532 | 22q13.32 |
| hsa-miR-10396a-5p | 6.04782 × 10−5 | 1.791 | 0.001968724 | 21p11.2 |
| hsa-miR-3940-5p | 2.45816 × 10-5 | 1.78 | 0.001216297 | 19p13.3 |
| hsa-miR-6808-5p | 0.000904459 | 1.76 | 0.009562526 | 1p36.33 |
| hsa-miR-1228-5p | 0.000103027 | 1.76 | 0.002591924 | 12q13.3 |
| hsa-miR-6774-5p | 6.90461 × 10−6 | 1.74 | 0.000731907 | 16q24.1 |
| hsa-miR-3180-3p | 0.000507427 | 1.74 | 0.006677529 | 16p13.11 |
| hsa-miR-3178 | 2.33963 × 10−5 | 1.73 | 0.001205886 | 16p13.3 |
| hsa-miR-3126-5p | 7.5417 × 10−5 | 1.69 | 0.00227243 | 2p13.3 |
| hsa-miR-4687-5p | 3.28134 × 10−5 | 1.67 | 0.001292429 | 11p15.4 |
| hsa-miR-663a | 8.50531 × 10−5 | 1.66 | 0.002355633 | 20p11.1 |
| hsa-miR-3158-5p | 6.85365 × 10−6 | 1.63 | 0.000731907 | 10q24.32 |
| hsa-miR-762 | 0.000107378 | 1.62 | 0.00260445 | 16p11.2 |
| hsa-miR-6715b-5p | 0.00066953 | 1.62 | 0.007813291 | 10q25.2 |
| hsa-miR-1247-3p | 1.33234 × 10−5 | 1.60 | 0.000867423 | 14q32.31 |
| hsa-miR-4476 | 4.84072 × 10−5 | 1.58 | 0.001814535 | 9p13.2 |
| hsa-miR-4749-5p | 8.89695 × 10−5 | 1.56 | 0.002355633 | 19q13.33 |
| hsa-miR-575 | 0.00018907 | 1.56 | 0.003712369 | 4q21.22 |
| hsa-miR-6724-5p | 8.70242 × 10−5 | 1.55 | 0.002355633 | 21p11.2 |
| hsa-miR-12114 | 0.000812774 | 1.51 | 0.009140012 | 22q13.33 |
| Downregulated | ||||
| hsa-miR-7114-5p | 0.000857372 | 0.65 | 0.009469368 | 9q34.3 |
| hsa-miR-197-5p | 0.000179571 | 0.65 | 0.003697546 | 1p13.3 |
| hsa-miR-3907 | 1.10865 × 10−5 | 0.65 | 0.000867423 | 7q36.1 |
| hsa-miR-1972 | 0.000596743 | 0.63 | 0.00723697 | 16p13.11 |
| hsa-miR-6735-5p | 9.42138 × 10−5 | 0.63 | 0.002427969 | 1p34.2 |
| hsa-miR-130b-3p | 7.89931E-05 | 0.63 | 0.00227243 | 22q11.21 |
| hsa-miR-487b-3p | 2.90019 × 10−5 | 0.63 | 0.001292429 | 14q32.31 |
| hsa-miR-6872-3p | 0.000677028 | 0.60 | 0.007826946 | 3p21.31 |
| hsa-miR-10524-5p | 4.7563 × 10−7 | 0.60 | 0.000349993 | 6q14.1 |
| hsa-miR-4260 | 0.000425744 | 0.60 | 0.00587887 | 1q32.2 |
| hsa-miR-3177-3p | 8.95026 × 10−5 | 0.60 | 0.002355633 | 16p13.3 |
| hsa-miR-874-5p | 0.000111442 | 0.59 | 0.002651026 | 5q31.2 |
| hsa-miR-4287 | 0.000214498 | 0.56 | 0.003919721 | 8p21.1 |
| hsa-miR-4448 | 1.41712 × 10−5 | 0.56 | 0.000876488 | 3q27.1 |
| hsa-miR-7843-5p | 5.23471 × 10−5 | 0.54 | 0.001850097 | 14q24.2 |
| hsa-miR-3189-5p | 0.000538484 | 0.54 | 0.00693859 | 19p13.11 |
| hsa-miR-106b-3p | 3.19133 × 10−5 | 0.53 | 0.001292429 | 7q22.1 |
| hsa-miR-4521 | 7.10015 × 10−6 | 0.47 | 0.000731907 | 17p13.1 |
| hsa-miR-431-3p | 0.00037795 | 0.46 | 0.005427912 | 14q32.2 |
| hsa-miR-4451 | 0.000147211 | 0.40 | 0.003194727 | 4q21.23 |
| hsa-miR-4632-5p | 9.73972 × 10−7 | 0.39 | 0.000388745 | 1p36.22 |
| hsa-miR-6880-5p | 2.78965 × 10−6 | 0.31 | 0.000450491 | 12q24.31 |
| hsa-miR-1292-5p | 0.000166801 | 0.29 | 0.003557465 | 20p13 |
| Author | Total Patients | Treatment Line | Complete Remission | Partial Remission | Stable Disease | Mixed Response | Progression | |
|---|---|---|---|---|---|---|---|---|
| Jefremow A et al. [48] | 7 | Second line | 2 | 0 | 1 | 1 | 0 | 0 |
| Third line | 3 | 0 | 2 | 0 | 1 | 0 | ||
| Fourth line | 2 | 0 | 1 | 1 | 0 | 0 | ||
| Chen YY et al. [15] | 40 | Second line | 20 | 1 | 5 | 7 | 0 | 7 |
| Third line | 10 | 0 | 4 | 4 | 0 | 2 | ||
| Fourth line | 10 | 0 | 1 | 5 | 0 | 4 | ||
| Tomonari T et al. [16] | 19 | Second line | 9 | 1 | 2 | 5 | 0 | 1 |
| Third line | 10 | 0 | 2 | 8 | 0 | 0 | ||
| 66 | 2 | 18 | 31 | 1 | 14 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, T.; Iwama, H.; Fujita, K.; Kobara, H.; Nishiyama, N.; Fujihara, S.; Goda, Y.; Yoneyama, H.; Morishita, A.; Tani, J.; et al. Evaluating the Effect of Lenvatinib on Sorafenib-Resistant Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 13071. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313071
Shi T, Iwama H, Fujita K, Kobara H, Nishiyama N, Fujihara S, Goda Y, Yoneyama H, Morishita A, Tani J, et al. Evaluating the Effect of Lenvatinib on Sorafenib-Resistant Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2021; 22(23):13071. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313071
Chicago/Turabian StyleShi, Tingting, Hisakazu Iwama, Koji Fujita, Hideki Kobara, Noriko Nishiyama, Shintaro Fujihara, Yasuhiro Goda, Hirohito Yoneyama, Asahiro Morishita, Joji Tani, and et al. 2021. "Evaluating the Effect of Lenvatinib on Sorafenib-Resistant Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 22, no. 23: 13071. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313071