
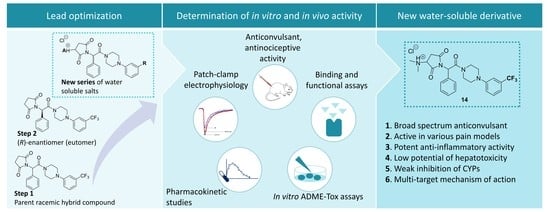
Identification of New Compounds with Anticonvulsant and Antinociceptive Properties in a Group of 3-substituted (2,5-dioxo-pyrrolidin-1-yl)(phenyl)-Acetamides

 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
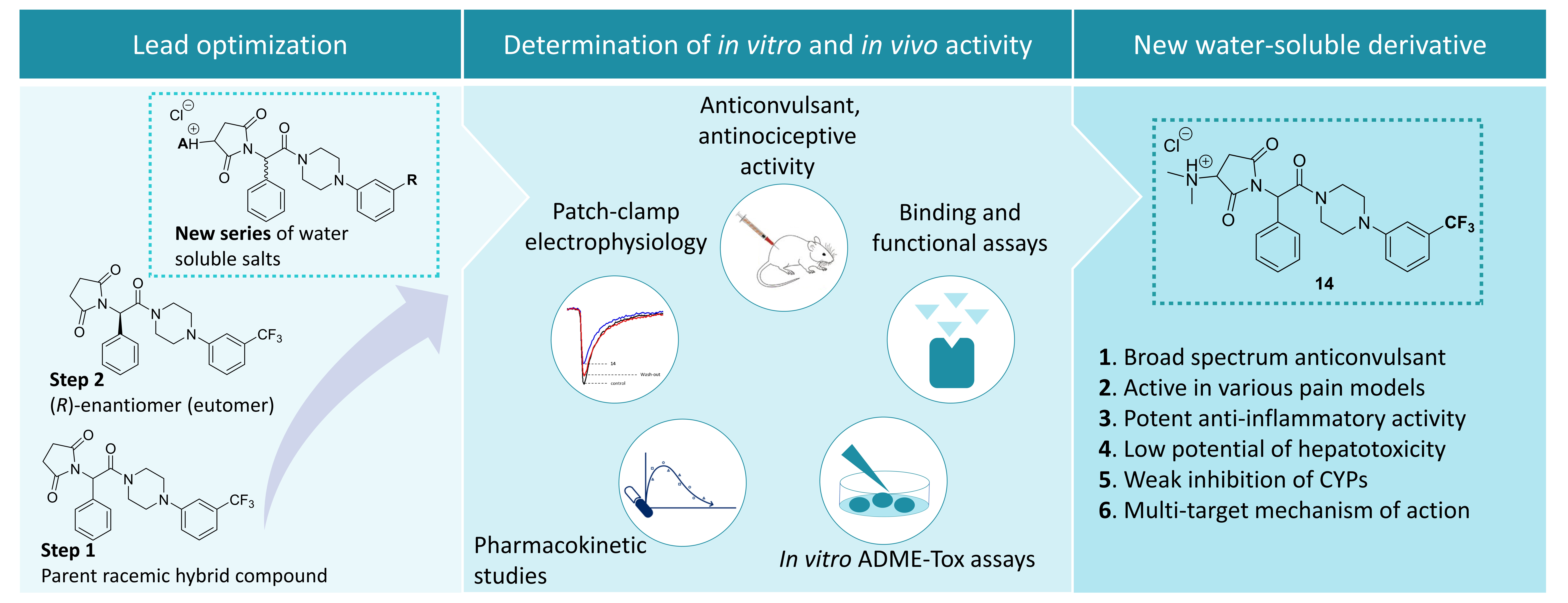
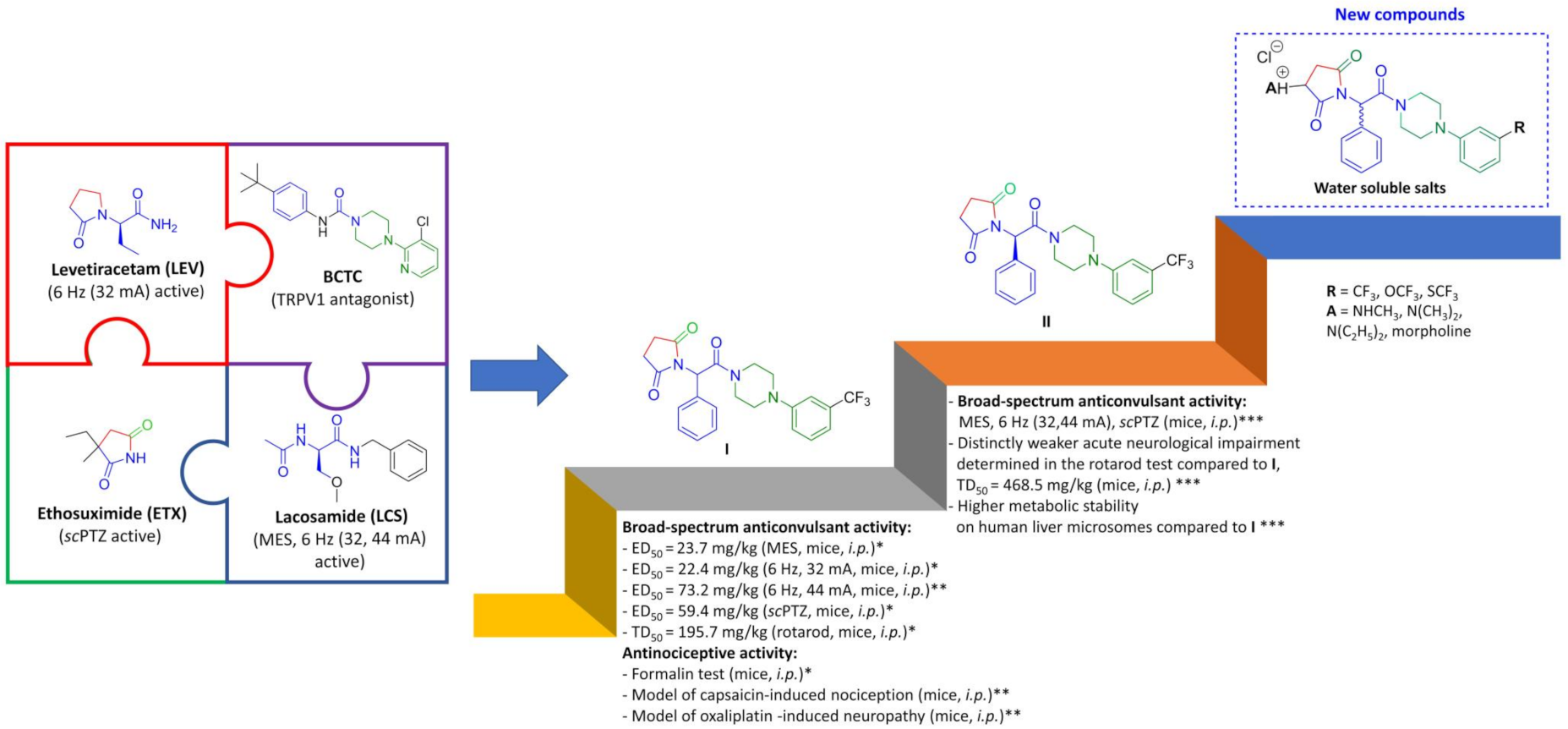
2. Results and Discussion
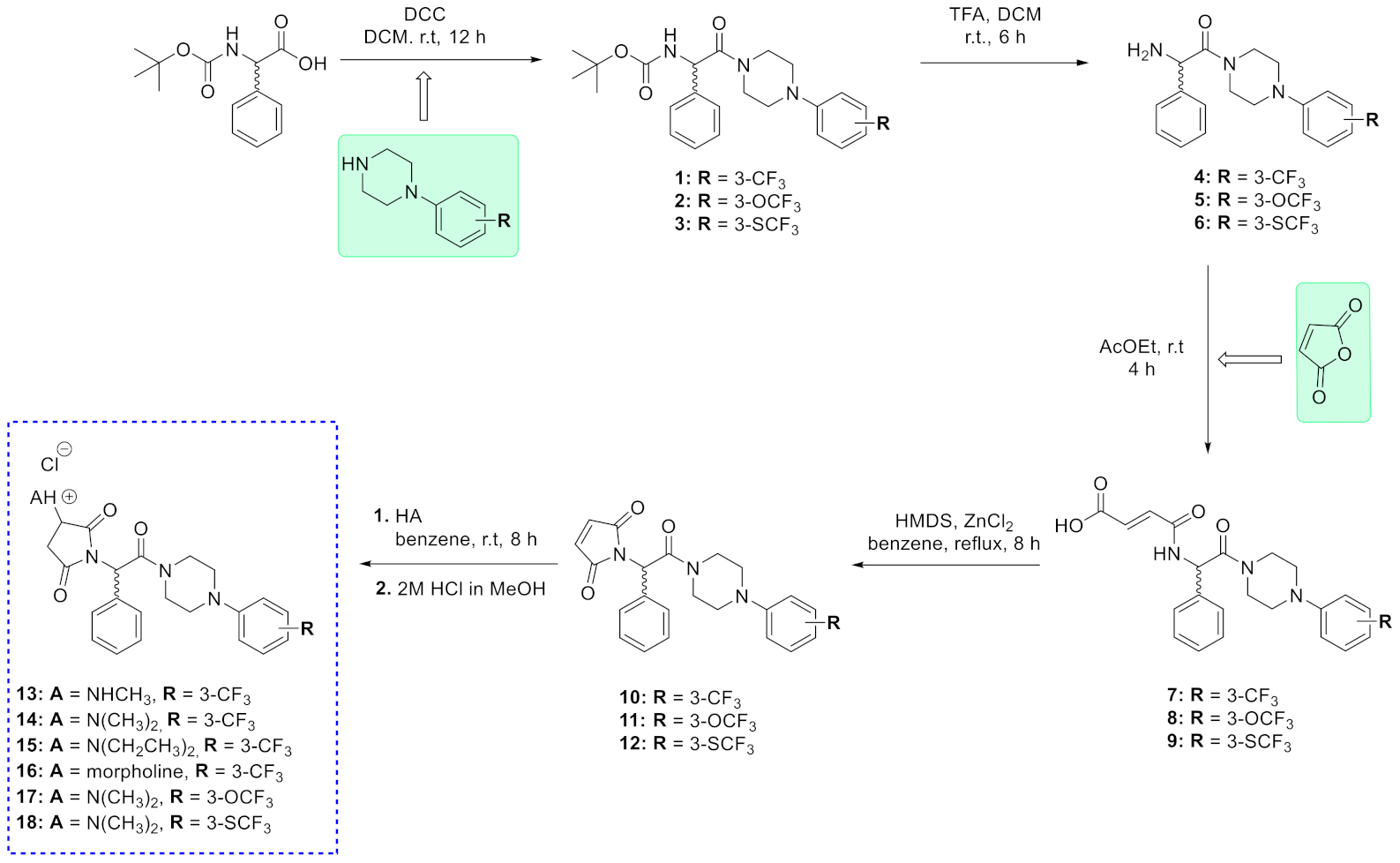
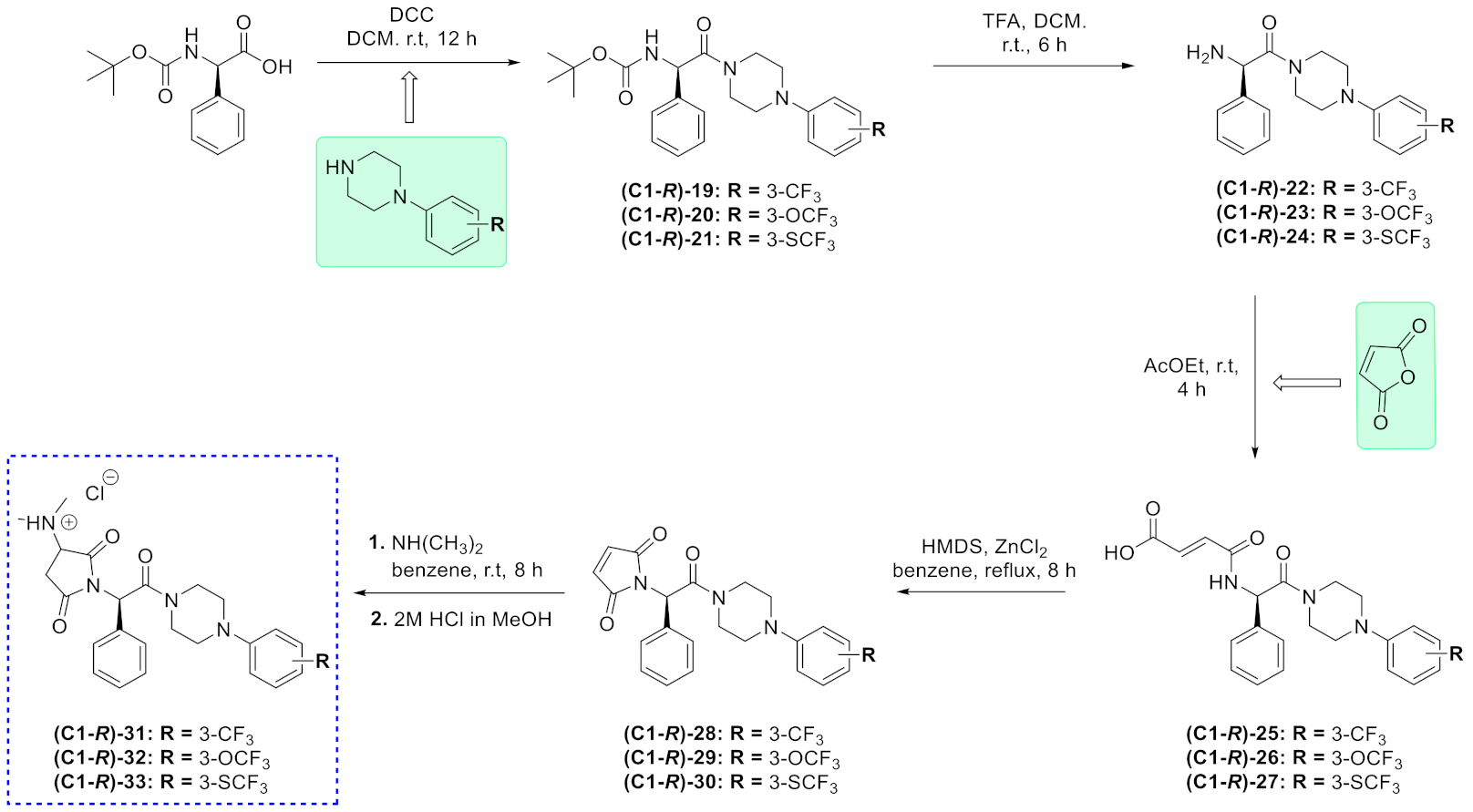
2.1. Chemistry
2.2. Anticonvulsant Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
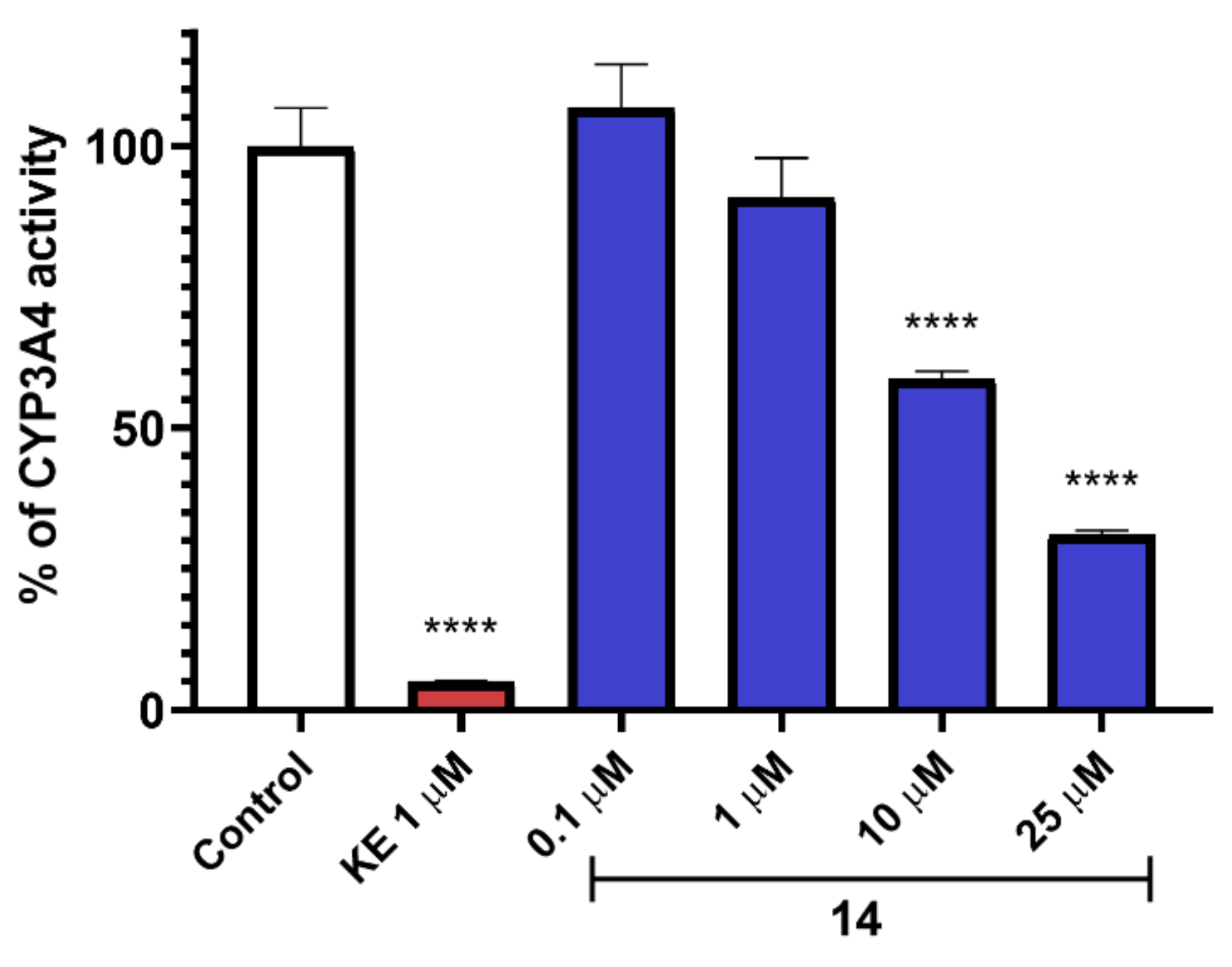{kind=link}
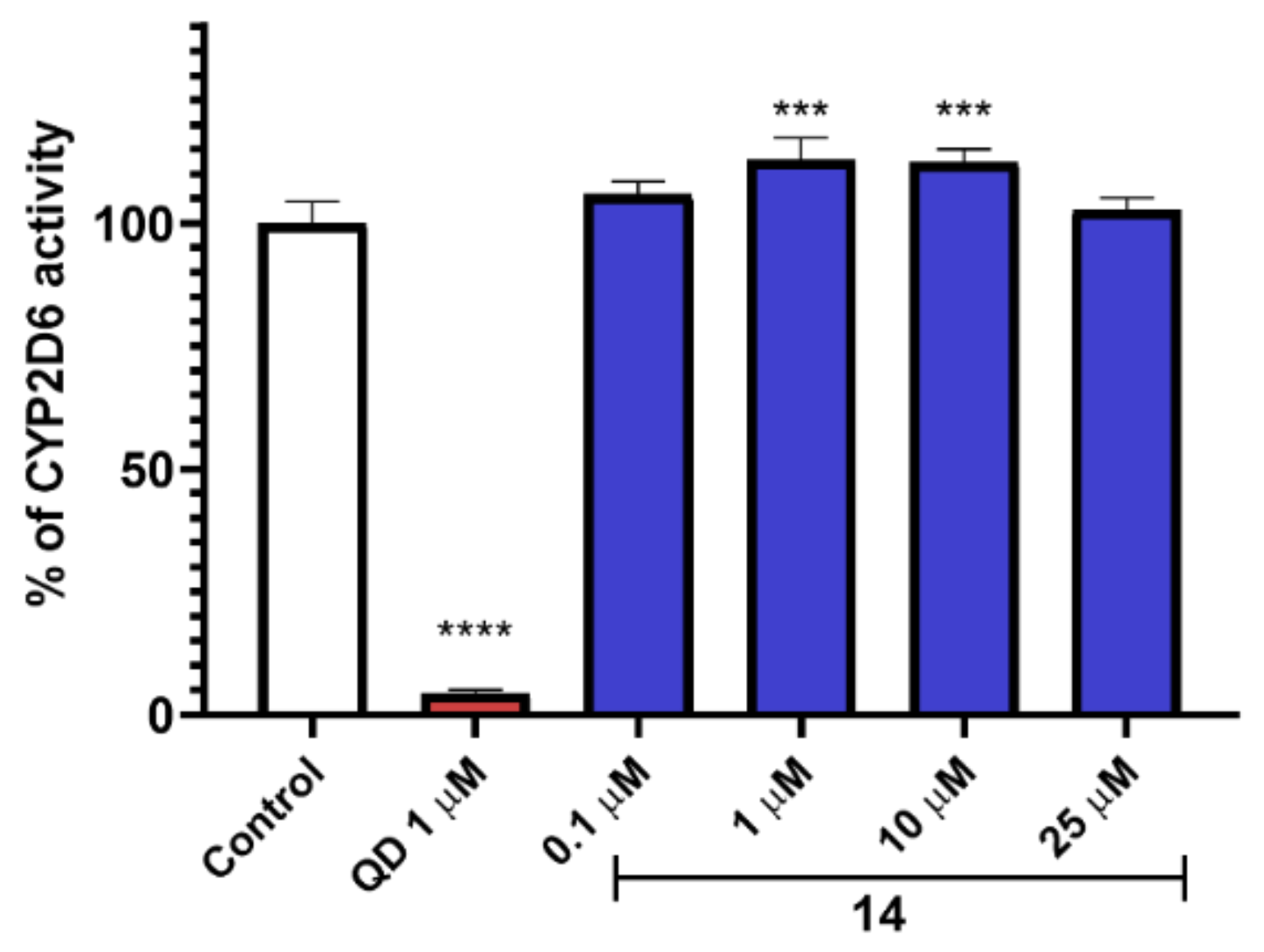{kind=link}
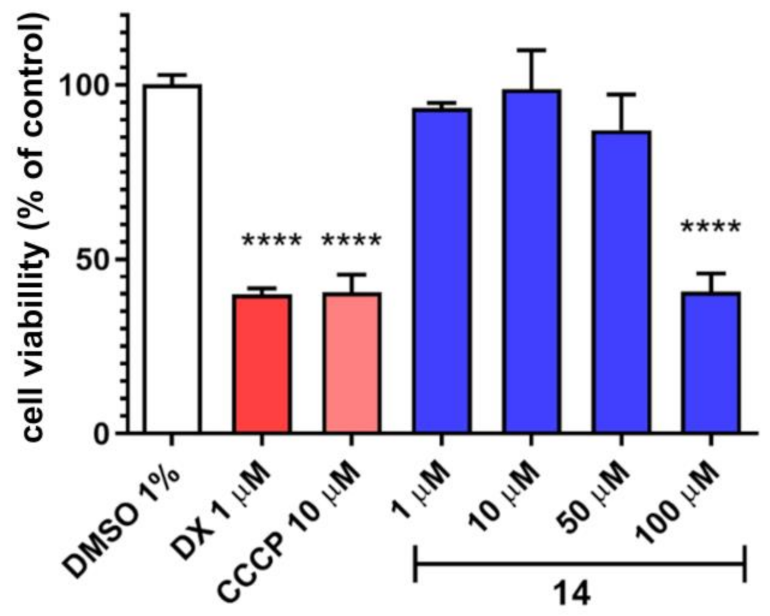{kind=link}
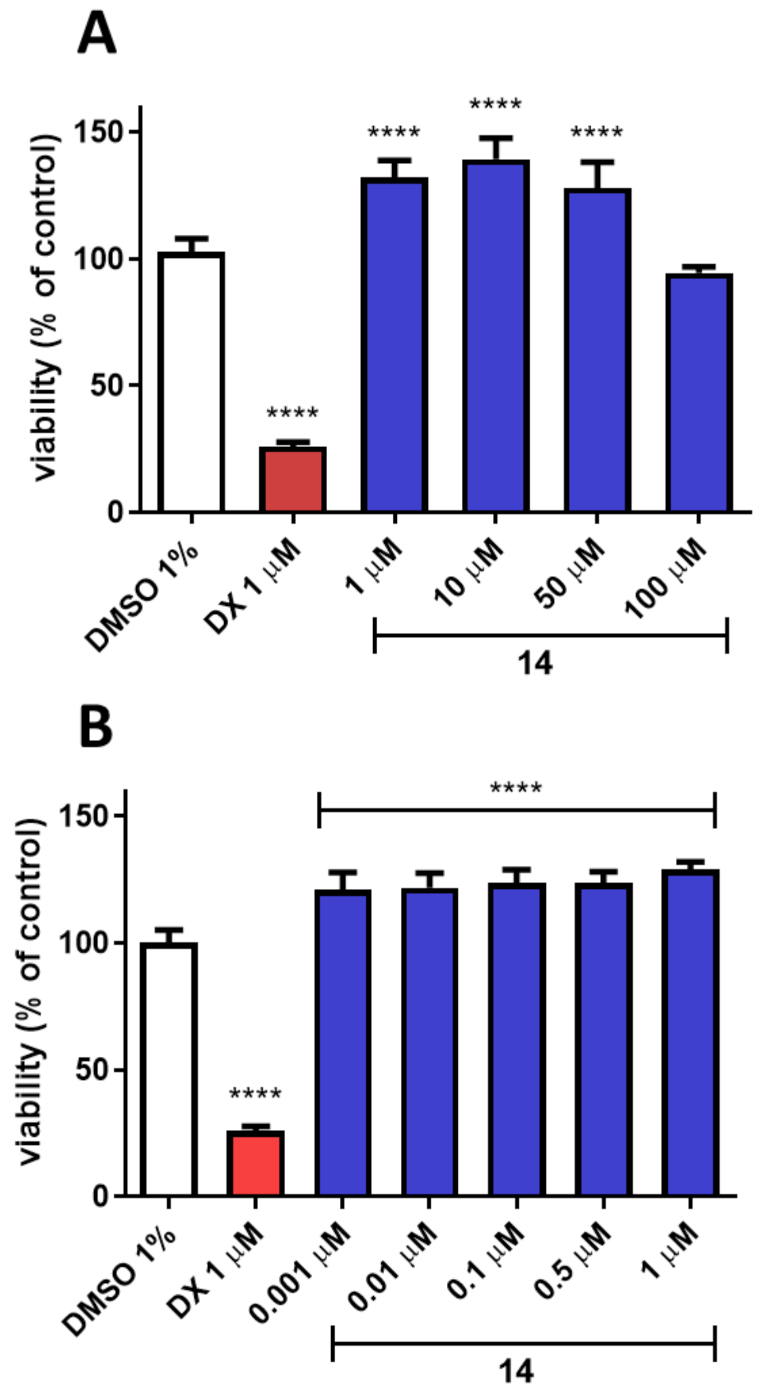{kind=link}
{kind=link}
{kind=link}
| Compound | TPE (h) a | ED50 MES (mg/kg) b | ED50 6 Hz (32 mA) (mg/kg) c | ED50 scPTZ (mg/kg) d | TD50 (mg/kg) e | PI (TD50/ED50) f |
|---|---|---|---|---|---|---|
| 13 | 0.5 | - | 93.6 (77.2–113.4) | 85.6 (57.1–128.4) | 160.1 (143.1–179.0 | 1.7 (6 Hz) 1.9 (scPTZ) |
| 14 | 0.5 | 49.6 (44.3–55.7) | 31.3 (18.2–53.9) | 67.4 (58.2–92.1) | 168.7 (146.3–194.5) | 3.4 (MES) 5.4 (6 Hz) 1.4 (scPTZ) |
| 15 | 0.5 | 77.5 (73.8-81.4) | 80.4 (70.5–91.7) | - | 246.6 (214.9–282.9) | 3.2 (MES) 1.8 (6 Hz) |
| 17 | 0.5 | 33.3 (28.9–38.4) | 28.2 (16.9–47.2) | 31.3 (18.2–53.9) | 69.7 (52.0–93.5) | 1.6 (MES) 3.7 (6 Hz) 2.2 (scPTZ) |
| (C1-R)-31 | 0.5 | 57.7 (33.9–97.9) | 50.9 (45.3–57.1)) | 65.5 (47.9–89.6) | 94.9 (75.3–119.4) | 1.6 (MES) 1.9 (6 Hz) 1.5 (scPTZ) |
| (C1-S)-31 | 0.5 | - | >130 | - | 188.5 (181.3–195.9) | n.c. |
| (C1-R)-32 | 0.5 | 26.3 (18.9–36.8) | 28.3 (20.0–40.1) | 44.9 (37.2–54.3) | 72.0 (64.5–80.4) | 2.7 (MES) 2.5 (6 Hz) 1.6 (scPTZ) |
| I * | 0.5 | 23.7 (18.4–31.2) | 22.4 (17.4–28.8) | 59.4 (37.5–94.1) | 195.7 (132.7–288.6) | 8.2 (MES) 8.7 (6 Hz) 3.3 (scPTZ) |
| II ** | 0.5 | 36.0 (31.4–41.2) | 39.2 (31.6–48.6) | 54.8 (43.5–69.1) | 468.5 (397.0–553.0) | 13.0 (MES) 12.0 (6 Hz) 8.5 (scPTZ) |
| ETX g | 0.25 | n.a. | >200 | 140.4 (115.8–170.2) | 318.0 (295.8–341.9) | 2.3 (scPTZ) |
| LCS g | 0.5 | 9.2 (8.5–10.0) | 5.3 (3.5–7.8) | n.a. | 46.2 (44.5–48.0) | 5.0 (MES) 8.8 (6 Hz) |
| LEV g | 1.0 | >500 | 15.7 (10.4–23.7) | n.a. | >500 | >31.8 (6 Hz) |
| VPA g | 0.5 | 252.7 (220.1–290.2) | 130.6 (117.6–145.2) | 239.4 (209.2–274.1) | 430.7 (407.9–454.9) | 1.7 (MES) 3.3 (6 Hz) 1.8 (scPTZ) |
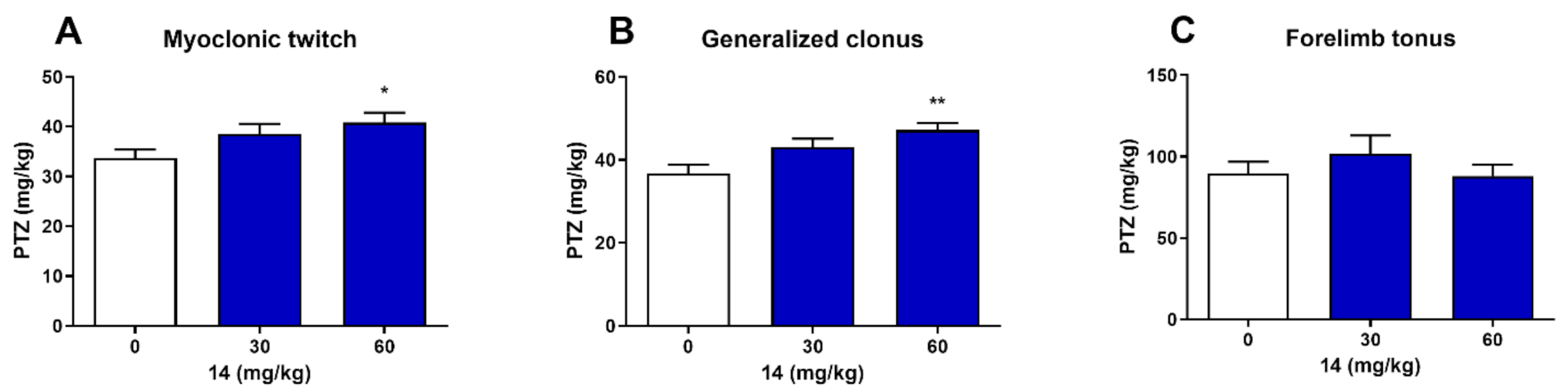
2.3. Effect on the Seizure Threshold in the ivPTZ Test in Mice
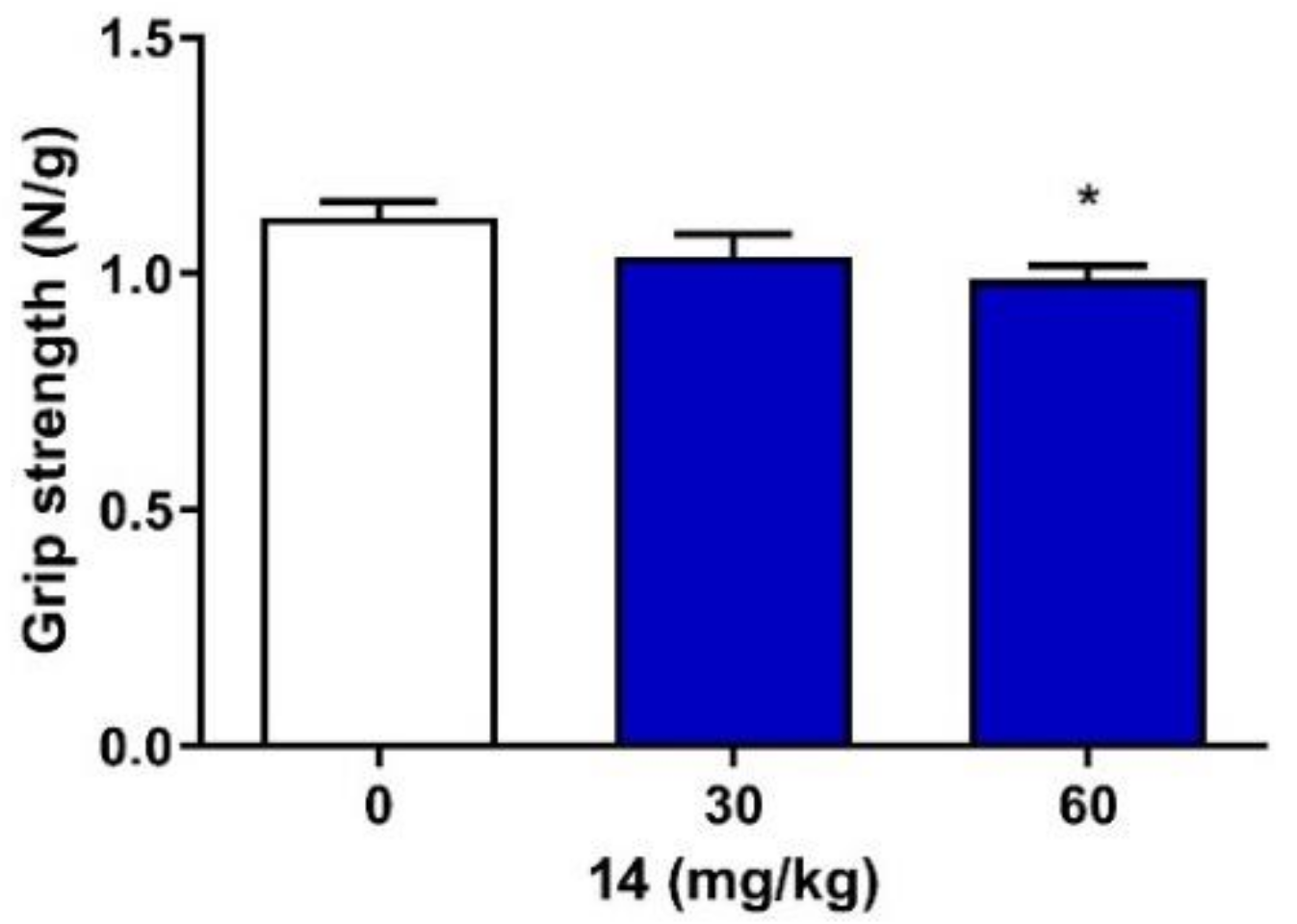
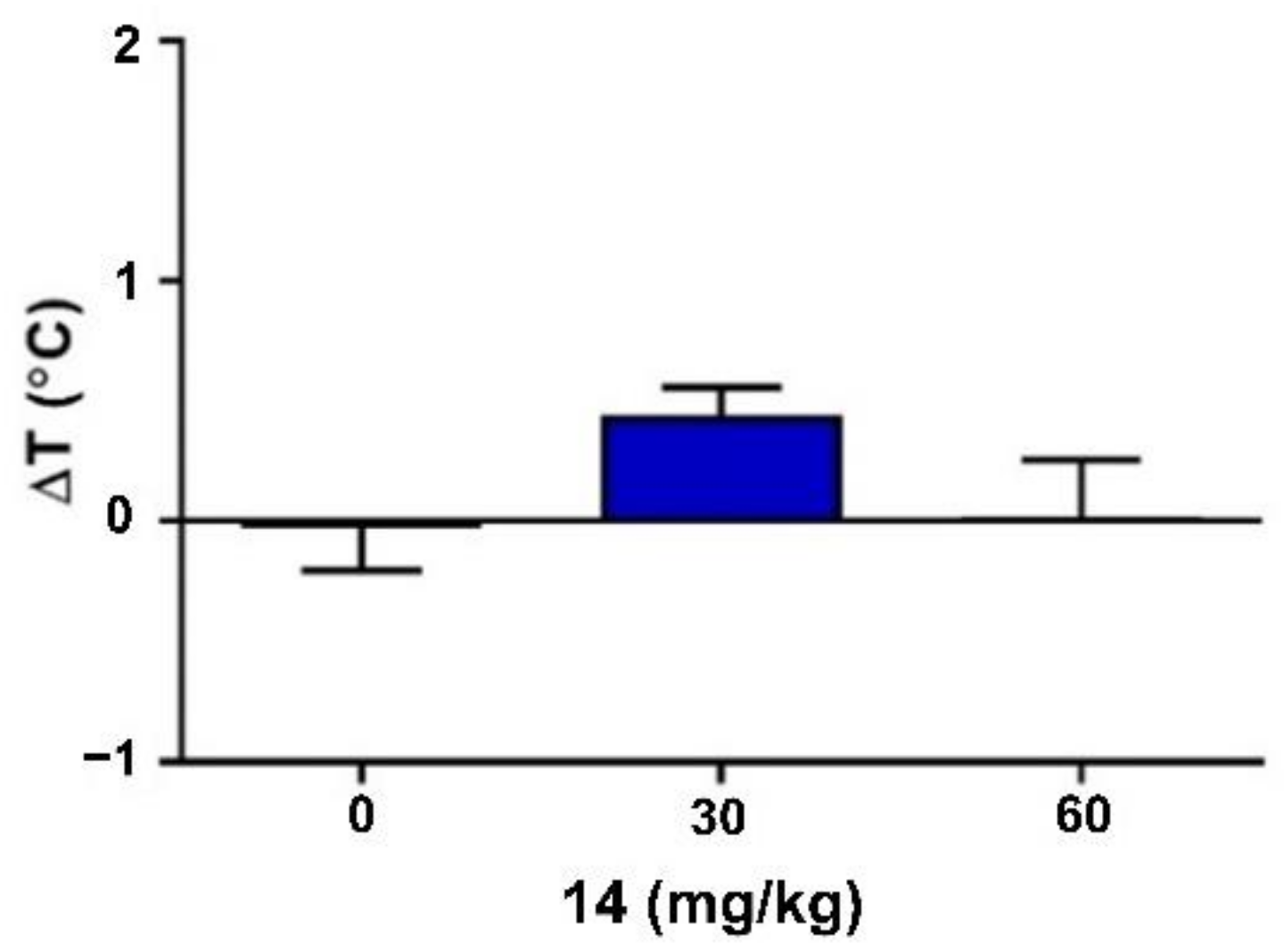
2.4. Neuromuscular Strength and Rectal Temperature


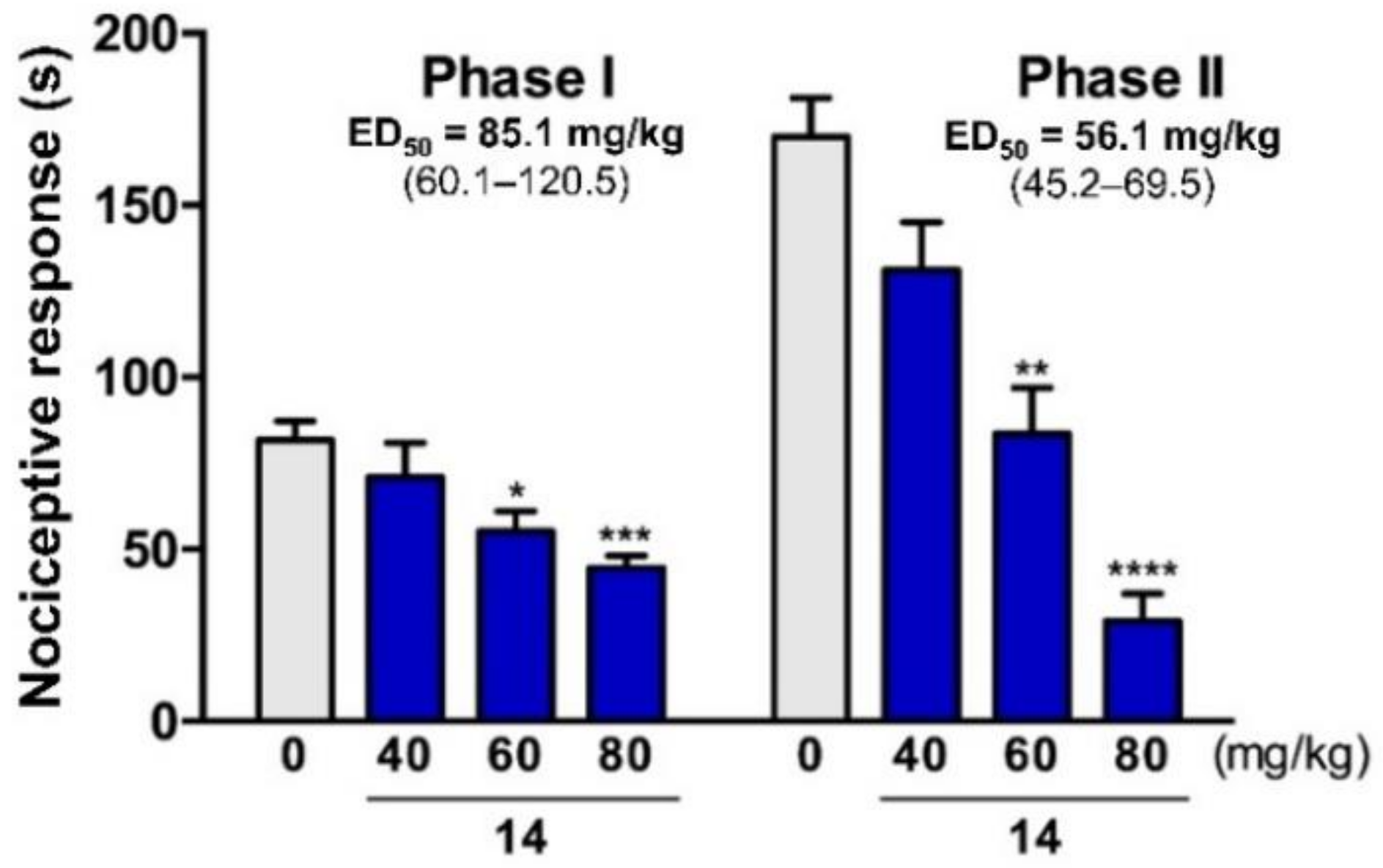
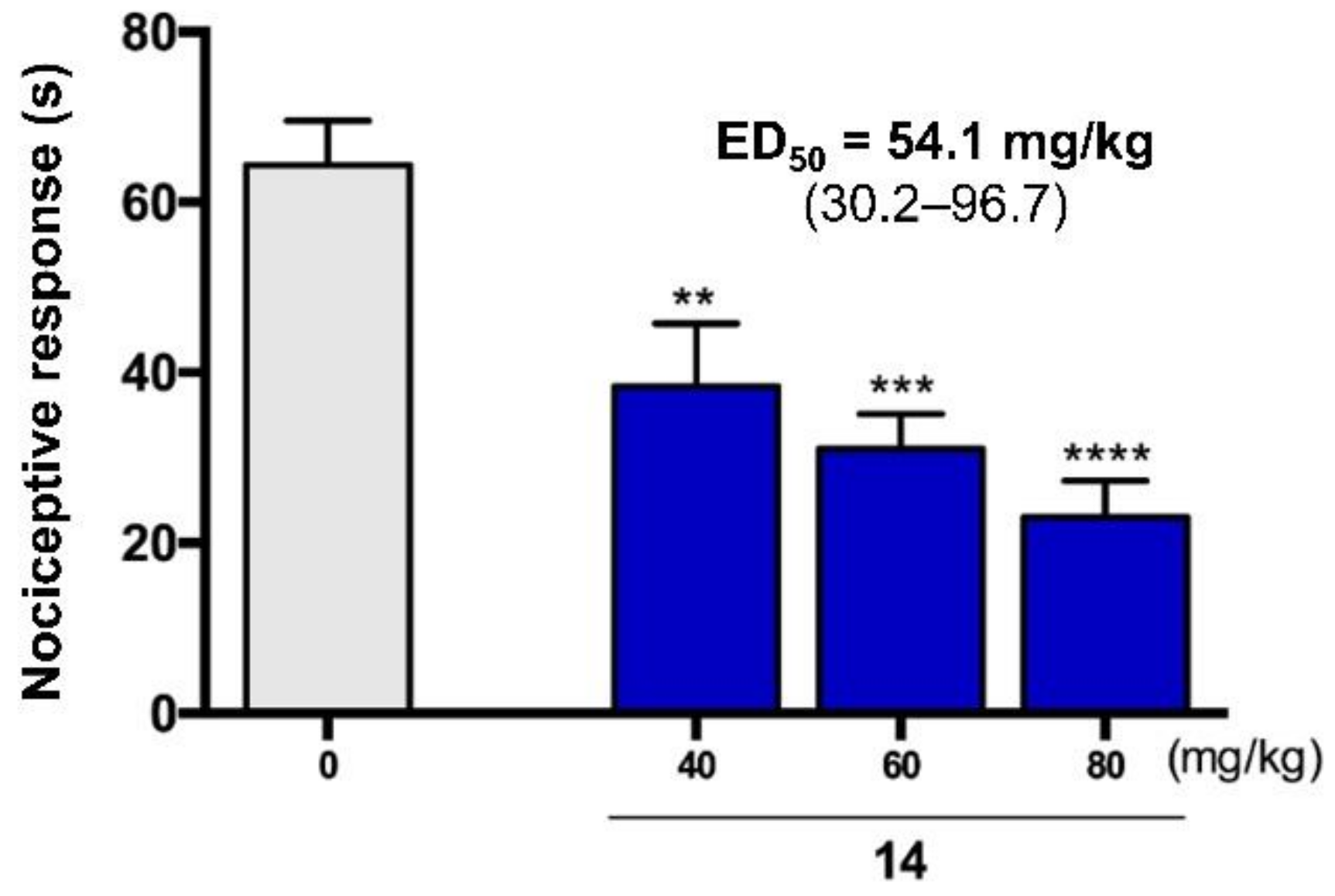
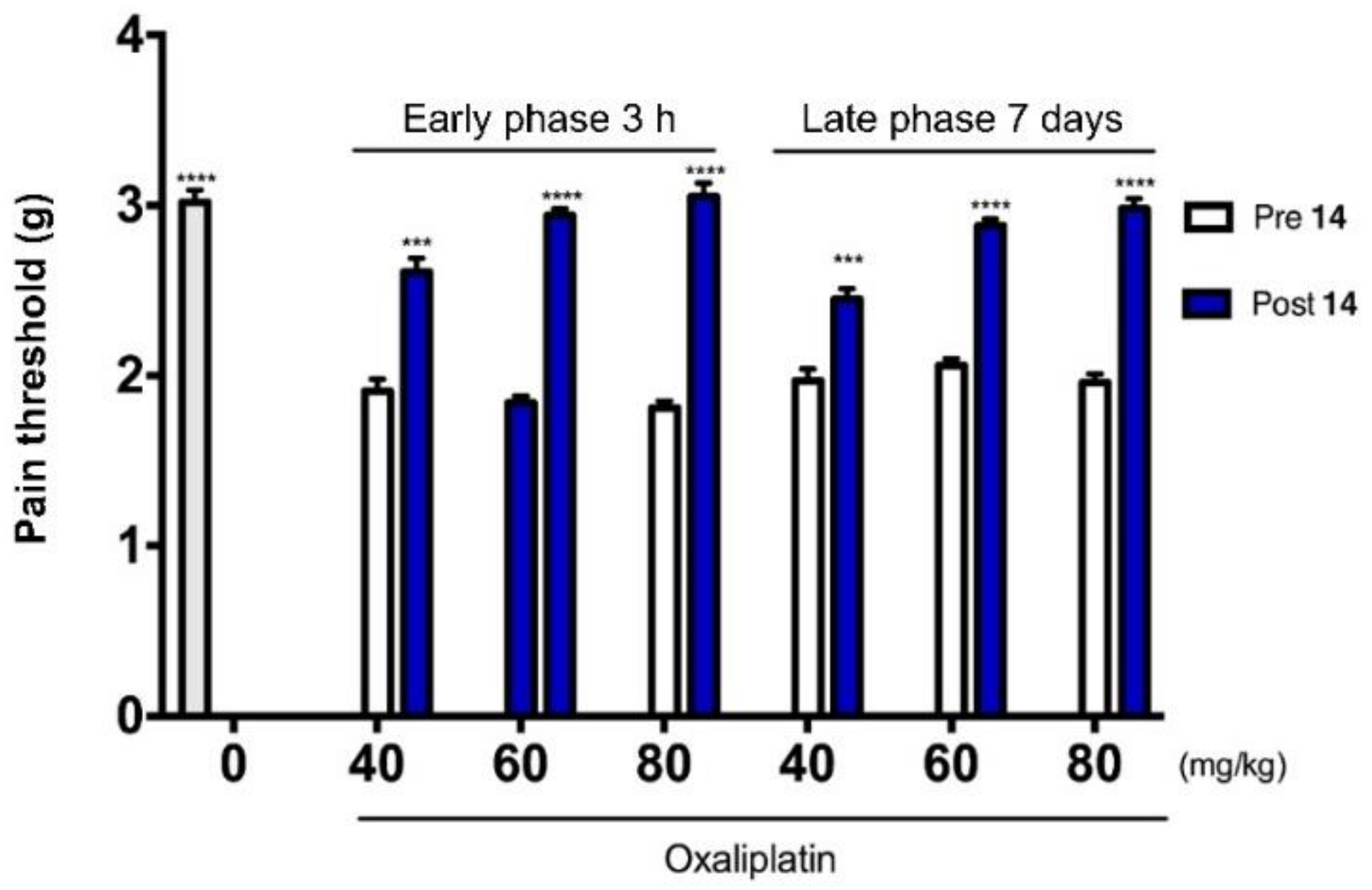
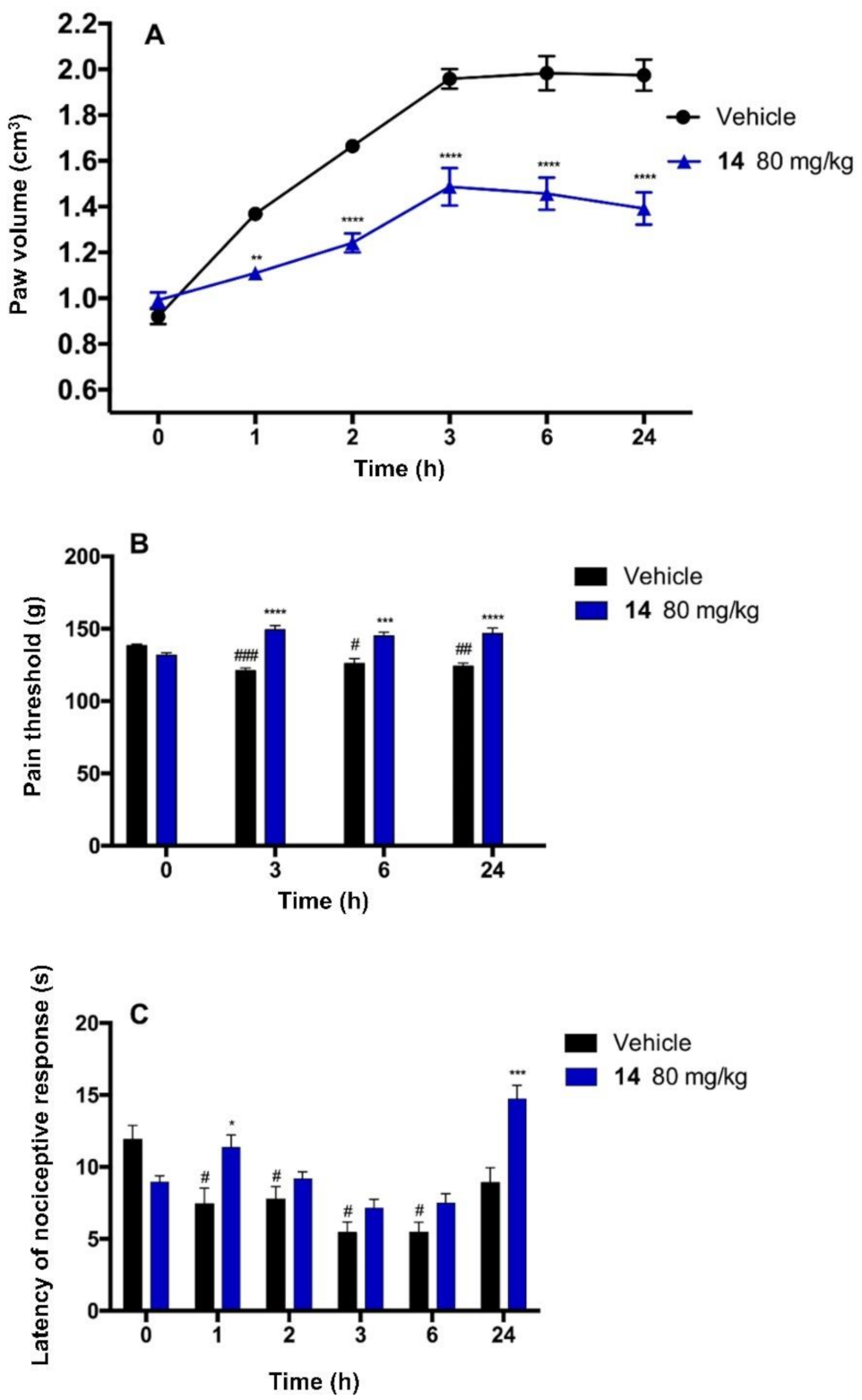
2.5. Antinociceptive Activity
2.6. In Vitro Radioligand Binding Studies and Functional Assays
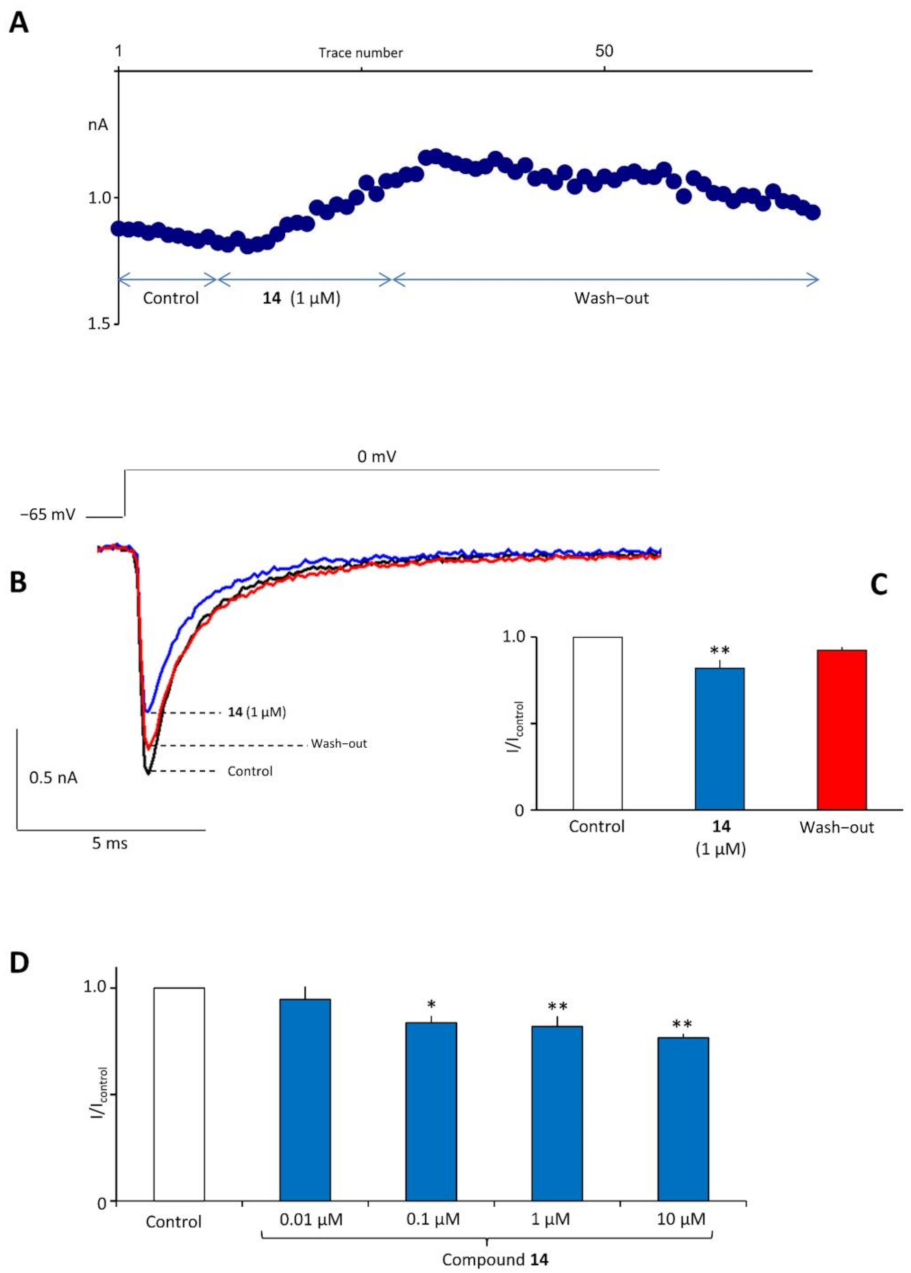
2.7. In Vitro Electrophysiological Studies
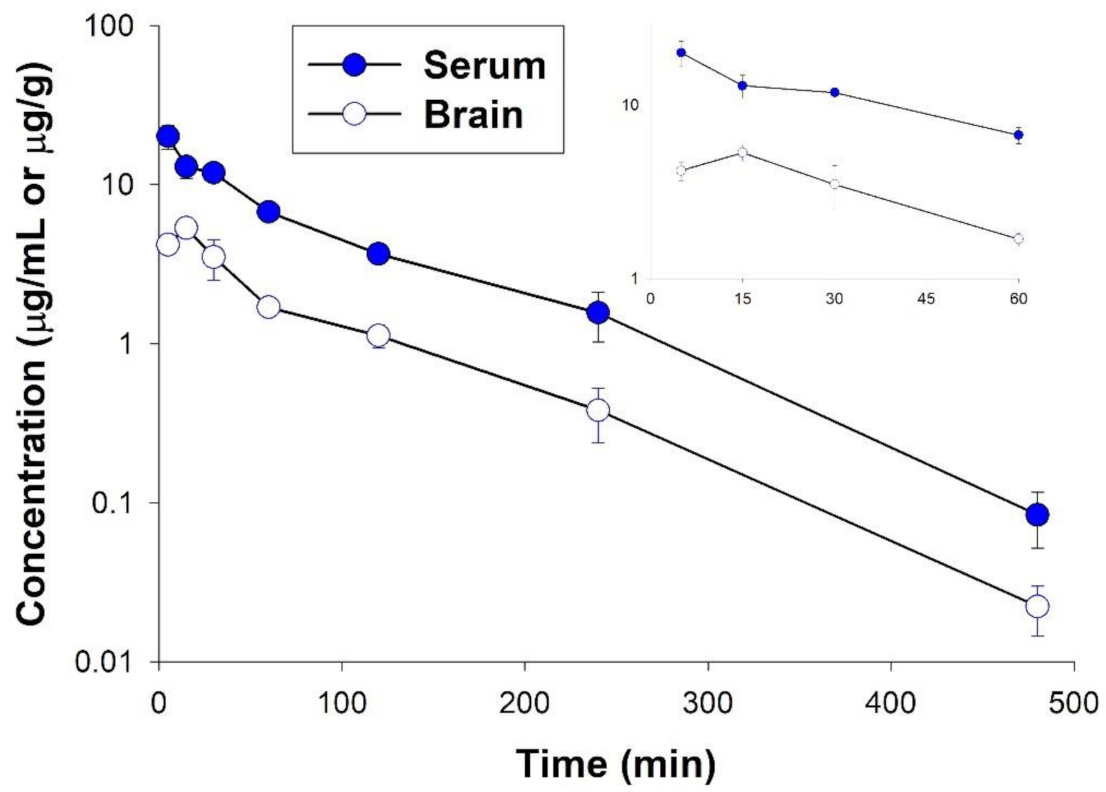
2.8. Pharmacokinetic Studies
2.9. In Vitro ADME-Tox Assays
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthetic Procedure for Boc-Protected Compounds (1–3 and (C1-R)-19–(C1-R)-21) and (C1-S)-19
3.1.2. Synthetic Procedure for Amines 4–6, (C1-R)-22–(C1-R)-24) and (C1-S)-22
3.1.3. Synthetic Procedure for Maleamic Acids (7–9, (C1-R)-25–(C1-R)-27 and (C1-S)-25)
3.1.4. Synthetic Procedure for Unsaturated pyrrolidine-2,5-dione Derivative (10–12, (C1-R)-28–(C1-R)-30) and (C1-S)-28
3.1.5. Synthetic Procedure for Target Hydrochlorides (13–18, (C1-R)-31–(C1-R)-33) and (C1-S)-31
3.2. Anticonvulsant Activity and Acute Neurotoxicity
Data Analysis—Anticonvulsant Activity and Neurotoxicity Studies
3.3. Intravenous (iv) Pentylenetetrazole (PTZ) Seizure Threshold Test, Rectal Temperature Measurement and Grip Strength Test
3.4. Antinociceptive Activity
Data Analysis—Antinociceptive Activity Studies
3.5. Pharmacokinetic Study
3.5.1. Analytical Method
3.5.2. Standard Solutions
3.5.3. Sample Preparation
3.6. In Vitro Pharmacology and ADME-Tox Studies
3.6.1. Radioligand Binding/Functional Assays
3.6.2. In Vitro Electrophysiological Studies
3.6.3. In Vitro Toxicity Studies
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ADME-Tox | Absorption, distribution, metabolism, excretion, toxicity |
| AEDs | Antiseizure drugs |
| CCCP | 3-Chlorophenylhydrazone |
| DCC | Dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DX | Doxorubicin |
| ETX | Ethosuximide |
| HLMs | Human liver microsomes |
| HMDS | Hexamethyldisilazane |
| 6 Hz | 6 Hz seizure test |
| LCS | Lacosamide |
| LEV | Levetiracetam |
| MeCN | Acetonitrile |
| MES | Maximal electroshock seizure test |
| MeOH | Methanol |
| OXPT | Oxaliplatin |
| PI | Protective index (TD50/ED50) |
| scPTZ | Subcutaneous pentylenetetrazole seizure test |
| SV2A | Synaptic vesicle glycoprotein 2A |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TPE | Time of peak effect |
| TRPV1 | Transient receptor potential cation channel vanilloid type 1 |
| VPA | Valproic acid |
References
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, R.; Sugano, K.; Higashida, A.; Hayashi, Y.; Machida, M.; Aso, Y.; Yamashita, S. Oral Absorption of Poorly Water-Soluble Drugs: Computer Simulation of Fraction Absorbed in Humans from a Miniscale Dissolution Test. Pharm. Res. 2006, 23, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-like Property Concepts in Pharmaceutical Design. Curr. Pharm. Des. 2009, 15, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Fish, P.V.; Mano, T. Bridging Solubility between Drug Discovery and Development. Drug Discov. Today 2012, 17, 486–495. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef]
- Abram, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Lubelska, A.; Kamiński, R.M.; Kamiński, K. Multitargeted Compounds Derived from (2,5-Dioxopyrrolidin-1-Yl)(Phenyl)-Acetamides as Candidates for Effective Anticonvulsant and Antinociceptive Agents. ACS Chem. Neurosci. 2020, 11, 1996–2008. [Google Scholar] [CrossRef]
- Kamiński, K.; Mogilski, S.; Abram, M.; Rapacz, A.; Latacz, G.; Szulczyk, B.; Walczak, M.; Kuś, K.; Matyjaszczyk, K.; Kamiński, R.M. KA-104, a New Multitargeted Anticonvulsant with Potent Antinociceptive Activity in Preclinical Models. Epilepsia 2020, 61, 2119–2128. [Google Scholar] [CrossRef]
- Abram, M.; Rapacz, A.; Latacz, G.; Szulczyk, B.; Kalinowska-Tłuścik, J.; Otto-Ślusarczyk, D.; Struga, M.; Kamiński, R.M.; Kamiński, K. Asymmetric Synthesis and in Vivo/in Vitro Characterization of New Hybrid Anticonvulsants Derived from (2,5-Dioxopyrrolidin-1-Yl)Phenylacetamides. Bioorganic Chem. 2021, 109, 104751. [Google Scholar] [CrossRef]
- Zhao, Z.; Yue, J.; Ji, X.; Nian, M.; Kang, K.; Qiao, H.; Zheng, X. Research Progress in Biological Activities of Succinimide Derivatives. Bioorganic Chem. 2021, 108, 104557. [Google Scholar] [CrossRef]
- Wróbel, M.Z.; Chodkowski, A.; Herold, F.; Marciniak, M.; Dawidowski, M.; Siwek, A.; Starowicz, G.; Stachowicz, K.; Szewczyk, B.; Nowak, G.; et al. Synthesis and Biological Evaluation of New Multi-Target 3-(1H-Indol-3-Yl)Pyrrolidine-2,5-Dione Derivatives with Potential Antidepressant Effect. Eur. J. Med. Chem. 2019, 183, 111736. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, M.Z.; Chodkowski, A.; Marciniak, M.; Dawidowski, M.; Maksymiuk, A.; Siwek, A.; Nowak, G.; Turło, J. Synthesis of New 4-Butyl-Arylpiperazine-3-(1H-Indol-3-Yl)Pyrrolidine-2,5-Dione Derivatives and Evaluation for Their 5-HT1A and D2 Receptor Affinity and Serotonin Transporter Inhibition. Bioorg. Chem. 2020, 97, 103662. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.; Jakubiec, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Kamiński, R.M.; Kamiński, K. The Search for New Anticonvulsants in a Group of (2,5-Dioxopyrrolidin-1-Yl)(Phenyl)Acetamides with Hybrid Structure—Synthesis and In Vivo/In Vitro Studies. Int. J. Mol. Sci. 2020, 21, 8780. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.; Zagaja, M.; Mogilski, S.; Andres-Mach, M.; Latacz, G.; Baś, S.; Łuszczki, J.J.; Kieć-Kononowicz, K.; Kamiński, K. Multifunctional Hybrid Compounds Derived from 2-(2,5-Dioxopyrrolidin-1-Yl)-3-Methoxypropanamides with Anticonvulsant and Antinociceptive Properties. J. Med. Chem. 2017, 60, 8565–8579. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, K.; Zagaja, M.; Łuszczki, J.J.; Rapacz, A.; Andres-Mach, M.; Latacz, G.; Kieć-Kononowicz, K. Design, Synthesis, and Anticonvulsant Activity of New Hybrid Compounds Derived from 2-(2,5-Dioxopyrrolidin-1-Yl)Propanamides and 2-(2,5-Dioxopyrrolidin-1-Yl)Butanamides. J. Med. Chem. 2015, 58, 5274–5286. [Google Scholar] [CrossRef]
- Kamiński, K.; Zagaja, M.; Rapacz, A.; Łuszczki, J.J.; Andres-Mach, M.; Abram, M.; Obniska, J. New Hybrid Molecules with Anticonvulsant and Antinociceptive Activity Derived from 3-Methyl- or 3,3-Dimethyl-1-[1-Oxo-1-(4-Phenylpiperazin-1-Yl)Propan-2-Yl]Pyrrolidine-2,5-Diones. Bioorganic Med. Chem. 2016, 24, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. Critical Review of Current Animal Models of Seizures and Epilepsy Used in the Discovery and Development of New Antiepileptic Drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Castel-Branco, M.M.; Alves, G.L.; Figueiredo, I.V.; Falcao, A.C.; Caramona, M.M. The Maximal Electroshock Seizure (MES) Model in the Preclinical Assessment of Potential New Antiepileptic Drugs. Methods Find. Exp. Clin. Pharmacol. 2009, 31, 101. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, K.; Kaminski, R.M. Genetic Background of Mice Strongly Influences Treatment Resistance in the 6 Hz Seizure Model. Epilepsia 2015, 56, 310–318. [Google Scholar] [CrossRef]
- Golyala, A.; Kwan, P. Drug Development for Refractory Epilepsy: The Past 25 Years and Beyond. Seizure 2017, 44, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Litchfield, J.T.; Wilcoxon, F. A Simplified Method of Evaluating Dose-Effect Experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar]
- Smith, M.; Wilcox, K.S.; White, H.S. Discovery of Antiepileptic Drugs. Neurotherapeutics 2007, 4, 12–17. [Google Scholar] [CrossRef]
- Metcalf, C.S.; West, P.J.; Thomson, K.; Edwards, S.; Smith, M.D.; White, H.S.; Wilcox, K.S. Development and Pharmacological Characterization of the Rat 6 Hz Model of Partial Seizures. Epilepsia 2017, 58, 1073–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcox, K.S.; Dixon-Salazar, T.; Sills, G.J.; Ben-Menachem, E.; White, H.S.; Porter, R.J.; Dichter, M.A.; Moshé, S.L.; Noebels, J.L.; Privitera, M.D.; et al. Issues Related to Development of New Anti-Seizure Treatments. Epilepsia 2013, 54, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Klein, P. The Pharmacology and Clinical Efficacy of Antiseizure Medications: From Bromide Salts to Cenobamate and Beyond. CNS Drugs 2021, 35, 935–963. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Khanna, R. Specific Binding of Lacosamide to Collapsin Response Mediator Protein 2 (CRMP2) and Direct Impairment of Its Canonical Function: Implications for the Therapeutic Potential of Lacosamide. Mol. Neurobiol. 2015, 51, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socała, K.; Nieoczym, D.; Kowalczuk-Vasilev, E.; Wyska, E.; Wlaź, P. Increased Seizure Susceptibility and Other Toxicity Symptoms Following Acute Sulforaphane Treatment in Mice. Toxicol Appl. Pharmacol. 2017, 326, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Abarca, A.B.; Avila-Rojas, S.H.; Barragán-Iglesias, P.; Pineda-Farias, J.B.; Granados-Soto, V. Formalin Injection Produces Long-Lasting Hypersensitivity with Characteristics of Neuropathic Pain. Eur. J. Pharmacol. 2017, 797, 83–93. [Google Scholar] [CrossRef]
- Hama, A.; Natsume, T.; Ogawa, S.; Higo, N.; Hayashi, I.; Takamatsu, H. Gaps in Understanding Mechanism and Lack of Treatments: Potential Use of a Nonhuman Primate Model of Oxaliplatin-Induced Neuropathic Pain. Pain Res. Manag. 2018, 2018, 1630709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viatchenko-Karpinski, V.; Ling, J.; Gu, J.G. Down-Regulation of Kv4.3 Channels and a-Type K+ Currents in V2 Trigeminal Ganglion Neurons of Rats Following Oxaliplatin Treatment. Mol. Pain 2018, 14, 1744806917750995. [Google Scholar] [CrossRef] [Green Version]
- Łażewska, D.; Bajda, M.; Kaleta, M.; Zaręba, P.; Doroz-Płonka, A.; Siwek, A.; Alachkar, A.; Mogilski, S.; Saad, A.; Kuder, K.; et al. Rational Design of New Multitarget Histamine H3 Receptor Ligands as Potential Candidates for Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2020, 207, 112743. [Google Scholar] [CrossRef] [PubMed]
- Mogilski, S.; Kubacka, M.; Łażewska, D.; Więcek, M.; Głuch-Lutwin, M.; Tyszka-Czochara, M.; Bukowska-Strakova, K.; Filipek, B.; Kieć-Kononowicz, K. Aryl-1,3,5-Triazine Ligands of Histamine H4 Receptor Attenuate Inflammatory and Nociceptive Response to Carrageen, Zymosan and Lipopolysaccharide. Inflamm. Res. 2017, 66, 79–95. [Google Scholar] [CrossRef] [Green Version]
- Łażewska, D.; Mogilski, S.; Hagenow, S.; Kuder, K.; Głuch-Lutwin, M.; Siwek, A.; Więcek, M.; Kaleta, M.; Seibel, U.; Buschauer, A.; et al. Alkyl Derivatives of 1,3,5-Triazine as Histamine H4 Receptor Ligands. Bioorganic Med. Chem. 2019, 27, 1254–1262. [Google Scholar] [CrossRef]
- Fornasari, D. Pharmacotherapy for Neuropathic Pain: A Review. Pain Ther. 2017, 6, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazıroğlu, M. TRPV1 Channel: A Potential Drug Target for Treating Epilepsy. Curr. Neuropharmacol. 2015, 13, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Carletti, F.; Gambino, G.; Rizzo, V.; Ferraro, G.; Sardo, P. Involvement of TRPV1 Channels in the Activity of the Cannabinoid WIN 55,212-2 in an Acute Rat Model of Temporal Lobe Epilepsy. Epilepsy Res. 2016, 122, 56–65. [Google Scholar] [CrossRef]
- De Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef]
- Roca-Lapirot, O.; Radwani, H.; Aby, F.; Nagy, F.; Landry, M.; Fossat, P. Calcium Signalling through L-type Calcium Channels: Role in Pathophysiology of Spinal Nociceptive Transmission. Br. J. Pharmacol. 2018, 175, 2362–2374. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jo, Y.Y.; Chung, G.; Jung, J.H.; Kim, Y.H.; Park, C.-K. Functional Importance of Transient Receptor Potential (TRP) Channels in Neurological Disorders. Front. Cell Dev. Biol. 2021, 9, 410. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.E.; Klein, B.D.; Wolf, H.H.; White, H.S. Pharmacological Characterization of the 6 Hz Psychomotor Seizure Model of Partial Epilepsy. Epilepsy Res. 2001, 47, 217–227. [Google Scholar] [CrossRef]
- Radwani, H.; Lopez-Gonzalez, M.J.; Cattaert, D.; Roca-Lapirot, O.; Dobremez, E.; Bouali-Benazzouz, R.; Eiríksdóttir, E.; Langel, Ü.; Favereaux, A.; Errami, M.; et al. Cav1.2 and Cav1.3 L-type Calcium Channels Independently Control Short- and Long-term Sensitization to Pain. J. Physiol. 2016, 594, 6607–6626. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Tabrizi, M.; Baraldi, P.G.; Baraldi, S.; Gessi, S.; Merighi, S.; Borea, P.A. Medicinal Chemistry, Pharmacology, and Clinical Implications of TRPV1 Receptor Antagonists. Med. Res. Rev. 2017, 37, 936–983. [Google Scholar] [CrossRef]
- Tate, S.K.; Depondt, C.; Sisodiya, S.M.; Cavalleri, G.L.; Schorge, S.; Soranzo, N.; Thom, M.; Sen, A.; Shorvon, S.D.; Sander, J.W.; et al. Genetic Predictors of the Maximum Doses Patients Receive during Clinical Use of the Anti-Epileptic Drugs Carbamazepine and Phenytoin. Proc. Natl. Acad. Sci. USA 2005, 102, 5507–5512. [Google Scholar] [CrossRef] [Green Version]
- Kwan, P.; Brodie, M.J. Phenobarbital for the Treatment of Epilepsy in the 21st Century: A Critical Review. Epilepsia 2004, 45, 1141–1149. [Google Scholar] [CrossRef]
- Sadeque, A.J.; Fisher, M.B.; Korzekwa, K.R.; Gonzalez, F.J.; Rettie, A.E. Human CYP2C9 and CYP2A6 Mediate Formation of the Hepatotoxin 4-Ene-Valproic Acid. J. Pharmacol. Exp. Ther. 1997, 283, 698–703. [Google Scholar]
- Tomson, T.; Tybring, G.; Bertilsson, L. Single-Dose Kinetics and Metabolism of Carbamazepine-10,11-Epoxide. Clin. Pharmacol. Ther. 1983, 33, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.U.; Nelson, D.L.; Button, D.; Cole, H.W.; Baez, M.B.; Lucaites, V.L.; Wainscott, D.B.; Whitesitt, C.; Reel, J.; Simon, R.; et al. A Novel Class of 5-HT2A Receptor Antagonists: Aryl Aminoguanidines. Life Sci. 1996, 59, 1259–1268. [Google Scholar] [CrossRef]
- Socała, K.; Mogilski, S.; Pieróg, M.; Nieoczym, D.; Abram, M.; Szulczyk, B.; Lubelska, A.; Latacz, G.; Doboszewska, U.; Wlaź, P.; et al. KA-11, a Novel Pyrrolidine-2,5-Dione Derived Broad-Spectrum Anticonvulsant: Its Antiepileptogenic, Antinociceptive Properties and in Vitro Characterization. ACS Chem. Neurosci. 2019, 10, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mach, M.; Szewczyk, A.; Zagaja, M.; Szala-Rycaj, J.; Lemieszek, M.K.; Maj, M.; Abram, M.; Kaminski, K. Preclinical Assessment of a New Hybrid Compound C11 Efficacy on Neurogenesis and Cognitive Functions after Pilocarpine Induced Status Epilepticus in Mice. Int. J. Mol. Sci. 2021, 22, 3240. [Google Scholar] [CrossRef]
- Wojda, E.; Wlaz, A.; Patsalos, P.N.; Luszczki, J.J. Isobolographic Characterization of Interactions of Levetiracetam with the Various Antiepileptic Drugs in the Mouse 6 Hz Psychomotor Seizure Model. Epilepsy Res. 2009, 86, 163–174. [Google Scholar] [CrossRef]
- Dunham, N.W.; Miya, T.S.; Edwards, L.D. The Pharmacological Activity of a Series of Basic Esters of Mono- and Dialkylmalonic Acids. J. Am. Pharm. Assoc. Am. Pharm. Assoc. 1957, 46, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Mogilski, S.; Kubacka, M.; Redzicka, A.; Kazek, G.; Dudek, M.; Malinka, W.; Filipek, B. Antinociceptive, Anti-Inflammatory and Smooth Muscle Relaxant Activities of the Pyrrolo[3,4-d]Pyridazinone Derivatives: Possible Mechanisms of Action. Pharmacol. Biochem. Behav. 2015, 133, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Szulczyk, B.; Nurowska, E. Valproic Acid Inhibits TTX-Resistant Sodium Currents in Prefrontal Cortex Pyramidal Neurons. Biochem. Biophys. Res. Commun. 2017, 491, 291–295. [Google Scholar] [CrossRef] [PubMed]














| Compound | TPE (h) a | ED50 6 Hz (44 mA) (mg/kg) b | TD50 (mg/kg) c | PI (TD50/ED50) d |
|---|---|---|---|---|
| 14 | 0.5 | 63.2 (37.6–106.4) | 168.7 (146.3–194.5) | 2.7 |
| I * | 0.5 | 73.2 (57.4–93.4) | 195.7 (132.7–288.6) | 2.7 |
| LCS * | 0.5 | 6.9 (5.4–8.6) | 46.2 (44.5–48.0) | 6.7 |
| LEV * | 1.0 | >500 | >500 | n.c. |
| VPA * | 0.5 | 183.1 (143.5–233.7) | 430.7 (407.9–454.9) | 2.3 |
| % Inhibition of Control-Specific Binding (Concentration µM) | ||
|---|---|---|
| Compd | TTX-sensitive Na+ channel * | Cav1.2 channel (dihydropyridine site, antagonist radioligand) * |
| 14 | 90.3 (50) 31.0 (10) | 75.4 (50) 6.0 (10) |
| (C1-R)-32 | 101.9 (50) 33.4 (10) | 88.9 (50) 18.6 (10) |
| % Inhibition of Control Agonist Response (Concentration µM) | ||
| Compd | TRPV1 (VR1) (h) (antagonist effect) ** | Cav1.2 (h) calcium ion channel cell based antagonist calcium flux assay ** |
| 14 | 88.8 (50) 17.3 (10) | 29.0 (10) |
| (C1-R)-32 | 44.6 (50) 8.5 (10) | 38.0 (10) |
| Parameter | Serum | Brain |
|---|---|---|
| tmax (min) | 5 | 15 |
| Cmax (µg/mL(g)) | 20.06 | 5.32 |
| λz (min−1) | 0.011 | 0.011 |
| t0.5λz (min) | 64.65 | 66.14 |
| V/F (L/kg) | 2.57 | - |
| CL/F (L/min/kg) | 0.027 | - |
| AUC0-∞ (µg⋅min/mL(g)) | 1451.22 | 413.17 |
| MRT (min) | 93.46 | 89.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abram, M.; Jakubiec, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Szulczyk, B.; Szafarz, M.; Socała, K.; Nieoczym, D.; Wyska, E.; et al. Identification of New Compounds with Anticonvulsant and Antinociceptive Properties in a Group of 3-substituted (2,5-dioxo-pyrrolidin-1-yl)(phenyl)-Acetamides. Int. J. Mol. Sci. 2021, 22, 13092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313092
Abram M, Jakubiec M, Rapacz A, Mogilski S, Latacz G, Szulczyk B, Szafarz M, Socała K, Nieoczym D, Wyska E, et al. Identification of New Compounds with Anticonvulsant and Antinociceptive Properties in a Group of 3-substituted (2,5-dioxo-pyrrolidin-1-yl)(phenyl)-Acetamides. International Journal of Molecular Sciences. 2021; 22(23):13092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313092
Chicago/Turabian StyleAbram, Michał, Marcin Jakubiec, Anna Rapacz, Szczepan Mogilski, Gniewomir Latacz, Bartłomiej Szulczyk, Małgorzata Szafarz, Katarzyna Socała, Dorota Nieoczym, Elżbieta Wyska, and et al. 2021. "Identification of New Compounds with Anticonvulsant and Antinociceptive Properties in a Group of 3-substituted (2,5-dioxo-pyrrolidin-1-yl)(phenyl)-Acetamides" International Journal of Molecular Sciences 22, no. 23: 13092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222313092