
Autism-Related Transcription Factors Underlying the Sex-Specific Effects of Prenatal Bisphenol A Exposure on Transcriptome-Interactome Profiles in the Offspring Prefrontal Cortex

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
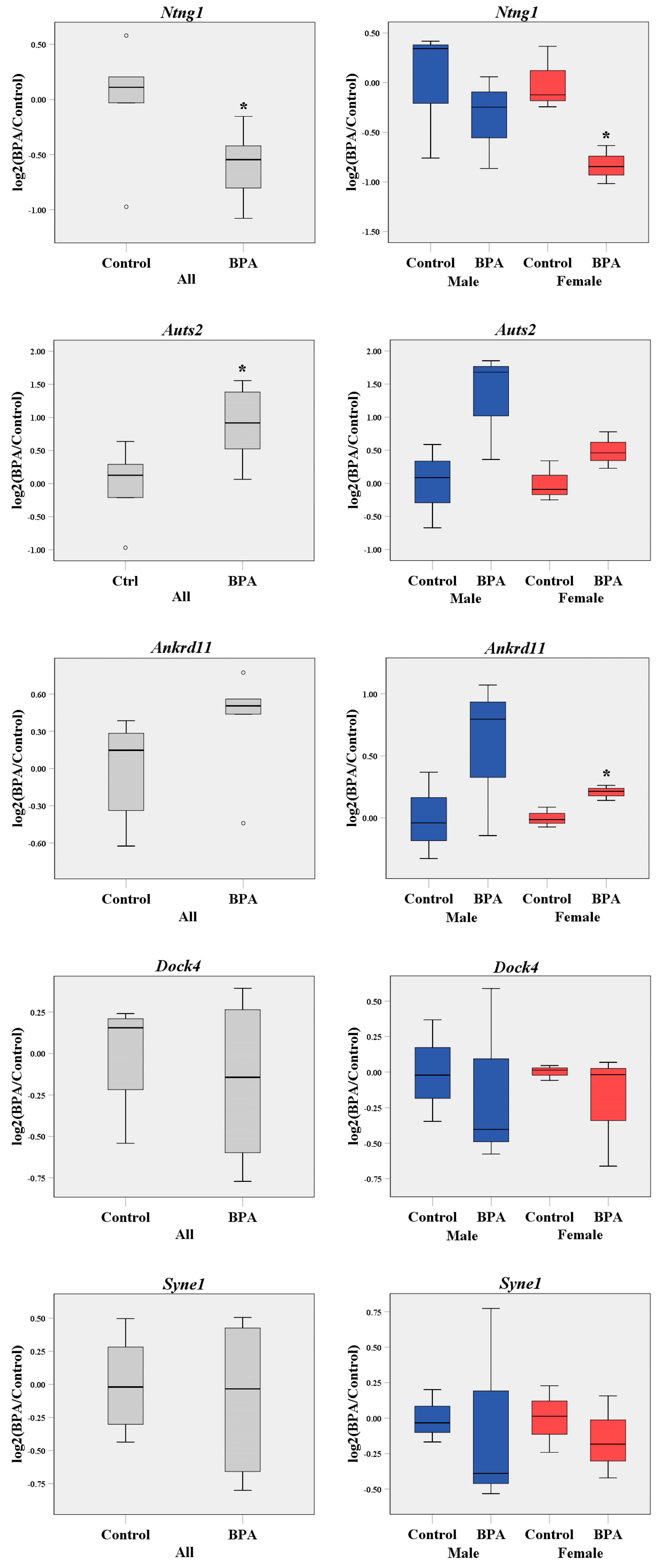
2.1. Prenatal BPA Exposure Disrupts the Transcriptome Profiles of the Offspring’s Prefrontal Cortex in a Sex-Dependent Manner
2.2. BPA-Responsive Genes in the Prefrontal Cortex Are Associated with ASD and Related Neurological Functions and Pathways
2.3. Known ASD Candidate Genes Are Significantly Enriched in the Lists of BPA-Responsive Genes in the Prefrontal Cortex
2.4. BPA-Responsive Genes Are Significantly Associated with DEGs in Postmortem Brain Tissues from ASD Cases
2.5. Identification of Upstream Regulators of DEGs in Response to Prenatal BPA Exposure
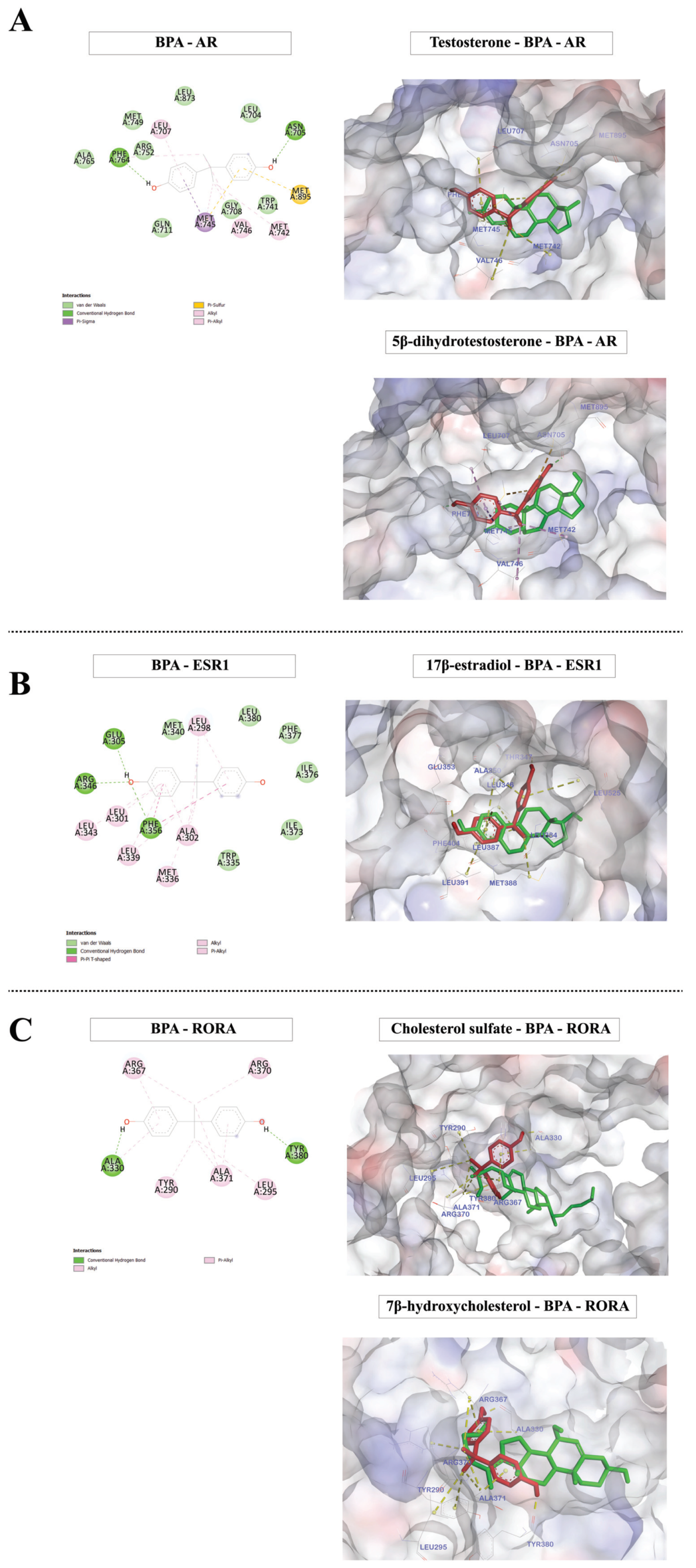
2.6. Molecular Docking Analysis of BPA and ASD-Related Transcription Factors of Which the Targets Are Over-Represented among BPA-Responsive Genes
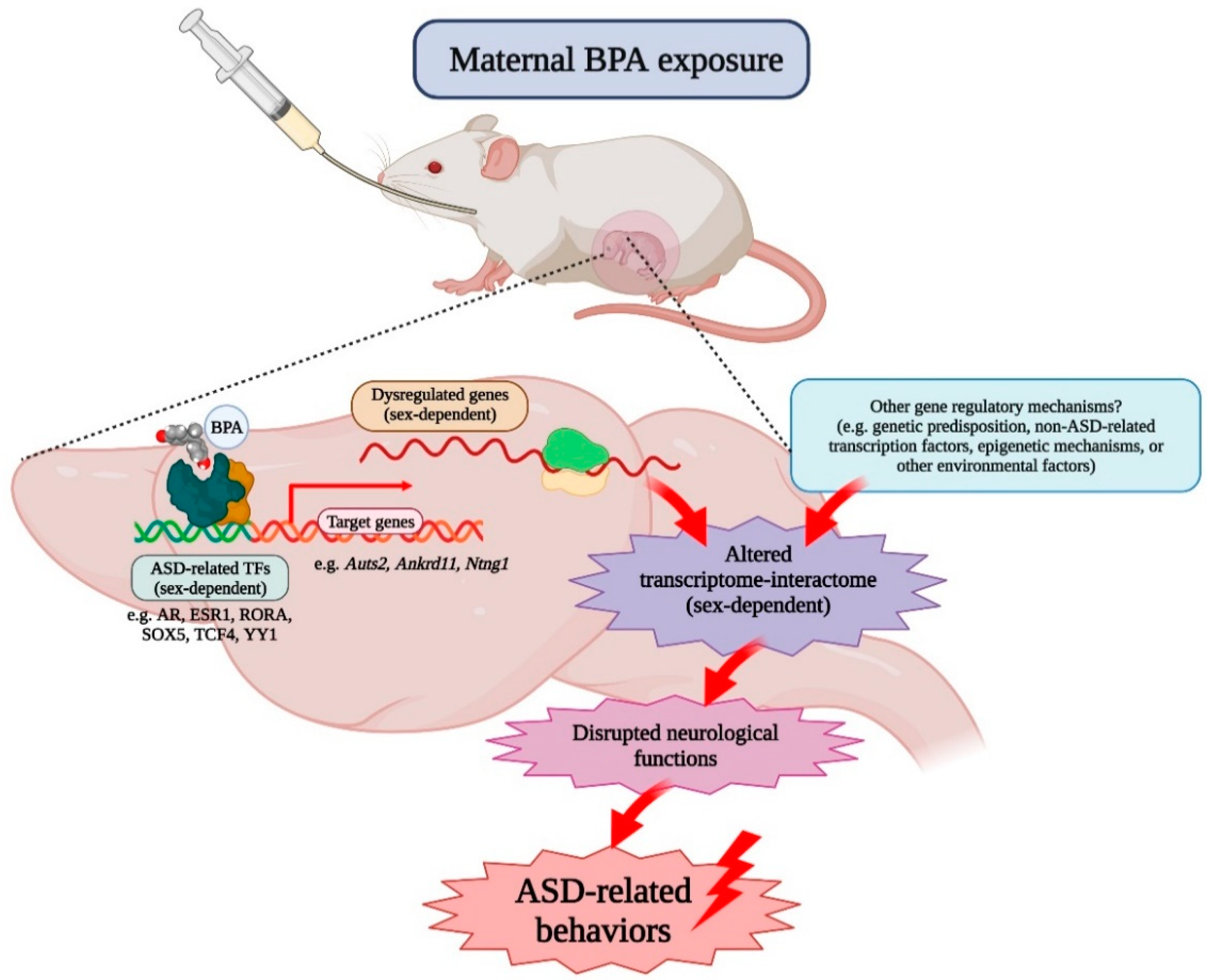
3. Discussion
4. Materials and Methods
4.1. Animal Husbandry and Treatment
4.2. Tissue Dissection
4.3. RNA Isolation
4.4. Transcriptome Profiling Analysis
4.5. qRT-PCR Analysis
4.6. Prediction of Biological Functions, Disorders, Canonical Pathways, and Interactome Networks Associated with DEGs
4.7. Transcriptome Profiling Analysis of Postmortem Brain Tissues from ASD and Unaffected Individuals
4.8. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Pub: Washington, DC, USA, 2013. [Google Scholar]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huguet, G.; Ey, E.; Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genom. Hum. Genet. 2013, 14, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Somogyi, E.; Coulon, N.; Kermarrec, S.; Cohen, D.; Bronsard, G.; Bonnot, O.; Weismann-Arcache, C.; Botbol, M.; Lauth, B.; et al. Gene × Environment Interactions in Autism Spectrum Disorders: Role of Epigenetic Mechanisms. Front. Psychiatry 2014, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosa, A.; Shu, H.; Sarachana, T.; Hu, V.W. Are endocrine disrupting compounds environmental risk factors for autism spectrum disorder? Horm. Behav. 2018, 101, 13–21. [Google Scholar] [CrossRef]
- Pichitpunpong, C.; Thongkorn, S.; Kanlayaprasit, S.; Yuwattana, W.; Plaingam, W.; Sangsuthum, S.; Aizat, W.M.; Baharum, S.N.; Tencomnao, T.; Hu, V.W.; et al. Phenotypic subgrouping and multi-omics analyses reveal reduced diazepam-binding inhibitor (DBI) protein levels in autism spectrum disorder with severe language impairment. PLoS ONE 2019, 14, e0214198. [Google Scholar] [CrossRef] [Green Version]
- Saeliw, T.; Tangsuwansri, C.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Integrated genome-wide Alu methylation and transcriptome profiling analyses reveal novel epigenetic regulatory networks associated with autism spectrum disorder. Mol. Autism 2018, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangsuwansri, C.; Saeliw, T.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Investigation of epigenetic regulatory networks associated with autism spectrum disorder (ASD) by integrated global LINE-1 methylation and gene expression profiling analyses. PLoS ONE 2018, 13, e0201071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellanger, M.; Demeneix, B.; Grandjean, P.; Zoeller, R.T.; Trasande, L. Neurobehavioral deficits, diseases, and associated costs of exposure to endocrine-disrupting chemicals in the European Union. J. Clin. Endocrinol. Metab. 2015, 100, 1256–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, M.; Ghisari, M.; Kjeldsen, L.; Wielsøe, M.; Nørgaard-Pedersen, B.; Mortensen, E.L.; Abdallah, M.W.; Bonefeld-Jørgensen, E.C. Autism spectrum disorders, endocrine disrupting compounds, and heavy metals in amniotic fluid: A case-control study. Mol. Autism 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Romdalvik, J.; Ramanujam, V.M.S.; Legator, M.S. Mercury, lead, and zinc in baby teeth of children with autism versus controls. J. Toxicol. Environ. Health Part A 2007, 70, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Toxicological status of children with autism vs. neurotypical children and the association with autism severity. Biol. Trace Elem. Res. 2013, 151, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; Kaur, B.; Lawson, A.; Ebert, J.; Nahhas, R.J.E.D. Secondhand smoke exposure is associated with autism spectrum disorder in US males but not in females: Results from the National Survey on Children’s Health. Environ. Dis. 2018, 3, 8. [Google Scholar] [CrossRef]
- Becerra, T.A.; Wilhelm, M.; Olsen, J.; Cockburn, M.; Ritz, B. Ambient air pollution and autism in Los Angeles County, California. Environ. Health Perspect. 2013, 121, 380–386. [Google Scholar] [CrossRef]
- Thongkorn, S.; Kanlayaprasit, S.; Jindatip, D.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Sex differences in the effects of prenatal bisphenol a exposure on genes associated with autism spectrum disorder in the hippocampus. Sci. Rep. 2019, 9, 3038. [Google Scholar] [CrossRef] [PubMed]
- Thongkorn, S.; Kanlayaprasit, S.; Panjabud, P.; Saeliw, T.; Jantheang, T.; Kasitipradit, K.; Sarobol, S.; Jindatip, D.; Hu, V.W.; Tencomnao, T.; et al. Sex differences in the effects of prenatal bisphenol A exposure on autism-related genes and their relationships with the hippocampus functions. Sci. Rep. 2021, 11, 1241. [Google Scholar] [CrossRef]
- Panesar, H.K.; Kennedy, C.L.; Keil Stietz, K.P.; Lein, P.J. Polychlorinated Biphenyls (PCBs): Risk factors for autism spectrum disorder? Toxics 2020, 8, 70. [Google Scholar] [CrossRef]
- Kim, J.I.; Lee, J.; Lee, K.-S.; Lee, Y.A.; Shin, C.H.; Hong, Y.-C.; Kim, B.-N.; Lim, Y.-H. Association of phthalate exposure with autistic traits in children. Environ. Int. 2021, 157, 106775. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I. Polybrominated diphenyl ethers (PBDES) in relation to autism and developmental delay. Epidemiology 2007, 18, S192. [Google Scholar] [CrossRef]
- Rist, S.; Carney Almroth, B.; Hartmann, N.B.; Karlsson, T.M. A critical perspective on early communications concerning human health aspects of microplastics. Sci. Total Environ. 2018, 626, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Campanale, C.; Massarelli, C.; Savino, I.; Locaputo, V.; Uricchio, V.F. A detailed review study on potential effects of microplastics and additives of concern on human health. Int. J. Environ. Res. Public Health 2020, 17, 1212. [Google Scholar] [CrossRef] [Green Version]
- Brotons, J.A.; Olea-Serrano, M.F.; Villalobos, M.; Pedraza, V.; Olea, N. Xenoestrogens released from lacquer coatings in food cans. Environ. Health Perspect. 1995, 103, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Habib, C.M.; Kugel, G. Estrogenicity of resin-based composites and sealants in dentistry. Environ. Health Perspect. 1996, 104, 808. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.V.; Stathis, P.; Permuth, S.F.; Tokes, L.; Feldman, D. Bisphenol-A: An estrogenic substance is released from polycarbonate flasks during autoclaving. Endocrinology 1993, 132, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Konieczna, A.; Rutkowska, A.; Rachoń, D. Health risk of exposure to Bisphenol A (BPA). Roczniki Państwowego Zakładu Higieny 2015, 66, 5–11. [Google Scholar]
- Thayer, K.A.; Doerge, D.R.; Hunt, D.; Schurman, S.H.; Twaddle, N.C.; Churchwell, M.I.; Garantziotis, S.; Kissling, G.E.; Easterling, M.R.; Bucher, J.R.; et al. Pharmacokinetics of bisphenol A in humans following a single oral administration. Environ. Int. 2015, 83, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Welshons, W.V.; Nagel, S.C.; vom Saal, F.S. Large effects from small exposures. III. Endocrine Mechanisms mediating effects of bisphenol a at levels of human exposure. Endocrinology 2006, 147, s56–s69. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Chahoud, I.; Heindel, J.J.; Padmanabhan, V.; Paumgartten, F.J.R.; Schoenfelder, G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to Bisphenol A. Environ. Health Perspect. 2010, 118, 1055–1070. [Google Scholar] [CrossRef] [Green Version]
- Braniste, V.; Audebert, M.; Zalko, D.; Houdeau, E. Bisphenol A in the gut: Another break in the wall. In Multi-System Endocrine Disruption; Bourguignon, J.-P., Jégou, B., Kerdelhué, B., Toppari, J., Christen, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 127–144. [Google Scholar]
- Nishikawa, M.; Iwano, H.; Yanagisawa, R.; Koike, N.; Inoue, H.; Yokota, H. Placental transfer of conjugated Bisphenol A and subsequent reactivation in the rat fetus. Environ. Health Perspect. 2010, 118, 1196–1203. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Henare, K.; Thorstensen, E.B.; Ponnampalam, A.P.; Mitchell, M.D. Transfer of bisphenol A across the human placenta. Am. J. Obstet. Gynecol. 2010, 202, 393.e391–393.e397. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, E.; van Schijndel, M.D.M.; Voulgaris, D.; Di Criscio, M.; Ramsbottom, K.A.; Rigden, D.J.; Herland, A.; Rüegg, J. Bisphenol A inhibits the transporter function of the blood-brain barrier by directly interacting with the ABC transporter breast cancer resistance protein (BCRP). Int. J. Mol. Sci. 2021, 22, 5534. [Google Scholar] [CrossRef]
- Sun, Y.; Nakashima, M.N.; Takahashi, M.; Kuroda, N.; Nakashima, K. Determination of Bisphenol A in rat brain by microdialysis and column switching high-performance liquid chromatography with fluorescence detection. Biomed. Chromatogr. 2002, 16, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Domoradzki, J.Y.; Pottenger, L.H.; Thornton, C.M.; Hansen, S.C.; Card, T.L.; Markham, D.A.; Dryzga, M.D.; Shiotsuka, R.N.; Waechter, J.M., Jr. Metabolism and pharmacokinetics of Bisphenol A (BPA) and the embryo-fetal distribution of BPA and BPA-monoglucuronide in CD sprague-dawley rats at three gestational stages. Toxicol. Sci. 2003, 76, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef]
- Liao, C.; Kannan, K. Widespread occurrence of Bisphenol A in paper and paper products: Implications for human exposure. Environ. Sci. Technol. 2011, 45, 9372–9379. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Ye, X.; Wong, L.-Y.; Reidy, J.A.; Needham, L.L. Exposure of the US population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.B.; Bilenberg, N.; Timmermann, C.A.G.; Jensen, R.C.; Frederiksen, H.; Andersson, A.-M.; Kyhl, H.B.; Jensen, T.K. Prenatal exposure to Bisphenol A and autistic- and ADHD-related symptoms in children aged 2 and5 years from the Odense Child Cohort. Environ. Health 2021, 20, 24. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Wu, Y.; Zhao, Y.; Luo, F.; Li, S.; Yang, L.; Moez, E.K.; Dinu, I.; Martin, J.W. Bisphenol A metabolites and Bisphenol S in paired maternal and cord serum. Environ. Sci. Technol. 2017, 51, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Edlow, A.G.; Chen, M.; Smith, N.A.; Lu, C.; McElrath, T.F. Fetal bisphenol A exposure: Concentration of conjugated and unconjugated bisphenol A in amniotic fluid in the second and third trimesters. Reprod. Toxicol. 2012, 34, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schönfelder, G.; Wittfoht, W.; Hopp, H.; Talsness, C.E.; Paul, M.; Chahoud, I. Parent Bisphenol A accumulation in the human maternal-fetal-placental unit. Environ. Health Perspect. 2002, 110, A703–A707. [Google Scholar] [CrossRef] [PubMed]
- Todaka, E.; Mori, C. Necessity to establish new risk assessment and risk communication for human fetal exposure to multiple endocrine disruptors in Japan. Congenit. Anom. 2002, 42, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kuruto-Niwa, R.; Tateoka, Y.; Usuki, Y.; Nozawa, R. Measurement of Bisphenol A concentrations in human colostrum. Chemosphere 2007, 66, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Kuklenyik, Z.; Needham, L.L.; Calafat, A.M. Measuring environmental phenols and chlorinated organic chemicals in breast milk using automated on-line column-switching–high performance liquid chromatography–isotope dilution tandem mass spectrometry. J. Chromatogr. B 2006, 831, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Kondolot, M.; Ozmert, E.N.; Ascı, A.; Erkekoglu, P.; Oztop, D.B.; Gumus, H.; Kocer-Gumusel, B.; Yurdakok, K. Plasma phthalate and bisphenol a levels and oxidant-antioxidant status in autistic children. Environ. Toxicol. Pharmacol. 2016, 43, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kardas, F.; Bayram, A.K.; Demirci, E.; Akin, L.; Ozmen, S.; Kendirci, M.; Canpolat, M.; Oztop, D.B.; Narin, F.; Gumus, H.; et al. Increased serum phthalates (MEHP, DEHP) and Bisphenol A concentrations in children with autism spectrum disorder: The role of endocrine disruptors in autism etiopathogenesis. J. Child. Neurol. 2016, 31, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.P.; Schluter, M.D.; Steer, R.A.; Guo, L.; Ming, X. Bisphenol A exposure in children with autism spectrum disorders. Autism Res. 2015, 8, 272–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United States Food and Drug Administration. 2014 Updated Safety Assessment of Bisphenol A (BPA) for Use in Food Contact Applications; The United States Food and Drug Administration: Silver Spring, MD, USA, 2014.
- Prins, G.S.; Hu, W.-Y.; Xie, L.; Shi, G.-B.; Hu, D.-P.; Birch, L.; Bosland, M.C. Evaluation of Bisphenol A (BPA) exposures on prostate stem cell homeostasis and prostate cancer risk in the NCTR-sprague-dawley rat: An NIEHS/FDA CLARITY-BPA Consortium Study. Environ. Health Perspect. 2018, 126, 117001. [Google Scholar] [CrossRef] [PubMed]
- Lejonklou, M.H.; Dunder, L.; Bladin, E.; Pettersson, V.; Rönn, M.; Lind, L.; Waldén, T.B.; Lind, P.M. Effects of low-dose developmental Bisphenol A exposure on metabolic parameters and gene expression in male and female Fischer 344 rat offspring. Environ. Health Perspect. 2017, 125, 067018. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Miyazaki, W.; Koibuchi, N.; Katoh, T. The effects of low-dose Bisphenol A and Bisphenol F on neural differentiation of a fetal brain-derived neural progenitor cell line. Front. Endocrinol. 2018, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekizawa, J. Low-dose effects of bisphenol A: A serious threat to human health? J. Toxicol. Sci. 2008, 33, 389–403. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, L.N.; Ehrlich, S.; Belcher, S.M.; Ben-Jonathan, N.; Dolinoy, D.C.; Hugo, E.R.; Hunt, P.A.; Newbold, R.R.; Rubin, B.S.; Saili, K.S.; et al. Low dose effects of bisphenol A. Endocr. Disruptors 2013, 1, e26490. [Google Scholar] [CrossRef]
- Schirmer, E.; Schuster, S.; Machnik, P. Bisphenols exert detrimental effects on neuronal signaling in mature vertebrate brains. Commun. Biology 2021, 4, 465. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Yaoi, T.; Fushiki, S. Bisphenol A, an endocrine-disrupting chemical, and brain development. Neuropathology 2012, 32, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, J.S.; Yin, L.; Measel, E.; Liang, S.; Yu, X. Effects of bisphenol A and its analogs on reproductive health: A mini review. Reprod. Toxicol. 2018, 79, 96–123. [Google Scholar] [CrossRef]
- Xu, J.; Huang, G.; Guo, T.L. Developmental Bisphenol A exposure modulates immune-related diseases. Toxics 2016, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Rui, M.; Nie, Y.; Lu, G. Influence of gastrointestinal tract on metabolism of bisphenol A as determined by in vitro simulated system. J. Hazard. Mater. 2018, 355, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Tagami, T.; Akamizu, T.; Usui, T.; Saijo, M.; Kanamoto, N.; Hataya, Y.; Shimatsu, A.; Kuzuya, H.; Nakao, K. Thyroid hormone action is disrupted by Bisphenol A as an antagonist. J. Clin. Endocrinol. Metab. 2002, 87, 5185–5190. [Google Scholar] [CrossRef] [PubMed]
- Thoene, M.; Rytel, L.; Dzika, E.; Włodarczyk, A.; Kruminis-Kaszkiel, E.; Konrad, P.; Wojtkiewicz, J. Bisphenol A causes liver damage and selectively alters the neurochemical coding of intrahepatic parasympathetic nerves in juvenile porcine models under physiological conditions. Int. J. Mol. Sci. 2017, 18, 2726. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, H.-S. Impact of Bisphenol A on the cardiovascular system—Epidemiological and experimental evidence and molecular mechanisms. Int. J. Environ. Res. Public Health 2014, 11, 8399–8413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Parra, E.; Herrero, J.A.; Elewa, U.; Bosch, R.J.; Arduán, A.O.; Egido, J. Bisphenol A in chronic kidney disease. Int. J. Nephrol. 2013, 2013, 437857. [Google Scholar] [CrossRef]
- Ben-Jonathan, N.; Hugo, E.R.; Brandebourg, T.D. Effects of Bisphenol A on adipokine release from human adipose tissue: Implications for the metabolic syndrome. Mol. Cell. Endocrinol. 2009, 304, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Kimura, E.; Matsuyoshi, C.; Miyazaki, W.; Benner, S.; Hosokawa, M.; Yokoyama, K.; Kakeyama, M.; Tohyama, C. Prenatal exposure to bisphenol A impacts neuronal morphology in the hippocampal CA1 region in developing and aged mice. Arch. Toxicol. 2016, 90, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolstenholme, J.T.; Rissman, E.F.; Connelly, J.J. The role of Bisphenol A in shaping the brain, epigenome and behavior. Horm. Behav. 2011, 59, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Stoner, R.; Chow, M.L.; Boyle, M.P.; Sunkin, S.M.; Mouton, P.R.; Roy, S.; Wynshaw-Boris, A.; Colamarino, S.A.; Lein, E.S.; Courchesne, E. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 2014, 370, 1209–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Itoh, K.; Yaoi, T.; Fujiwara, Y.; Sugimoto, T.; Fushiki, S. Murine neocortical histogenesis is perturbed by prenatal exposure to low doses of Bisphenol A. J. Neurosci. Res. 2006, 84, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, J.T.; Edwards, M.; Shetty, S.R.J.; Gatewood, J.D.; Taylor, J.A.; Rissman, E.F.; Connelly, J.J. Gestational exposure to Bisphenol A produces transgenerational changes in behaviors and gene expression. Endocrinology 2012, 153, 3828–3838. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, U.; Tinsley, B.; Sen, Y.; Stein, J.; Palacios, Y.; Ceballos, A.; Welch, C.; Nzenkue, K.; Penn, A.; Murphy, L.; et al. Exposure to Bisphenol A differentially impacts neurodevelopment and behavior in Drosophila melanogaster from distinct genetic backgrounds. NeuroToxicology 2021, 82, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Nolte, E.L.R.; Wang, Y.; Margolis, A.E.; Calafat, A.M.; Wang, S.; Garcia, W.; Hoepner, L.A.; Peterson, B.S.; Rauh, V.; et al. Bisphenol A exposure and symptoms of anxiety and depression among inner city children at 10–12 years of age. Environ. Res. 2016, 151, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Rochester, J.R.; Bolden, A.L.; Kwiatkowski, C.F. Prenatal exposure to bisphenol A and hyperactivity in children: A systematic review and meta-analysis. Environ. Int. 2018, 114, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Tewar, S.; Auinger, P.; Braun, J.M.; Lanphear, B.; Yolton, K.; Epstein, J.N.; Ehrlich, S.; Froehlich, T.E. Association of Bisphenol A exposure and attention-deficit/hyperactivity disorder in a national sample of U.S. children. Environ. Res. 2016, 150, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.H.; Tanimura, Y.; Lee, L.W.; Bodfish, J.W. Animal models of restricted repetitive behavior in autism. Behav. Brain Res. 2007, 176, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olexová, L.; Štefánik, P.; Kršková, L. Increased anxiety-like behaviour and altered GABAergic system in the amygdala and cerebellum of VPA rats—An animal model of autism. Neurosci. Lett. 2016, 629, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Przewłocki, R. Behavioral alterations in rats prenatally exposed to valproic acid: Animal model of autism. Neuropsychopharmacology 2005, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Seese, R.R.; Maske, A.R.; Lynch, G.; Gall, C.M. Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology 2014, 39, 1664–1673. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.; Sánchez, P.; Torres, J.M.; Ortega, E. Effects of adult exposure to Bisphenol A on genes involved in the physiopathology of rat prefrontal cortex. PLoS ONE 2013, 8, e0073584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arambula, S.E.; Belcher, S.M.; Planchart, A.; Turner, S.D.; Patisaul, H.B. Impact of low dose oral exposure to Bisphenol A (BPA) on the neonatal rat hypothalamic and hippocampal transcriptome: A CLARITY-BPA consortium study. Endocrinology 2016, 157, 3856–3872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathisen, G.H.; Yazdani, M.; Rakkestad, K.E.; Aden, P.K.; Bodin, J.; Samuelsen, M.; Nygaard, U.C.; Goverud, I.L.; Gaarder, M.; Løberg, E.M.; et al. Prenatal exposure to Bisphenol A interferes with the development of cerebellar granule neurons in mice and chicken. Int. J. Dev. Neurosci. 2013, 31, 762–769. [Google Scholar] [CrossRef]
- Arambula, S.E.; Jima, D.; Patisaul, H.B. Prenatal bisphenol A (BPA) exposure alters the transcriptome of the neonate rat amygdala in a sex-specific manner: A CLARITY-BPA consortium study. NeuroToxicology 2018, 65, 207–220. [Google Scholar] [CrossRef]
- Bicks, L.K.; Koike, H.; Akbarian, S.; Morishita, H. Prefrontal cortex and social cognition in mouse and man. Front. Psychol. 2015, 6, 1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Raine, A. Prefrontal structural and functional brain imaging findings in antisocial, violent, and psychopathic individuals: A meta-analysis. Psychiatry Res. Neuroimaging 2009, 174, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Parker, A.; Derrington, A.; Blakemore, C.; Miller, E.K.; Freedman, D.J.; Wallis, J.D. The prefrontal cortex: Categories, concepts and cognition. Philos. Trans. R. Soc. B Biol. Sci. 2002, 357, 1123–1136. [Google Scholar] [CrossRef] [Green Version]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef]
- Pitskel, N.B.; Bolling, D.Z.; Kaiser, M.D.; Pelphrey, K.A.; Crowley, M.J. Neural systems for cognitive reappraisal in children and adolescents with autism spectrum disorder. Dev. Cogn. Neurosci. 2014, 10, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Sarachana, T.; Xu, M.; Wu, R.-C.; Hu, V.W. Sex hormones in autism: Androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. PloS ONE 2011, 6, e0017116. [Google Scholar] [CrossRef]
- Hu, V.W.; Sarachana, T.; Sherrard, R.M.; Kocher, K.M. Investigation of sex differences in the expression of RORA and its transcriptional targets in the brain as a potential contributor to the sex bias in autism. Mol. Autism 2015, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarachana, T.; Hu, V.W. Differential recruitment of coregulators to the RORA promoter adds another layer of complexity to gene (dys) regulation by sex hormones in autism. Mol. Autism 2013, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Bando, Y.; Sato, K. Involvement of netrins and their receptors in neuronal migration in the cerebral cortex. Front. Cell Dev. Biol. 2021, 8, 590009. [Google Scholar] [CrossRef]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell. Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lei, K.; Yuan, X.; Wu, X.; Zhuang, Y.; Xu, T.; Xu, R.; Han, M. SUN1/2 and Syne/Nesprin-1/2 Complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 2009, 64, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, D.; Voronova, A.; Zander, M.A.; Cancino, G.I.; Bramall, A.; Krause, M.P.; Abad, C.; Tekin, M.; Neilsen, P.M.; Callen, D.F.; et al. Ankrd11 Is a chromatin regulator involved in autism that is essential for neural development. Dev. Cell 2015, 32, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ka, M.; Kim, W.-Y. ANKRD11 associated with intellectual disability and autism regulates dendrite differentiation via the BDNF/TrkB signaling pathway. Neurobiol. Dis. 2018, 111, 138–152. [Google Scholar] [CrossRef]
- Xiao, Y.; Peng, Y.; Wan, J.; Tang, G.; Chen, Y.; Tang, J.; Ye, W.-C.; Ip, N.Y.; Shi, L. The atypical guanine nucleotide exchange factor Dock4 regulates neurite differentiation through modulation of Rac1 GTPase and actin dynamics. J. Biol. Chem. 2013, 288, 20034–20045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Cang, X.; Liu, J. Molecular mechanism of Bisphenol A on androgen receptor antagonism. Toxicol. Vitr. 2019, 61, 104621. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Zhang, Y.; Niu, Y.; Yao, X.; Liu, H. The molecular mechanism of bisphenol A (BPA) as an endocrine disruptor by interacting with nuclear receptors: Insights from molecular dynamics (MD) simulations. PLoS ONE 2015, 10, e0120330. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.A.; Sheikh, I.A. Endocrine disruption: Molecular interactions of environmental bisphenol contaminants with thyroid hormone receptor and thyroxine-binding globulin. Toxicol. Ind. Health 2020, 36, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The human transcription factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matys, V.; Fricke, E.; Geffers, R.; Gössling, E.; Haubrock, M.; Hehl, R.; Hornischer, K.; Karas, D.; Kel, A.E.; Kel-Margoulis, O.V.; et al. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31, 374–378. [Google Scholar] [CrossRef]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef] [Green Version]
- Sarachana, T.; Hu, V.W. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol. Autism 2013, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallen, J.A.; Schlaeppi, J.-M.; Bitsch, F.; Geisse, S.; Geiser, M.; Delhon, I.; Fournier, B. X-Ray Structure of the hRORα LBD at 1.63 Å: Structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORα. Structure 2002, 10, 1697–1707. [Google Scholar] [CrossRef] [Green Version]
- Ronald, A.; Hoekstra, R.A. Autism spectrum disorders and autistic traits: A decade of new twin studies. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2011, 156, 255–274. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef]
- John, N.; Constantino, M.D.; Richard, D.; Todd, M.D. Genetic structure of reciprocal social behavior. Am. J. Psychiatry 2000, 157, 2043–2045. [Google Scholar] [CrossRef]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Edelson, L.R.; Saudino, K.J. Genetic and environmental influences on autistic-like behaviors in 2-year-old twins. Behav. Genet. 2009, 39, 255. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, R.A.; Bartels, M.; Verweij, C.J.H.; Boomsma, D.I. Heritability of autistic traits in the general population. Arch. Pediatrics Adolesc. Med. 2007, 161, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Zou, X.; Deng, H.; Li, J.; Tang, C.; Wang, X.; Guo, X. The relationship among genetic heritability, environmental effects, and autism spectrum disorders. J. Child Neurol. 2015, 30, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Miodovnik, A.; Engel, S.M.; Zhu, C.; Ye, X.; Soorya, L.V.; Silva, M.J.; Calafat, A.M.; Wolff, M.S. Endocrine disruptors and childhood social impairment. NeuroToxicology 2011, 32, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.F.; Kobrosly, R.W.; Barrett, E.S.; Thurston, S.W.; Calafat, A.M.; Weiss, B.; Stahlhut, R.; Yolton, K.; Swan, S.H. Prenatal Bisphenol A exposure and maternally reported behavior in boys and girls. NeuroToxicology 2014, 45, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Harley, K.G.; Gunier, R.B.; Kogut, K.; Johnson, C.; Bradman, A.; Calafat, A.M.; Eskenazi, B. Prenatal and early childhood bisphenol A concentrations and behavior in school-aged children. Environ. Res. 2013, 126, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, F.; Vishnevetsky, J.; Herbstman, J.B.; Calafat, A.M.; Xiong, W.; Rauh, V.; Wang, S. Prenatal Bisphenol A exposure and child behavior in an inner-city cohort. Environ. Health Perspect. 2012, 120, 1190–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roen, E.L.; Wang, Y.; Calafat, A.M.; Wang, S.; Margolis, A.; Herbstman, J.; Hoepner, L.A.; Rauh, V.; Perera, F.P. Bisphenol A exposure and behavioral problems among inner city children at 7–9 years of age. Environ. Res. 2015, 142, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippat, C.; Nakiwala, D.; Calafat, A.M.; Botton, J.; Agostini, M.D.; Heude, B.; Slama, R. Prenatal Exposure to nonpersistent endocrine disruptors and behavior in boys at 3 and 5 years. Environ. Health Perspect. 2017, 125, 097014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.-H.; Bae, S.; Kim, B.-N.; Shin, C.H.; Lee, Y.A.; Kim, J.I.; Hong, Y.-C. Prenatal and postnatal bisphenol A exposure and social impairment in 4-year-old children. Environ. Health 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, J.M.; Yolton, K.; Dietrich, K.N.; Hornung, R.; Ye, X.; Calafat, A.M.; Lanphear, B.P. Prenatal Bisphenol a exposure and early childhood behavior. Environ. Health Perspect. 2009, 117, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Kalkbrenner, A.E.; Calafat, A.M.; Yolton, K.; Ye, X.; Dietrich, K.N.; Lanphear, B.P. Impact of early-life Bisphenol A exposure on behavior and executive function in children. Pediatrics 2011, 128, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacy, S.L.; Papandonatos, G.D.; Calafat, A.M.; Chen, A.; Yolton, K.; Lanphear, B.P.; Braun, J.M. Early life Bisphenol A exposure and neurobehavior at 8 years of age: Identifying windows of heightened vulnerability. Environ. Int. 2017, 107, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.P.A.; Basson, M.A. The neuroanatomy of autism—A developmental perspective. J. Anat. 2017, 230, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmen, S.J.M.C.; van Engeland, H.; Hof, P.R.; Schmitz, C. Neuropathological findings in autism. Brain 2004, 127, 2572–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukjamnong, S.; Thongkorn, S.; Kanlayaprasit, S.; Saeliw, T.; Hussem, K.; Warayanon, W.; Hu, V.W.; Tencomnao, T.; Sarachana, T. Prenatal exposure to bisphenol A alters the transcriptome-interactome profiles of genes associated with Alzheimer’s disease in the offspring hippocampus. Sci. Rep. 2020, 10, 9487. [Google Scholar] [CrossRef]
- Volkmar, F.R.; Lord, C.; Bailey, A.; Schultz, R.T.; Klin, A. Autism and pervasive developmental disorders. J. Child Psychol. Psychiatry 2004, 45, 135–170. [Google Scholar] [CrossRef]
- Licari, M.K.; Alvares, G.A.; Varcin, K.; Evans, K.L.; Cleary, D.; Reid, S.L.; Glasson, E.J.; Bebbington, K.; Reynolds, J.E.; Wray, J.; et al. Prevalence of motor difficulties in autism spectrum disorder: Analysis of a population-based cohort. Autism Res. 2020, 13, 298–306. [Google Scholar] [CrossRef]
- Zheng, Z.; Zheng, P.; Zou, X. Association between schizophrenia and autism spectrum disorder: A systematic review and meta-analysis. Autism Res. 2018, 11, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Sahin, M. Autism spectrum disorder and epileptic encephalopathy: Common causes, many questions. J. Neurodev. Disord. 2017, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, B.A.; El Hokayem, J.; Artimovich, E.; Garcia-Serje, C.; Phillips, A.W.; Van Booven, D.; Nestor, J.E.; Wang, L.; Cuccaro, M.L.; Vance, J.M.; et al. Convergent pathways in idiopathic autism revealed by time course transcriptomic analysis of patient-derived neurons. Sci. Rep. 2018, 8, 8423. [Google Scholar] [CrossRef] [Green Version]
- Rademacher, S.; Eickholt, B.J. PTEN in autism and neurodevelopmental disorders. Cold Spring Harb. Perspect. Med. 2019, 9, a036780. [Google Scholar] [CrossRef] [Green Version]
- Jung, N.H.; Janzarik, W.G.; Delvendahl, I.; Münchau, A.; Biscaldi, M.; Mainberger, F.; Bäumer, T.; Rauh, R.; Mall, V. Impaired induction of long-term potentiation-like plasticity in patients with high-functioning autism and Asperger syndrome. Dev. Med. Child Neurol. 2013, 55, 83–89. [Google Scholar] [CrossRef]
- Hansel, C. Deregulation of synaptic plasticity in autism. Neurosci. Lett. 2019, 688, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.; Hyun, S.-A.; Ko, M.Y.; Kim, H.R.; Rho, J.; Kim, K.K.; Kim, W.-Y.; Ka, M. Maternal Bisphenol A (BPA) exposure alters cerebral cortical morphogenesis and synaptic function in mice. Cereb. Cortex 2021, 31, 5598–5612. [Google Scholar] [CrossRef]
- Schafer, S.T.; Paquola, A.C.M.; Stern, S.; Gosselin, D.; Ku, M.; Pena, M.; Kuret, T.J.M.; Liyanage, M.; Mansour, A.A.; Jaeger, B.N.; et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 2019, 22, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Belinson, H.; Tian, Y.; Freitas, B.C.; Fu, C.; Vadodaria, K.C.; Beltrao-Braga, P.C.; Trujillo, C.A.; Mendes, A.P.D.; Padmanabhan, K.; et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 2017, 22, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Quartier, A.; Chatrousse, L.; Redin, C.; Keime, C.; Haumesser, N.; Maglott-Roth, A.; Brino, L.; Le Gras, S.; Benchoua, A.; Mandel, J.-L.; et al. Genes and pathways regulated by androgens in human neural cells, potential candidates for the male excess in autism spectrum disorder. Biol. Psychiatry 2018, 84, 239–252. [Google Scholar] [CrossRef]
- Baron-Cohen, S.; Tsompanidis, A.; Auyeung, B.; Nørgaard-Pedersen, B.; Hougaard, D.M.; Abdallah, M.; Cohen, A.; Pohl, A. Foetal oestrogens and autism. Mol. Psychiatry 2020, 25, 2970–2978. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Novick, P.; Ferro-Novick, S. ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell Biol. 2012, 14, 707–716. [Google Scholar] [CrossRef]
- Breuss, M.W.; Nguyen, A.; Song, Q.; Nguyen, T.; Stanley, V.; James, K.N.; Musaev, D.; Chai, G.; Wirth, S.A.; Anzenberg, P.; et al. Mutations in LNPK, Encoding the endoplasmic reticulum junction stabilizer lunapark, cause a recessive neurodevelopmental syndrome. Am. J. Hum. Genet. 2018, 103, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, D.A.; Huang, Y.-C.; Swigut, T.; Mirick, A.L.; Garcia-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Potts, M.B.; Siu, J.J.; Price, J.D.; Salinas, R.D.; Cho, M.J.; Ramos, A.D.; Hahn, J.; Margeta, M.; Oldham, M.C.; Lim, D.A. Analysis of Mll1 Deficiency identifies neurogenic transcriptional modules and brn4 as a factor for direct astrocyte-to-neuron reprogramming. Neurosurgery 2014, 75, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Jakovcevski, M.; Ruan, H.; Shen, E.Y.; Dincer, A.; Javidfar, B.; Ma, Q.; Peter, C.J.; Cheung, I.; Mitchell, A.C.; Jiang, Y.; et al. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J. Neurosci. 2015, 35, 5097–5108. [Google Scholar] [CrossRef] [Green Version]
- Shen, E.Y.; Jiang, Y.; Javidfar, B.; Kassim, B.; Loh, Y.-H.E.; Ma, Q.; Mitchell, A.C.; Pothula, V.; Stewart, A.F.; Ernst, P.; et al. Neuronal deletion of Kmt2a/Mll1 Histone methyltransferase in ventral striatum is associated with defective spike-timing-dependent striatal synaptic plasticity, altered response to dopaminergic drugs, and increased anxiety. Neuropsychopharmacology 2016, 41, 3103–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.J.S.; Cytrynbaum, C.; Hoang, N.; Ambrozewicz, P.M.; Weksberg, R.; Drmic, I.; Ritzema, A.; Schachar, R.; Walker, S.; Uddin, M.; et al. Expanding the neurodevelopmental phenotypes of individuals with de novo KMT2A variants. NPJ Genom. Med. 2019, 4, 9. [Google Scholar] [CrossRef]
- Wilson, N.H.; Key, B. Neogenin interacts with RGMa and Netrin-1 to guide axons within the embryonic vertebrate forebrain. Dev. Biol. 2006, 296, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, D.P.; Cole, S.J.; Hammond, A.; Seaman, C.; Cooper, H.M. Characterization of neogenin-expressing neural progenitor populations and migrating neuroblasts in the embryonic mouse forebrain. Neuroscience 2006, 142, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Sun, X.-D.; Zhao, L.; Lee, D.-H.; Hu, J.-X.; Tang, F.-L.; Pan, J.-X.; Mei, L.; Zhu, X.-J.; Xiong, W.-C. Neogenin, a regulator of adult hippocampal neurogenesis, prevents depressive-like behavior. Cell Death Dis. 2018, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, W.-K.; Lam, C.-W.; Gao, W.-W.; Vincent Tang, H.-M.; Jin, D.-Y.; Mak, C.M. Unmasking a novel disease gene NEO1 associated with autism spectrum disorders by a hemizygous deletion on chromosome 15 and a functional polymorphism. Behav. Brain Res. 2016, 300, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Ho, W.-H.; Gurney, A.; Rosenthal, A. The netrin-G1 ligand NGL-1 promotes the outgrowth of thalamocortical axons. Nat. Neurosci. 2003, 6, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.L.W.; Correia, J.P.; Kennedy, T.E. Netrins: Versatile extracellular cues with diverse functions. Development 2011, 138, 2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Nakanishi, T.; Ueno, M.; Itohara, S.; Yamashita, T. Netrin-G1 Regulates microglial accumulation along axons and supports the survival of layer V neurons in the postnatal mouse brain. Cell Rep. 2020, 31, 107580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Goto, H.; Akiyoshi-Nishimura, S.; Prosselkov, P.; Sano, C.; Matsukawa, H.; Yaguchi, K.; Nakashiba, T.; Itohara, S. Diversification of behavior and postsynaptic properties by netrin-G presynaptic adhesion family proteins. Mol. Brain 2016, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, I.; Freude, K.; Kübart, S.; Hoffmann, K.; Menzel, C.; Laccone, F.; Firth, H.; Ferguson-Smith, M.A.; Tommerup, N.; Ropers, H.-H.; et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with rett syndrome. Eur. J. Hum. Genet. 2005, 13, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Bedogni, F.; Hodge, R.D.; Nelson, B.R.; Frederick, E.A.; Shiba, N.; Daza, R.A.; Hevner, R.F. Autism susceptibility candidate 2 (Auts2) encodes a nuclear protein expressed in developing brain regions implicated in autism neuropathology. Gene Expr. Patterns 2010, 10, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksenberg, N.; Ahituv, N. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 2013, 29, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagamani, S.C.S.; Erez, A.; Ben-Zeev, B.; Frydman, M.; Winter, S.; Zeller, R.; El-Khechen, D.; Escobar, L.; Stankiewicz, P.; Patel, A.; et al. Detection of copy-number variation in AUTS2 gene by targeted exonic array CGH in patients with developmental delay and autistic spectrum disorders. Eur. J. Hum. Genet. 2013, 21, 343–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhao, D.; Dong, R.; Yang, X.; Zhang, Y.; Tammimies, K.; Uddin, M.; Scherer, S.W.; Gai, Z. De novo exon 1 deletion of AUTS2 gene in a patient with autism spectrum disorder and developmental delay: A case report and a brief literature review. Am. J. Med. Genet. Part A 2015, 167, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-David, E.; Granot-Hershkovitz, E.; Monderer-Rothkoff, G.; Lerer, E.; Levi, S.; Yaari, M.; Ebstein, R.P.; Yirmiya, N.; Shifman, S. Identification of a functional rare variant in autism using genome-wide screen for monoallelic expression. Hum. Mol. Genet. 2011, 20, 3632–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, K.; Yamashiro, K.; Nagai, T.; Shan, W.; Egusa, S.F.; Shimaoka, K.; Kuniishi, H.; Sekiguchi, M.; Go, Y.; Tatsumoto, S.; et al. AUTS2 regulation of synapses for proper synaptic inputs and social communication. iScience 2020, 23, 101183. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemsen, M.H.; Fernandez, B.A.; Bacino, C.A.; Gerkes, E.; de Brouwer, A.P.M.; Pfundt, R.; Sikkema-Raddatz, B.; Scherer, S.W.; Marshall, C.R.; Potocki, L.; et al. Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur. J. Hum. Genet. 2010, 18, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucerzan, S.; Miclea, D.; Lazea, C.; Asavoaie, C.; Kulcsar, A.; Grigorescu-Sido, P. 16q24.3 microduplication in a patient with developmental delay, intellectual disability, short stature, and nonspecific dysmorphic features: Case Report and review of the literature. Front. Pediatr. 2020, 8, 390. [Google Scholar] [CrossRef] [PubMed]
- Crippa, M.; Rusconi, D.; Castronovo, C.; Bestetti, I.; Russo, S.; Cereda, A.; Selicorni, A.; Larizza, L.; Finelli, P. Familial intragenic duplication of ANKRD11 underlying three patients of KBG syndrome. Mol. Cytogenet. 2015, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acconcia, F.; Pallottini, V.; Marino, M. Molecular mechanisms of action of BPA. Dose Response 2015, 13, 1559325815610582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Zhou, W.; Sun, Z. Nuclear receptor corepressors in intellectual disability and autism. Mol. Psychiatry 2020, 25, 2220–2236. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Xu, Y. Nuclear receptor coactivators (NCOAs) and corepressors (NCORs) in the brain. Endocrinology 2020, 161, bqaa083. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, T.; Casingal, C.R.; Chun, S.H.; Shinohara, H.; Hiraoka, K.; Osumi, N. The role of Pax6 in brain development and its impact on pathogenesis of autism spectrum disorder. Brain Res. 2019, 1705, 95–103. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [CrossRef] [PubMed] [Green Version]
- Jin, H.-J.; Kim, J.; Yu, J. Androgen receptor genomic regulation. Transl. Androl. Urol. 2013, 2, 157–177. [Google Scholar] [CrossRef]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spijker, S. Dissection of rodent brain regions. In Neuroproteomics; Humana Press: Totowa, NJ, USA, 2011; pp. 13–26. [Google Scholar]
- Guo, W.; Patzlaff, N.E.; Jobe, E.M.; Zhao, X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat. Protoc. 2012, 7, 2005–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2020, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Howe, E.A.; Sinha, R.; Schlauch, D.; Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 2011, 27, 3209–3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Diseases or Disorders | p-Value (Number of Genes) | ||
|---|---|---|---|
| Both Sexes | Male | Female | |
| Autism or intellectual disability | 4.11 × 10−11 (233) | 1.89 × 10−13 (128) | 2.31 × 10−5 (105) |
| Schizophrenia spectrum disorder | NS | 3.64 × 10−8 (128) | 1.75 × 10−9 (139) |
| Mood disorders | 4.82 × 10−10 (227) | 3.40 × 10−8 (111) | 1.27 × 10−6 (110) |
| Pervasive developmental disorder | 2.05 × 10−7 (107) | 4.92 × 10−5 (50) | NA |
| Disorder of basal ganglia | 1.15 × 10−23 (516) | 1.60 × 10−12 (186) | 1.42 × 10−11 (191) |
| Movement disorders | 4.36 × 10−37 (760) | 5.32 × 10−16 (234) | 1.96 × 10−14 (239) |
| Amyotrophic lateral sclerosis | NA | 1.84 × 10−6 (59) | 3.57 × 10−9 (69) |
| Alzheimer disease | 6.74 × 10−19 (347) | 3.35 × 10−11 (163) | 1.28 × 10−11 (172) |
| Huntington’s disease | 6.54 × 10−19 (388) | 6.67 × 10−9 (126) | 2.30 × 10−10 (137) |
| Syndromic encephalopathy | 3.77 × 10−21 (358) | 1.49 × 10−7 (75) | 5.82 × 10−6 (73) |
| Gene List Category (No. of Genes) | Both Sexes | Male | Female | |||
|---|---|---|---|---|---|---|
| No. of Target Genes Detected in the Rat Frontal Cortex | No. of Overlapping Genes (p-Value) | No. of Target Genes Detected in the Rat Frontal Cortex | No. of Overlapping Genes (p-Value) | No. of Target Genes Detected in the Rat Frontal Cortex | No. of Overlapping Genes (p-Value) | |
| All genes (986) | 835 | 408 (4.44 × 10−3) | 847 | 243 (6.64 × 10−17) | 843 | 228 (1.70 × 10−10) |
| Syndromic (143) | 130 | 66 (0.085) | 133 | 49 (7.32 × 10−8) | 129 | 38 (1.54 × 10−3) |
| Score 1 High confidence (25) | 23 | 10 (0.615) | 23 | 10 (3.27 × 10−3) | 23 | 11 (1.25 × 10−3) |
| Score 2 Strong candidate (59) | 54 | 24 (0.552) | 55 | 18 (4.45 × 10−3) | 55 | 17 (1.77 × 10−2) |
| Score 3 Suggestive evidence (176) | 159 | 70 (0.572) | 163 | 53 (2.11 × 10−6) | 159 | 52 (1.10 × 10−5) |
| Score 4 Minimal evidence (406) | 321 | 162 (1.62 × 10−2) | 322 | 90 (1.70 × 10−6) | 324 | 84 (4.84 × 10−4) |
| Score 5 Hypothesized (157) | 133 | 68 (0.071) | 135 | 28 (0.186) | 136 | 30 (0.164) |
| Studies (Year) | Brain Region | Both Sexes (6284 Genes) | Male (2565 Genes) | Female (2706 Genes) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of DEGs Detected in the Rat Frontal Cortex | No. of Overlapping Genes | p-Value | No. of DEGs Detected in the Rat Frontal Cortex | No. of Overlapping Genes | p-Value | No. of DEGs Detected in the Rat Frontal Cortex | No. of Overlapping Genes | p-Value | ||
| Parikshak et al. (2015) | Frontal and temporal cortex | 954 | 462 | 5.61 × 10−3 | 956 | 221 | 2.71 × 10−6 | 958 | 219 | 2.28 × 10−4 |
| Voineagu et al. (2011) | Frontal cortex (BA9, BA44/45) | 384 | 177 | 0.269 | 383 | 89 | 2.22 × 10−3 | 385 | 86 | 0.029 |
| Chow et al. (2012) | Prefrontal cortex | 71 | 36 | 0.172 | 76 | 24 | 1.94 × 10−3 | 73 | 16 | 0.264 |
| Garbett et. al. (2008) | Temporal cortex | 101 | 58 | 5.72 × 10−3 | 102 | 21 | 0.238 | 101 | 29 | 7.72 × 10−3 |
| Voineagu et al. (2011) | Temporal cortex (BA41/42, 22) | 545 | 248 | 0.318 | 546 | 117 | 8.98 × 10−3 | 539 | 120 | 0.013 |
| Ginsberg et al. (2012) | Occipital lobe (BA19) | 269 | 116 | 0.690 | 270 | 60 | 0.026 | 277 | 56 | 0.245 |
| Ginsberg et al. (2012) | Cerebellum | 749 | 307 | 0.977 | 744 | 150 | 0.029 | 746 | 151 | 0.108 |
| Voineagu et al. (2011) | Cerebellum; vermis | 57 | 24 | 0.685 | 58 | 12 | 0.309 | 58 | 11 | 0.514 |
| Transcription Factors | Both Sexes (6284 Genes) | Male (2565 Genes) | Female (2706 Genes) | |||
|---|---|---|---|---|---|---|
| No. of Target Genes Detected in the Rat Frontal Cortex | No. of Overlapping Genes (p-Value) | No. of Target Genes Detected in the Rat Frontal Cortex | No. of Overlapping Genes (p-Value) | No. of Target Genes Detected in the Rat Frontal Cortex | No. of Overlapping Genes (p-Value) | |
| AR | 609 | 274 (0.403) | 614 | 140 (3.56 × 10−4) | 618 | 145 (8.48 × 10−4) |
| CUX1 | 360 | 161 (0.475) | 371 | 77 (0.056) | 366 | 82 (3.07 × 10−2) |
| EGR2 | 173 | 80 (0.342) | 170 | 37 (0.087) | 171 | 42 (2.74 × 10−2) |
| ESR1 | 372 | 178 (0.098) | 380 | 81 (2.93 × 10−2) | 378 | 82 (0.060) |
| MTF1 | 211 | 103 (0.111) | 220 | 54 (4.78 × 10−3) | 213 | 50 (3.79 × 10−2) |
| PAX5 | 137 | 67 (0.165) | 137 | 26 (0.355) | 138 | 32 (0.094) |
| PAX6 | 85 | 42 (0.206) | 85 | 15 (0.529) | 84 | 16 (0.489) |
| POU3F2 | 435 | 189 (0.679) | 437 | 96 (8.54 × 10−3) | 441 | 90 (0.156) |
| RORA | 264 | 109 (0.864) | 266 | 63 (5.84 × 10−3) | 266 | 69 (1.45 × 10−3) |
| SMAD4 | 196 | 86 (0.589) | 195 | 39 (0.200) | 194 | 31 (0.839) |
| SOX5 | 219 | 104 (0.198) | 214 | 56 (8.80 × 10−4) | 221 | 55 (1.01 × 10−2) |
| STAT1 | 304 | 130 (0.741) | 305 | 64 (0.063) | 306 | 64 (0.148) |
| TCF4 | 375 | 187 (1.83 × 10−2) | 379 | 96 (6.38 × 10−5) | 377 | 94 (9.40 × 10−4) |
| YY1 | 633 | 259 (0.969) | 645 | 145 (5.38 × 10−4) | 634 | 114 (0.641) |
| Protein ID | TFs | Name | Known Ligand | Mean Binding Free Energy ± SD (kcal/mol) | |
|---|---|---|---|---|---|
| Known Ligand | BPA Ligand | ||||
| PDB:2AM9 | AR | Androgen receptor | Testosterone | −11.17 ± 0.00 | −8.98 ± 0.01 |
| 5β-dihydrotestosterone | −11.07 ± 0.00 | ||||
| PDB: 1A52 | ESR1 | Estrogen receptor alpha | 17β-estradiol | −9.96 ± 0.00 | −7.49 ± 0.00 |
| PDB:1S0X | RORA | Retinoic acid-related orphan receptor-alpha | Cholesterol sulfate [104] | −12.64 ± 0.18 | −7.47 ± 0.04 |
| 7β-hydroxycholesterol [104] | −11.05 ± 0.16 | ||||
| PDB:1I11 | SOX5 | SRY-box 5 (DNA-binding domain) | NA | NA | −5.85 ± 0.01 |
| PDB:2KWF | TCF4 | Transcription factor 4 | NA | NA | −5.75 ± 0.04 |
| PDB:1UBD | YY1 | Yin Yang 1 transcriptional repressor protein (Zinc-finger domain) | NA | NA | −5.68 ± 0.02 |
| Alphafold ID: Q14872 | MTF1 | Metal regulatory transcription factor 1 | NA | NA | −4.33 ± 0.05 |
| Alphafold ID: P20265 | POU3F2 | POU domain, class 3, and transcription factor 2 | NA | NA | −4.33 ± 0.38 |
| Alphafold ID: P39880 | CUX1 | Homeobox protein cut-like 1 | NA | NA | −4.15 ± 0.37 |
| Alphafold ID: P11161 | EGR2 | E3 SUMO-protein ligase EGR2 | NA | NA | −3.66 ± 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanlayaprasit, S.; Thongkorn, S.; Panjabud, P.; Jindatip, D.; Hu, V.W.; Kikkawa, T.; Osumi, N.; Sarachana, T. Autism-Related Transcription Factors Underlying the Sex-Specific Effects of Prenatal Bisphenol A Exposure on Transcriptome-Interactome Profiles in the Offspring Prefrontal Cortex. Int. J. Mol. Sci. 2021, 22, 13201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413201
Kanlayaprasit S, Thongkorn S, Panjabud P, Jindatip D, Hu VW, Kikkawa T, Osumi N, Sarachana T. Autism-Related Transcription Factors Underlying the Sex-Specific Effects of Prenatal Bisphenol A Exposure on Transcriptome-Interactome Profiles in the Offspring Prefrontal Cortex. International Journal of Molecular Sciences. 2021; 22(24):13201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413201
Chicago/Turabian StyleKanlayaprasit, Songphon, Surangrat Thongkorn, Pawinee Panjabud, Depicha Jindatip, Valerie W. Hu, Takako Kikkawa, Noriko Osumi, and Tewarit Sarachana. 2021. "Autism-Related Transcription Factors Underlying the Sex-Specific Effects of Prenatal Bisphenol A Exposure on Transcriptome-Interactome Profiles in the Offspring Prefrontal Cortex" International Journal of Molecular Sciences 22, no. 24: 13201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413201