
Enhanced p53 Levels Are Involved in the Reduced Mineralization Capacity of Osteoblasts Derived from Shwachman–Diamond Syndrome Subjects

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
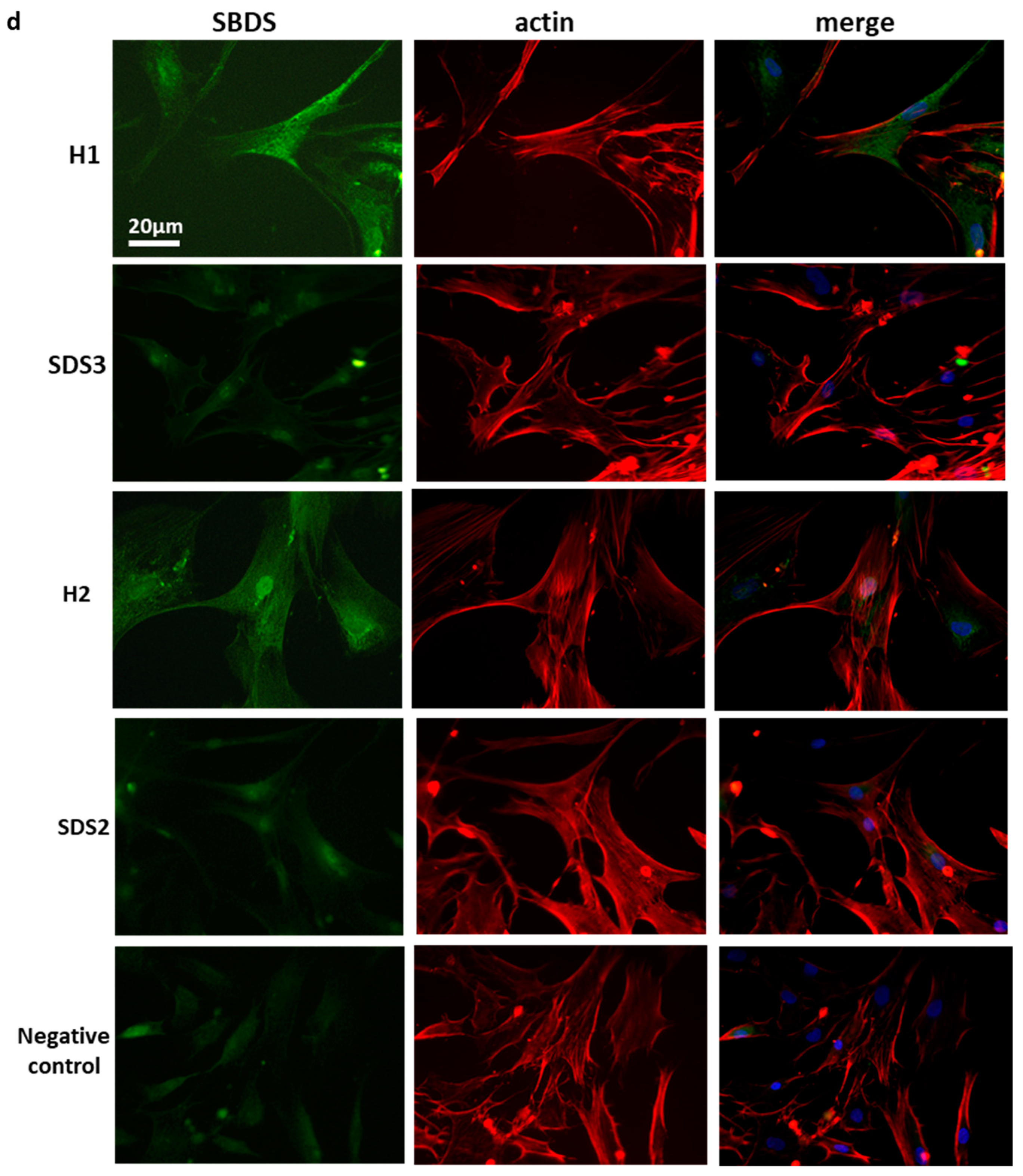
2.1. Low SBDS Levels in Osteoblasts Derived from SDS Patients
2.2. Mineralization of SDS-OBs Is Impaired
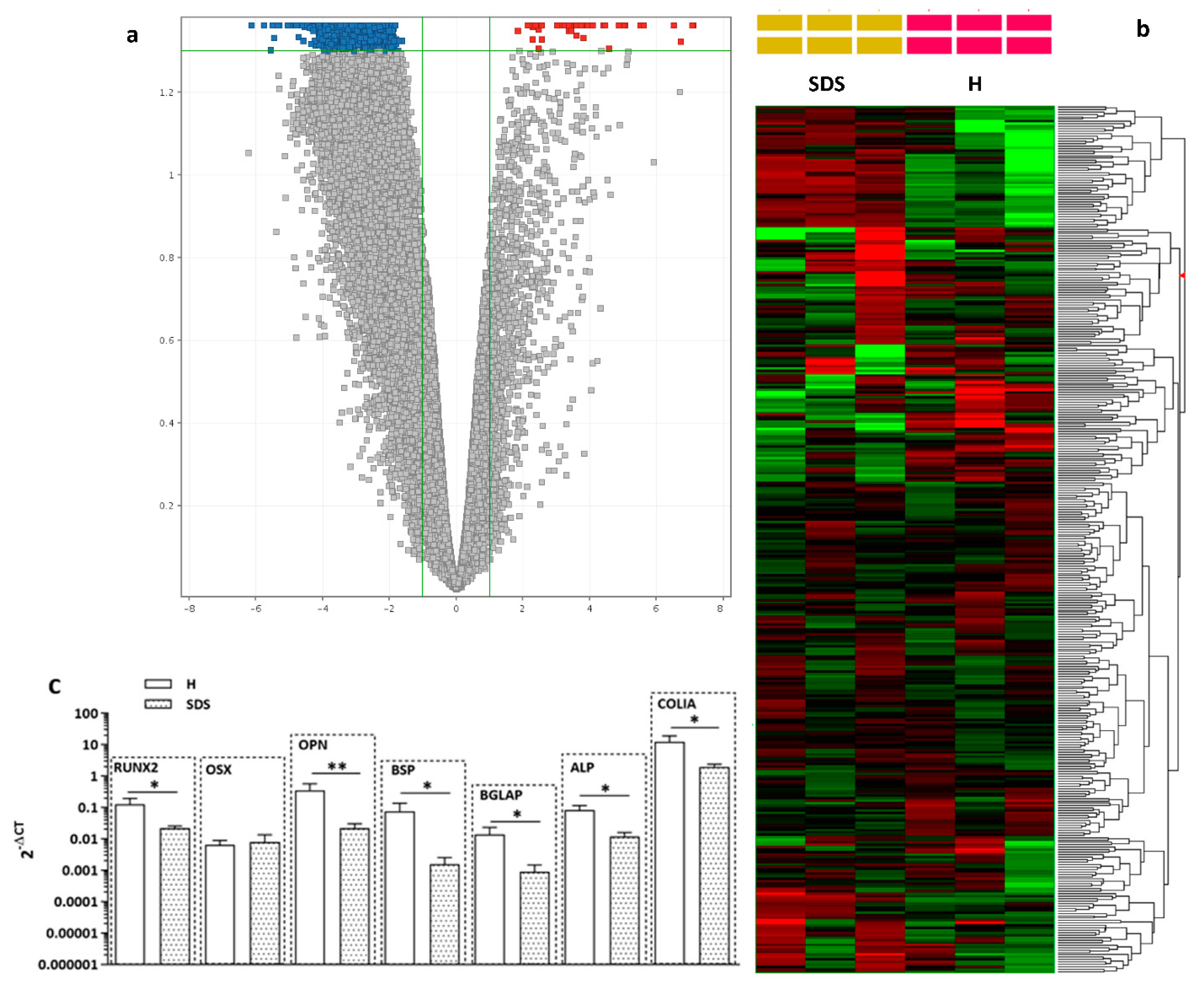
2.3. Osteogenesis-Related Gene and Relative Protein Expressions Are Reduced in SDS-OBs
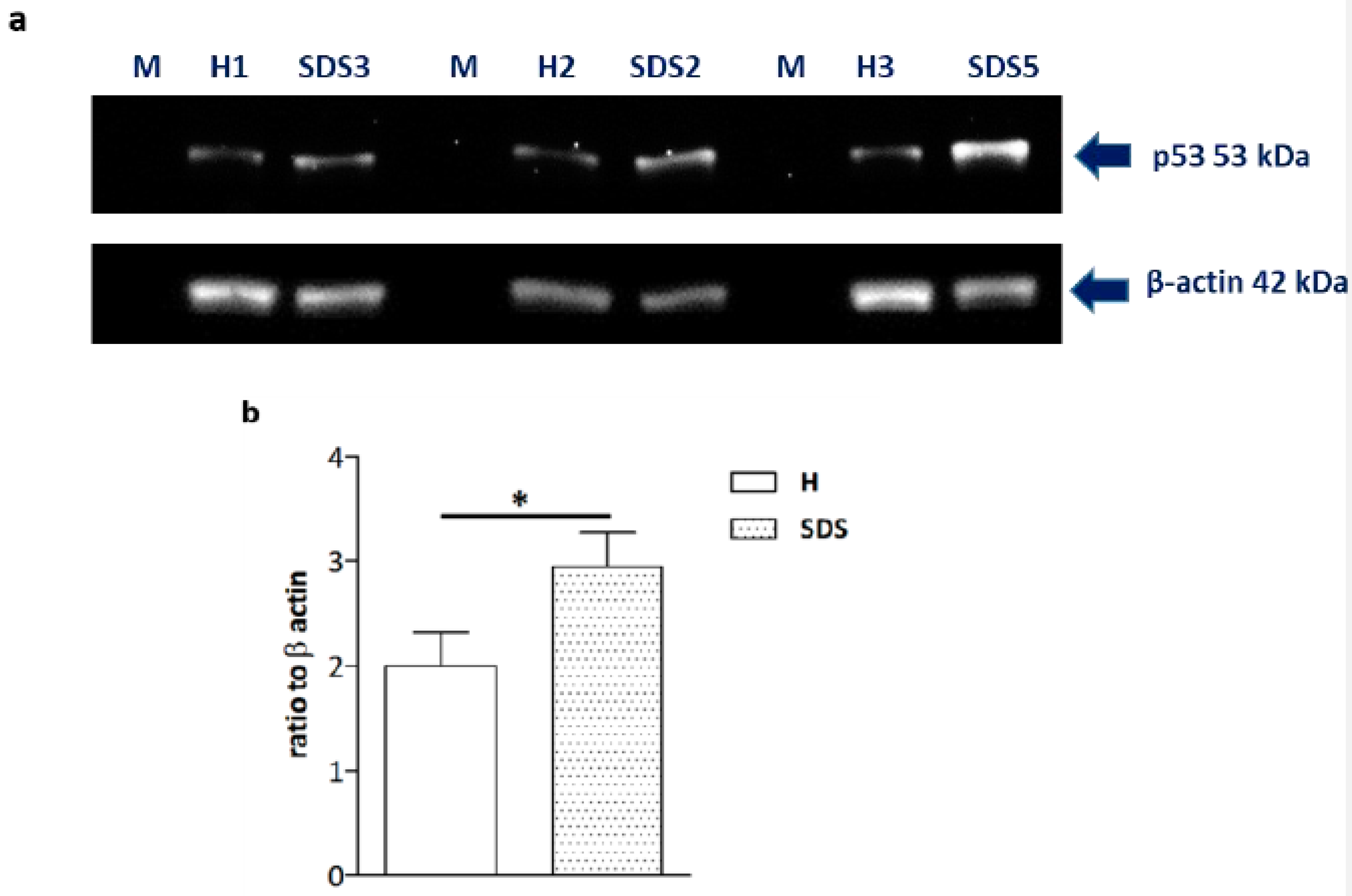
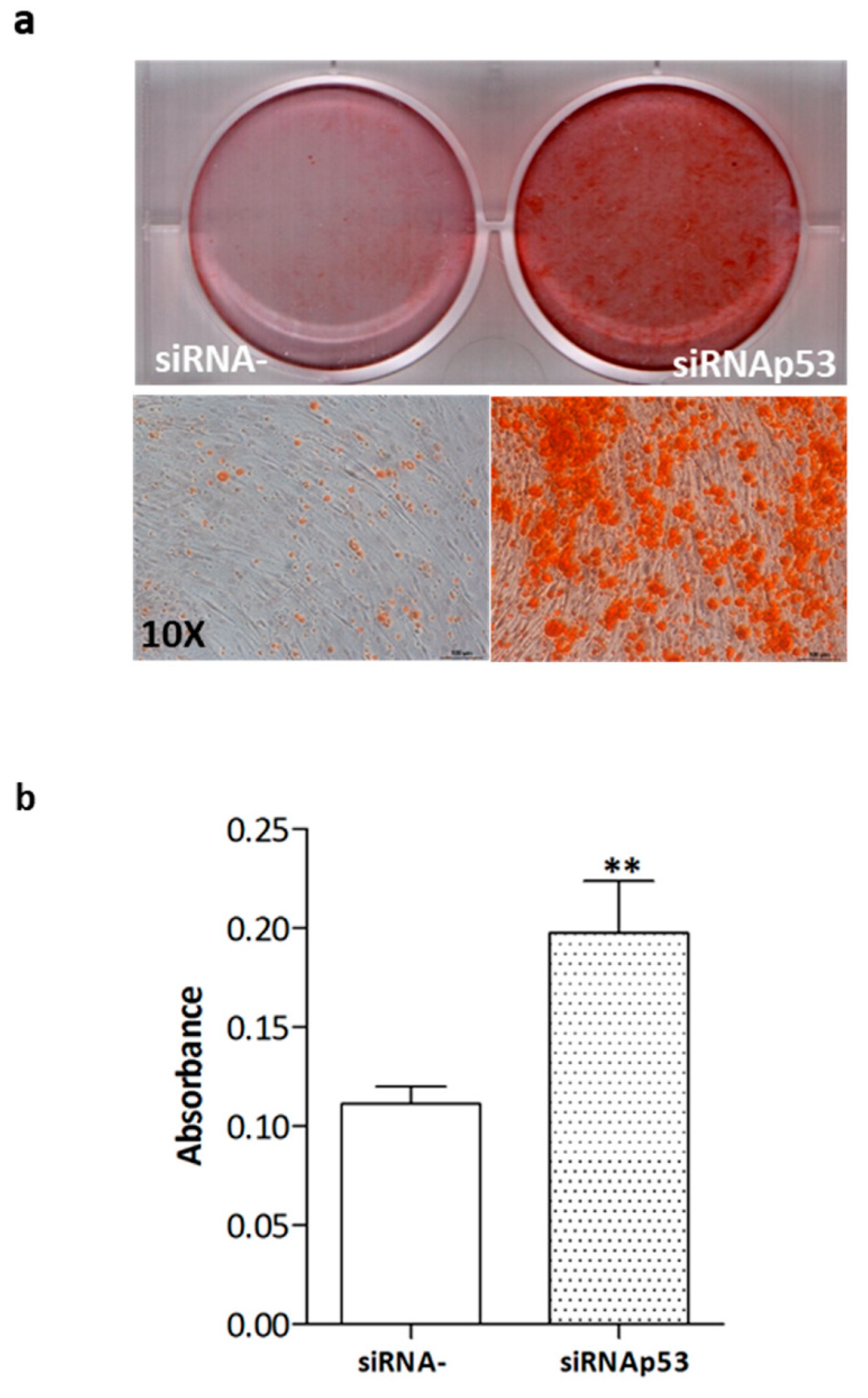
2.4. High p53 Levels Are Involved in SDS-OB Functional Impairment
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Proliferation Assay
4.3. Alizarin Red Staining
4.4. Immunofluorescence Staining
4.5. RNA Extraction, Reverse Transcription, and RT-qPCR
- SBDS_FW AGATAGAACGTGCTCACATGAGGC;
- SBDS_REV GGTGTCATTCAAATTTCTCATGTC;
- β-ACT_FW CATGTACGTTGCTATCCAGGC;
- β-ACT_REV CTCCTTAATGTCACGCACGAT,
- gSBDS_FW GGAACAGATGACCAAACTGAAATC;
- gSBDS_REV TCAATAAGGATCACGGTGTATGG,
4.6. One-Color Expression Arrays
4.7. Western Blot Analysis
4.8. Tp53 Mutation Analysis
- p53
- _93FW: GTGACACGCTTCCCTGGATT;
- p53
- p53_962REV: CACGCACCTCAAAGCTGTTC;
- p53
- p53_569FW: GTGCAGCTGTGGGTTGATT;
- p53
- p53_1067REV: GCAGTGCTCGCTTAGTGCTC;
- p53
- p53_1355REV: GCTGTCAGTGGGGAACAAGAA,
4.9. siRNA-Mediated p53 Gene Silencing
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shwachman, H.; Diamond, L.K.; Oski, F.A.; Khaw, K.-T. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J. Pediatr. 1964, 65, 645–663. [Google Scholar] [CrossRef]
- Bodian, M.; Sheldon, W.; Lightwood, R. Congenital Hypoplasia of the Exocrine Pancreas. Acta Paediatr. 1964, 53, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Valli, R.; Minelli, A.; Galbiati, M.; D’Amico, G.; Frattini, A.; Montalbano, G.; Khan, A.W.; Porta, G.; Millefanti, G.; Olivieri, C.; et al. Shwachman-Diamond syndrome with clonal interstitial deletion of the long arm of chromosome 20 in bone marrow: Haematological features, prognosis and genomic instability. Br. J. Haematol. 2018, 184, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.J. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv. Biol. Regul. 2018, 67, 109–127. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Shimamura, A. Genetic predisposition to MDS: Clinical features and clonal evolution. Blood 2019, 133, 1071–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsley, R.C.; Saber, W.; Mar, B.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Bezzerri, V.; Cipolli, M. Shwachman-Diamond Syndrome: Molecular Mechanisms and Current Perspectives. Mol. Diagn. Ther. 2018, 23, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Boocock, G.R.; Morrison, J.A.; Popovic, M.; Richards, N.; Ellis, L.; Durie, P.R.; Rommens, J.M. Mutations in SBDS are associated with Shwachman–Diamond syndrome. Nat. Genet. 2003, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Leary, R.J.; Shimamura, A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood 2005, 106, 1253–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Traynor, D.; Basse, N.; Kay, R.R.; Warren, A.J. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood 2011, 118, 4305–4312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provost, E.; Wehner, K.A.; Zhong, X.; Ashar, F.; Nguyen, E.; Green, R.; Parsons, M.J.; Leach, S.D. Ribosomal biogenesis genes play an essential and p53-independent role in zebrafish pancreas development. Development 2012, 139, 3232–3241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shi, M.; Hui, C.-C.; Rommens, J.M. Loss of the Mouse Ortholog of the Shwachman-Diamond Syndrome Gene (Sbds) Results in Early Embryonic Lethality. Mol. Cell. Biol. 2006, 26, 6656–6663. [Google Scholar] [CrossRef] [Green Version]
- Austin, K.M.; Gupta, M.L.; Coats, S.A.; Tulpule, A.; Mostoslavsky, G.; Balazs, A.B.; Mulligan, R.C.; Daley, G.; Pellman, D.; Shimamura, A. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J. Clin. Investig. 2008, 118, 1511–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orelio, C.; Verkuijlen, P.; Geissler, J.; van den Berg, T.K.; Kuijpers, T.W. SBDS Expression and Localization at the Mitotic Spindle in Human Myeloid Progenitors. PLoS ONE 2009, 4, e7084. [Google Scholar] [CrossRef] [Green Version]
- Morini, J.; Babini, G.; Mariotti, L.; Baiocco, G.; Nacci, L.; Maccario, C.; Rößler, U.; Minelli, A.; Savio, M.; Gomolka, M.; et al. Radiosensitivity in lymphoblastoid cell lines derived from Shwachman–Diamond syndrome patients. Radiat. Prot. Dosim. 2015, 166, 95–100. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Cao, Y.; Xu, H.; Wu, Y.; Wu, S.; Liu, D.; Zhao, Y.; Songyang, Z.; Ma, W. Shwachman-Diamond Syndrome Protein SBDS Maintains Human Telomeres by Regulating Telomerase Recruitment. Cell Rep. 2018, 22, 1849–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burwick, N.; Coats, S.A.; Nakamura, T.; Shimamura, A. Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood 2012, 120, 5143–5152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, A.J.; Hilcenko, C.; Basse, N.; Drynan, L.F.; Goyenechea, B.; Menne, T.F.; Fernández, G.; Simpson, P.; D’Santos, C.S.; Arends, M.J.; et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011, 25, 917–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, F.; Giudice, E.; Churcher, M.; Jin, L.; Hilcenko, C.; Wong, C.C.; Traynor, D.; Kay, R.R.; Warren, A.J. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat. Struct. Mol. Biol. 2015, 22, 914–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luviano, A.; Cruz-Castañeda, R.; Sánchez-Puig, N.; García-Hernández, E. Cooperative energetic effects elicited by the yeast Shwachman-Diamond syndrome protein (Sdo1) and guanine nucleotides modulate the complex conformational landscape of the elongation factor-like 1 (Efl1) GTPase. Biophys. Chem. 2019, 247, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, O.; Ellis, L.; Durie, P.R.; Morrison, J.A.; Sochett, E.B.; Rommens, J.M.; Cole, W.G. Skeletal phenotype in patients with Shwachman-Diamond syndrome and mutations in SBDS. Clin. Genet. 2004, 65, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, L.; Woolfrey, A.; Shimamura, A. Shwachman-Diamond Syndrome: A Review of the Clinical Presentation, Molecular Pathogenesis, Diagnosis, and Treatment. Hematol. Oncol. Clin. N. Am. 2009, 23, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Toiviainen-Salo, S.; Mäyränpää, M.K.; Durie, P.R.; Richards, N.; Grynpas, M.; Ellis, L.; Ikegawa, S.; Cole, W.G.; Rommens, J.; Marttinen, E.; et al. Shwachman–Diamond syndrome is associated with low-turnover osteoporosis. Bone 2007, 41, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Aggett, P.J.; Cavanagh, N.P.; Matthew, D.J.; Pincott, J.R.; Sutcliffe, J.; Harries, J.T. Shwachman’s syndrome: A review of 21 cases. Arch. Dis. Child. 1980, 55, 331–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginzberg, H.; Shin, J.; Ellis, L.; Morrison, J.; Ip, W.; Dror, Y.; Freedman, M.; Heitlinger, L.A.; Belt, M.A.; Corey, M.; et al. Shwachman syndrome: Phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J. Pediatr. 1999, 135, 81–88. [Google Scholar] [CrossRef]
- André, V.; Longoni, D.; Bresolin, S.; Cappuzzello, C.; Dander, E.; Galbiati, M.; Bugarin, C.; Di Meglio, A.; Nicolis, E.; Maserati, E.; et al. Mesenchymal stem cells from Shwachman–Diamond syndrome patients display normal functions and do not contribute to hematological defects. Blood Cancer J. 2012, 2, e94. [Google Scholar] [CrossRef]
- Zimmermann, E.A.; Riedel, C.; Schmidt, F.N.; Stockhausen, K.E.; Chushkin, Y.; Schaible, E.; Gludovatz, B.; Vettorazzi, E.; Zontone, F.; Püschel, K.; et al. Mechanical Competence and Bone Quality Develop During Skeletal Growth. J. Bone Miner. Res. 2019, 34, 1461–1472. [Google Scholar] [CrossRef] [Green Version]
- Trainor, P.A.; Merrill, A.E. Ribosome biogenesis in skeletal development and the pathogenesis of skeletal disorders. Biochim. Biophys. Acta 2013, 1842, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkitie, R.E.; Costantini, A.; Kämpe, A.; Alm, J.J.; Mäkitie, O. New Insights into Monogenic Causes of Osteoporosis. Front. Endocrinol. (Lausanne) 2019, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Elghetany, M.T.; Alter, B.P. p53 protein overexpression in bone marrow biopsies of patients with Shwachman-Diamond syndrome has a prevalence similar to that of patients with refractory anemia. Arch. Pathol. Lab. Med. 2002, 126, 452–455. [Google Scholar] [CrossRef]
- Tataria, M.; Quarto, N.; Longaker, M.T.; Sylvester, K.G. Absence of the p53 tumor suppressor gene promotes osteogenesis in mesenchymal stem cells. J. Pediatr. Surg. 2006, 41, 624–632. [Google Scholar] [CrossRef]
- Doffe, F.; Carbonnier, V.; Tissier, M.; Leroy, B.; Martins, I.; Mattsson, J.S.M.; Micke, P.; Pavlova, S.; Pospisilova, S.; Smardova, J.; et al. Identification and functional characterization of new missense SNPs in the coding region of the TP53 gene. Cell Death Differ. 2021, 28, 1477–1492. [Google Scholar] [CrossRef]
- Frischmeyer, P.A.; Dietz, H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999, 8, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Regulation of osteoblast differentiation by runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49. [Google Scholar]
- Ganss, B.; Kim, R.H.; Sodek, J. Bone sialoprotein. Crit. Rev. Oral Biol. Med. 1999, 10, 79–98. [Google Scholar] [CrossRef]
- Bouleftour, W.; Boudiffa, M.; Wade-Gueye, N.M.; Bouet, G.; Cardelli, M.; Laroche, N.; Vanden-Bossche, A.; Thomas, M.; Bonnelye, E.; Aubin, J.E.; et al. Skeletal Development of Mice Lacking Bone Sialoprotein (BSP)—Impairment of Long Bone Growth and Progressive Establishment of High Trabecular Bone Mass. PLoS ONE 2014, 9, e95144. [Google Scholar] [CrossRef]
- Osathanon, T.; Giachelli, C.M.; Somerman, M.J. Immobilization of alkaline phosphatase on microporous nanofibrous fibrin scaffolds for bone tissue engineering. Biomaterials 2009, 30, 4513–4521. [Google Scholar] [CrossRef] [Green Version]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kua, H.-Y.; Hu, Y.; Guo, K.; Zeng, Q.; Wu, Q.; Ng, H.H.; Karsenty, G.; De Crombrugghe, B.; Yeh, J.; et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J. Cell Biol. 2005, 172, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Robey, P.G.; Termine, J.D. Human bone cellsin vitro. Calcif. Tissue Int. 1985, 37, 453–460. [Google Scholar] [CrossRef]
- Dillon, J.; Waring-Green, V.; Taylor, A.; Wilson, P.; Birch, M.; Gartland, A.; Gallagher, J. Primary Human Osteoblast Cultures. In Bone Protocols; Hielfrich, M., Ralston, S., Eds.; Humana Press: New York, NY, USA, 2012; Volume 16. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | SBDS Mutations (m = Maternal; p = Paternal; u = Undetermined) |
|---|---|
| SDS1 | c.183_184TA > CT, p.K62X (m); c.258 + 2T > C, p.C84fsX3 (p) |
| SDS2 | c.183_184TA > CT, p.K62X (u); c.258 + 2T > C, p.C84fsX3 (u) |
| SDS3 | c.183_184TA >CT, p.K62X (m); c.258 + 2T > C, p.C84fsX3 (p) |
| SDS5 | c.183_184TA > CT, p.K62X (m); c.258 + 2T > C, p.C84fsX3 (p) |
| SDS6 | c.258 + 2T > C, p.C84fsX3 (m); c.183_184TA > CT, p.K62X (p) |
| SDS7 | c.258 + 2T > C, p.C84fsX3 (m); c.183_184TA > CT, p.K62X (p) |
| SDS8 | c.258 + 2T > C, p.C84fsX3 (u); c.92_93GC > AG, p.C31X (u) |
| SDS9 | HOMO c.258 + 2T > C, p.C84fsX3 (m;p) |
| SDS10 | c.258 + 2T > C, p.C84fsX3 (m); c.183_184TA > CT, p.K62X (p) c.258 + 2T > C, p.C84fsX3 (de novo mutation) |
| SDS11 | c.356G > A, p.V57I (m); c.258 + 2T > C, p.C84fsX3 (p) |
| SDS12 | c.258 + 2T > C p.C84fsX3 (m); c.183_184TA > CT p.K62X(p); c.258 + 2T > C, p.C84fsX3 (p) |
| SDS13 | c.258 + 2T > C, p.C84fsX3 (m); c.289_292delGATA (C139X) (p) |
| SDS14 | c.258 + 2T > C, p.C84fsX3 (m); c.183_184TA > C, p.K62X(p) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frattini, A.; Bolamperti, S.; Valli, R.; Cipolli, M.; Pinto, R.M.; Bergami, E.; Frau, M.R.; Cesaro, S.; Signo, M.; Bezzerri, V.; et al. Enhanced p53 Levels Are Involved in the Reduced Mineralization Capacity of Osteoblasts Derived from Shwachman–Diamond Syndrome Subjects. Int. J. Mol. Sci. 2021, 22, 13331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413331
Frattini A, Bolamperti S, Valli R, Cipolli M, Pinto RM, Bergami E, Frau MR, Cesaro S, Signo M, Bezzerri V, et al. Enhanced p53 Levels Are Involved in the Reduced Mineralization Capacity of Osteoblasts Derived from Shwachman–Diamond Syndrome Subjects. International Journal of Molecular Sciences. 2021; 22(24):13331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413331
Chicago/Turabian StyleFrattini, Annalisa, Simona Bolamperti, Roberto Valli, Marco Cipolli, Rita Maria Pinto, Elena Bergami, Maria Rita Frau, Simone Cesaro, Michela Signo, Valentino Bezzerri, and et al. 2021. "Enhanced p53 Levels Are Involved in the Reduced Mineralization Capacity of Osteoblasts Derived from Shwachman–Diamond Syndrome Subjects" International Journal of Molecular Sciences 22, no. 24: 13331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413331