
Comparative Mitogenomics of Fungal Species in Stachybotryaceae Provides Evolutionary Insights into Hypocreales


Abstract
:1. Introduction
2. Results
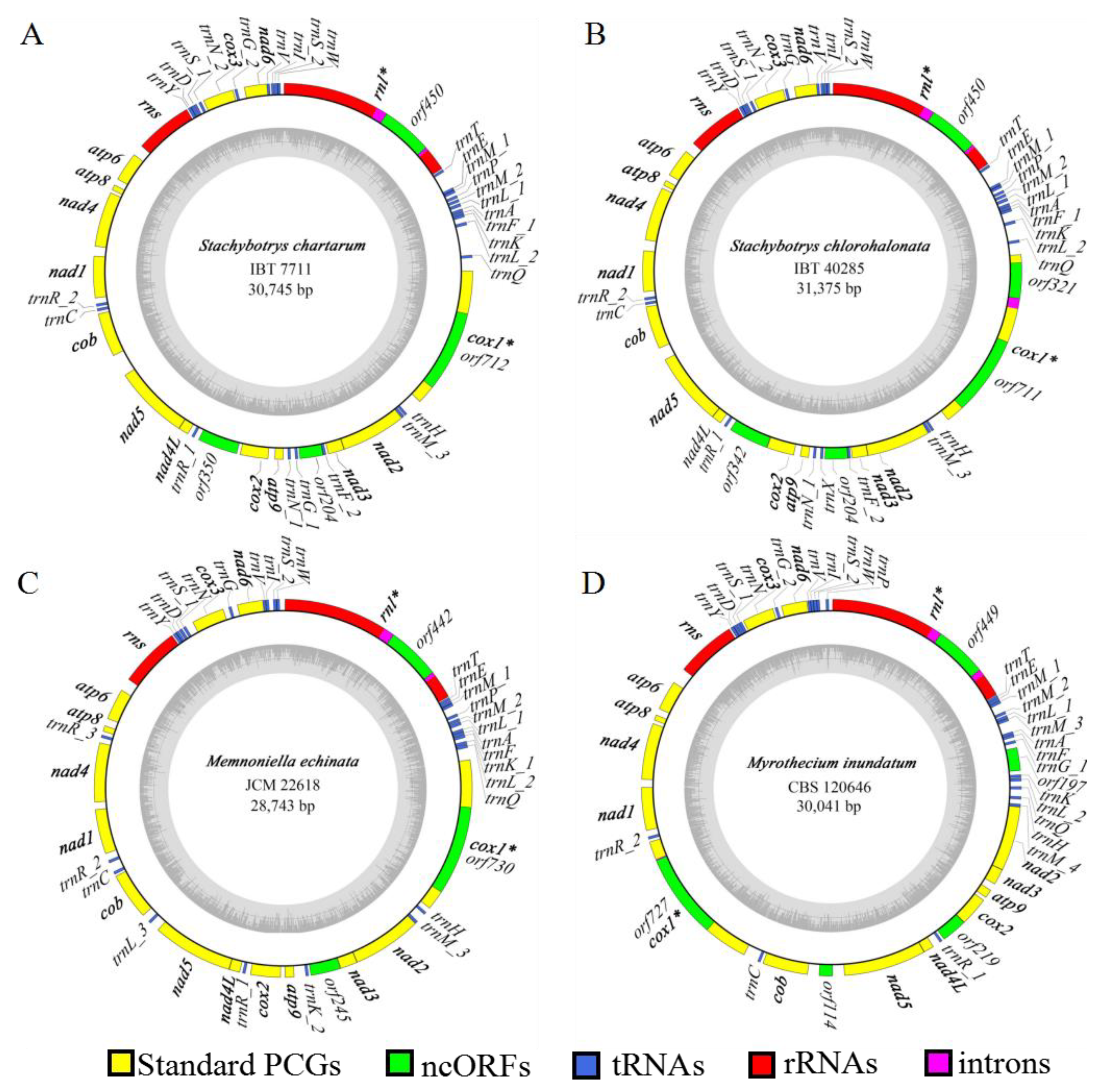
2.1. Organization of the S. chartarum Mitogenome
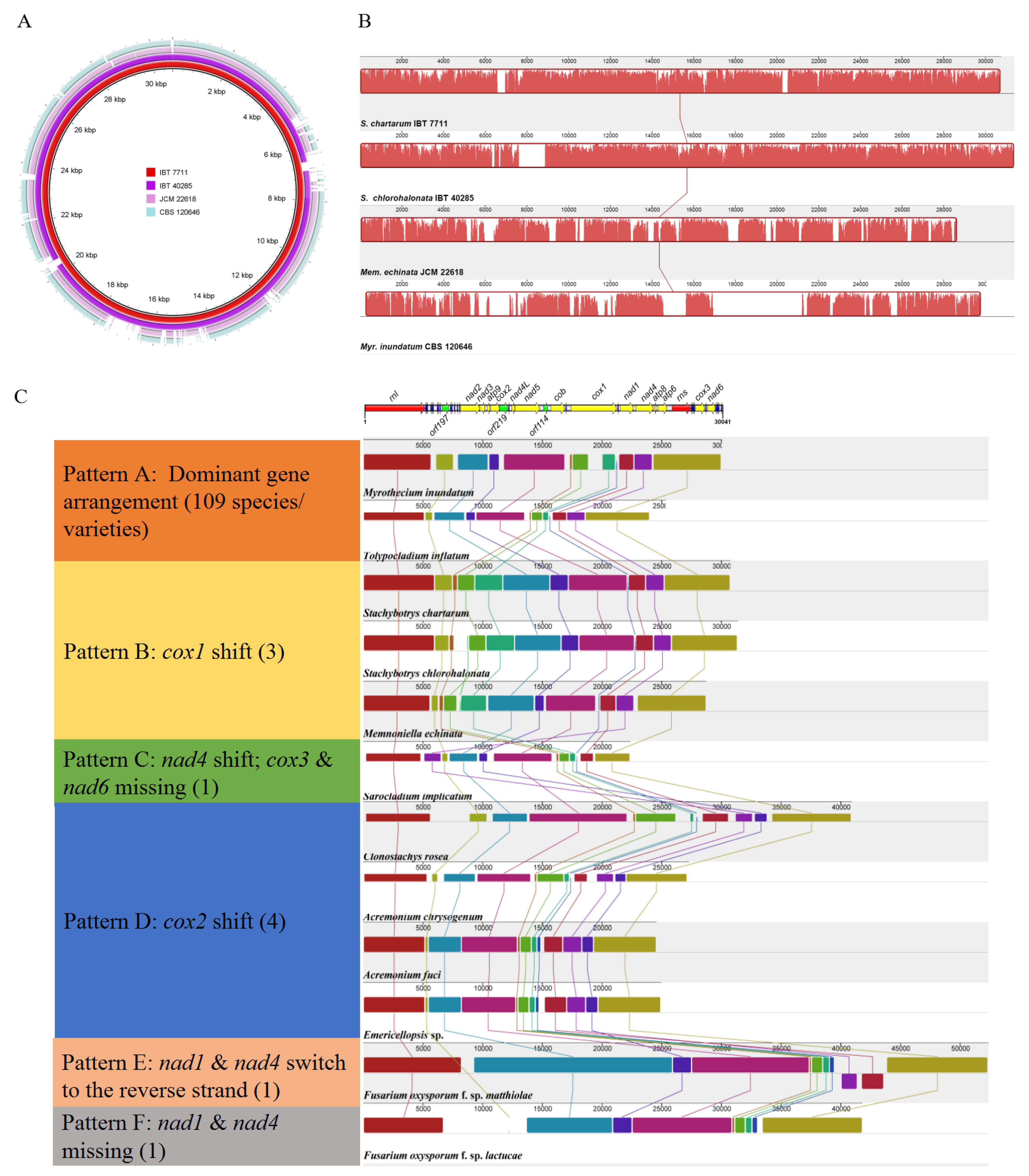
2.2. Comparison on Mitogenomes among Different Individuals of S. chartarum
2.3. Comparison on Mitogenomes among Different Species in Stachybotryaceae
2.4. Comparison of Mitogenomes among Different Species in Hypocreales
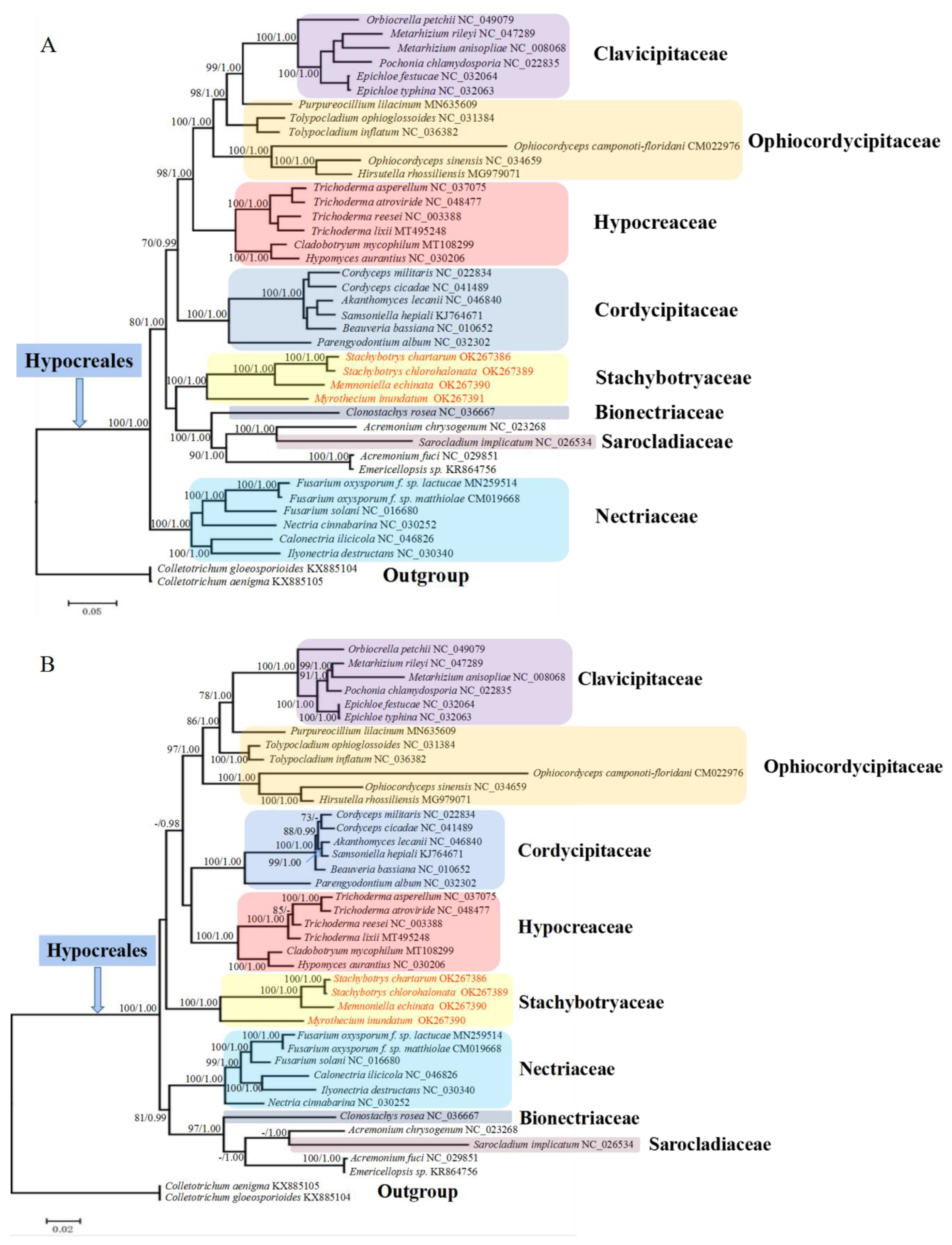
2.5. Phylogenetic Analyses of Hypocreales Species
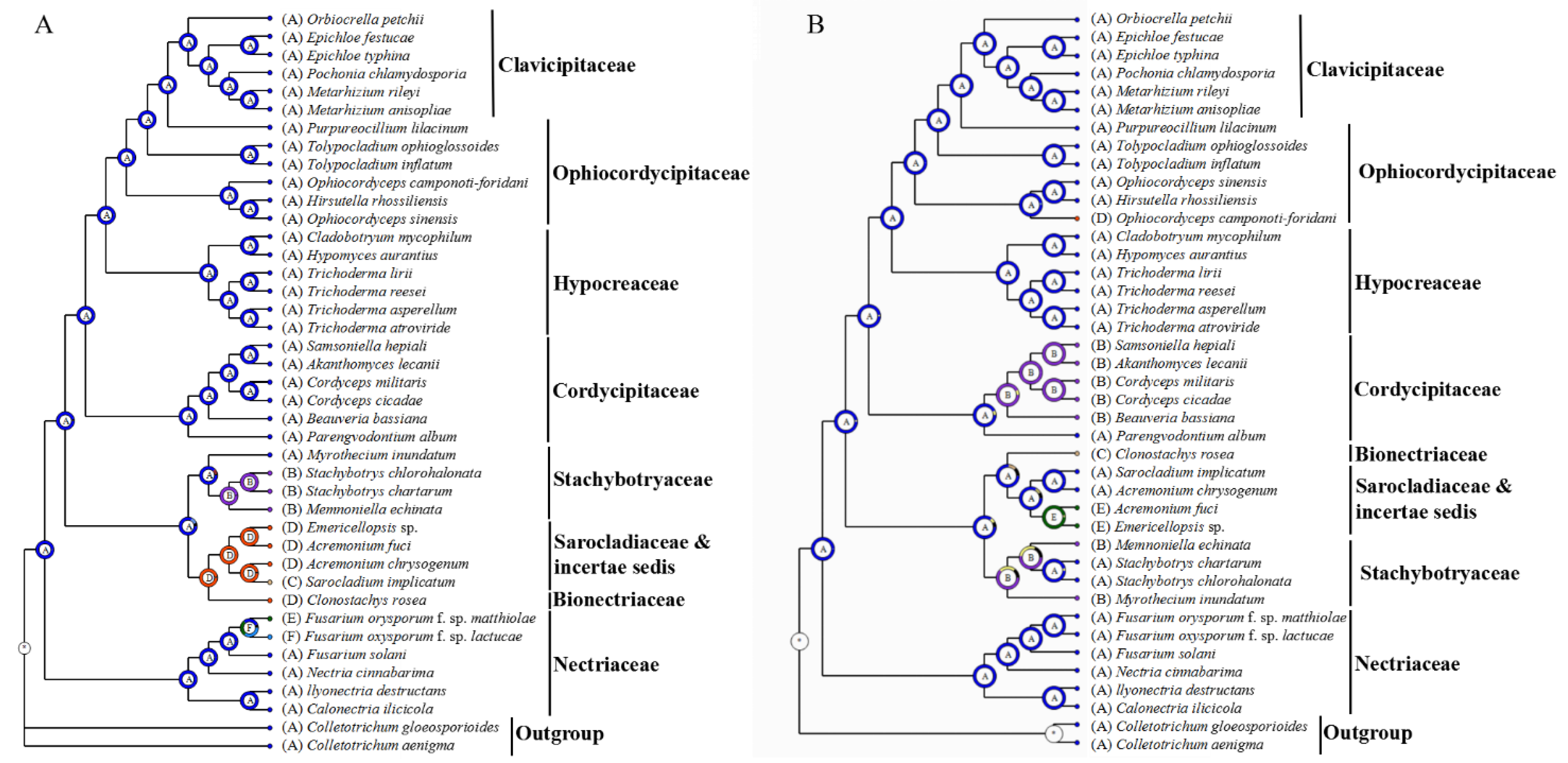
2.6. Ancestral State Estimation
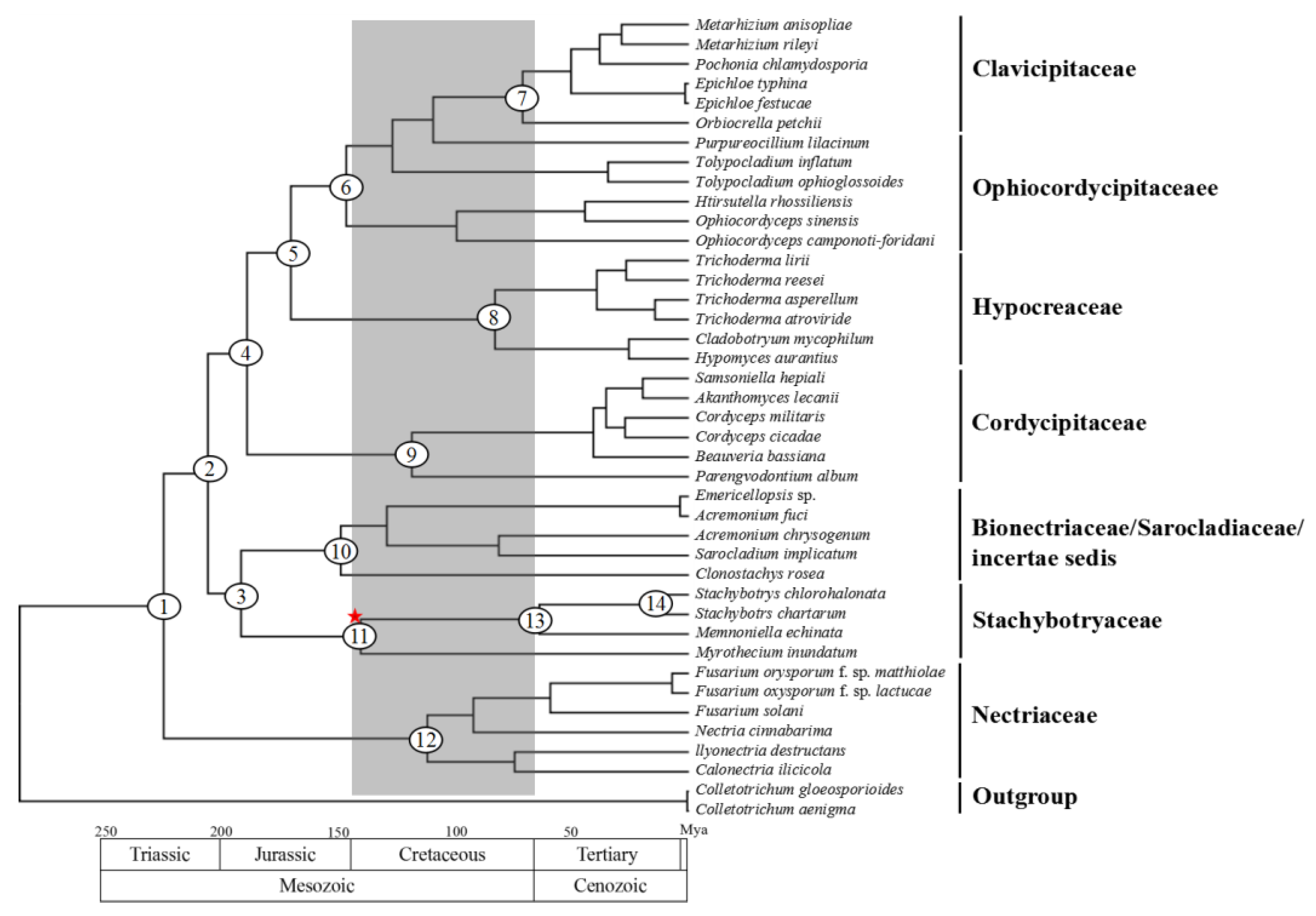
2.7. Divergence Time Estimation
3. Discussion
4. Materials and Methods
4.1. Assembly and Annotation of Mitogenomes
4.2. Comparison among Different Mitogenomes
4.3. Phylogenetic Tree Reconstruction
4.4. Ancestral State Inference
4.5. Divergence Time Estimation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Giezen, M.; Tovar, J.; Clark, C.G. Mitochondrion-derived organelles in protists and fungi. Int. Rev. Cytol. 2005, 244, 175–225. [Google Scholar]
- Hausner, G. Fungal mitochondrial genomes, plasmids and introns. In Applied Mycology and Biotechnology; Dilip, K.A., George, G.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 3, pp. 101–131. [Google Scholar]
- Lang, B.F. Mitochondrial Genomes in Fungi. In Molecular Life Sciences; Wells, R.D., Bond, J.S., Klinman, J., Masters, B.S.S., Eds.; Springer: New York, NY, USA, 2018; pp. 722–728. [Google Scholar]
- Zhang, Y.-J.; Zhang, H.-Y.; Liu, X.-Z.; Zhang, S. Mitochondrial genome of the nematode endoparasitic fungus Hirsutella vermicola reveals a high level of synteny in the family Ophiocordycipitaceae. Appl. Microbiol. Biotechnol. 2017, 101, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.-W.; Zhang, S.; Zhang, Y.-J. The complete mitochondrial genome of the Chan-hua fungus Isaria cicadae: A tale of intron evolution in Cordycipitaceae. Env. MicroBiol. 2019, 21, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Wang, L.; Cai, Y.; Tao, W.; Zhang, Y.-J.; Huang, B. Mitochondrial genome of the entomophthoroid fungus Conidiobolus heterosporus provides insights into evolution of basal fungi. Appl. Microbiol. Biotechnol. 2019, 103, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Yang, X.-Q.; Zhang, S.; Humber, R.A.; Xu, J. Genomic analyses reveal low mitochondrial and high nuclear diversity in the cyclosporin-producing fungus Tolypocladium inflatum. Appl. Microbiol. Biotechnol. 2017, 101, 8517–8531. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, S.; Li, J.-H.; Zhang, Y.-J. Mitochondrial genome, comparative analysis and evolutionary insights into the entomopathogenic fungus Hirsutella thompsonii. Environ. MicroBiol. 2018, 20, 3393–3405. [Google Scholar] [CrossRef]
- Lombard, L.; Houbraken, J.; Decock, C.; Samson, R.A.; Meijer, M.; Reblova, M.; Groenewald, J.Z.; Crous, P.W. Generic hyper-diversity in Stachybotriaceae. Persoonia 2016, 36, 156–246. [Google Scholar] [CrossRef] [Green Version]
- Hyde, K.D.; Norphanphoun, C.; Maharachchikumbura, S.S.N.; Bhat, D.J.; Jones, E.B.G.; Bundhun, D.; Chen, Y.J.; Bao, D.F.; Boonmee, S.; Calabon, M.S.; et al. Refined families of Sordariomycetes. Mycosphere 2020, 11, 305–1059. [Google Scholar] [CrossRef]
- Castlebury, L.A.; Rossman, A.Y.; Sung, G.H.; Hyten, A.S.; Spatafora, J.W. Multigene phylogeny reveals new lineage for Stachybotrys chartarum, the indoor air fungus. Mycol. Res. 2004, 108 Pt 8, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Hyde, K.D.; Al-Hatmi, A.M.S.; Andersen, B.; Boekhout, T.; Buzina, W.; Dawson, T.L.; Eastwood, D.C.; Jones, E.B.G.; de Hoog, S.; Kang, Y.; et al. The world’s ten most feared fungi. Fungal Divers. 2018, 93, 161–194. [Google Scholar] [CrossRef]
- Ding, Z.G.; Zhao, J.Y.; Ding, J.H.; Chunyu, W.X.; Li, M.G.; Gu, S.J.; Wang, F.; Wen, M.L. A novel phenylspirodrimane dimer from cultuRes. of the fungus Stachybotrys chartarum. Nat. Prod. Res. 2018, 32, 2370–2374. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.R.; Feng, H.L.; Yao, X.S. Stachybotranes A-D, phenylspirodrimanes from the wetland fungus Stachybotrys chartarum with cytotoxic activities. Nat Prod Res. 2021, 1–7. [Google Scholar] [CrossRef]
- Semeiks, J.; Borek, D.; Otwinowski, Z.; Grishin, N.V. Comparative genome sequencing reveals chemotype-specific gene clusters in the toxigenic black mold Stachybotrys. BMC Genom. 2014, 15, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancourt, D.A.; Dean, T.R.; Kim, J.; Levy, J. Genome sequence of Stachybotrys chartarum strain 51-11. Genome Announc. 2015, 3, e01114-15. [Google Scholar] [CrossRef] [Green Version]
- Sung, G.H.; Poinar, G.O., Jr.; Spatafora, J.W. The oldest fossil evidence of animal parasitism by fungi supports a Cretaceous diversification of fungal–arthropod symbioses. Mol. Phylogenet Evol. 2008, 49, 495–502. [Google Scholar] [CrossRef]
- Zhang, C.; Dai, Y.; Wang, G.; Wang, C.; Gao, Y.; Deng, W.; Li, T. Mitogenome of Tolypocladium guangdongense. Appl. MicroBiol. Biotechnol. 2020, 104, 9295–9308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Hao, A.J.; Zhao, Y.X.; Zhang, X.Y.; Zhang, Y.-J. Comparative mitochondrial genomics toward exploring molecular markers in the medicinal fungus Cordyceps militaris. Sci. Rep. 2017, 7, 40219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Liu, X.; Zhang, Y. Reanalysis of the mitochondrial genome of the nematophagous fungus Hirsutella rhossiliensis. Acta MicroBiol. Sin. 2019, 59, 280–290. [Google Scholar]
- Pantou, M.; Kouvelis, V.; Typas, M. The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: A novel gene order for Verticillium and a diagnostic tool for species identification. Curr. Genet. 2006, 50, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Gissi, C.; Reyes, A.; Larizza, A.; Sbisà, E.; Pesole, G. Mitochondrial DNA in metazoa: Degree of freedom in a frozen event. Gene 2002, 286, 3–12. [Google Scholar]
- Aguileta, G.; de Vienne, D.M.; Ross, O.N.; Hood, M.E.; Giraud, T.; Petit, E.; Gabaldon, T. High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 2014, 6, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, C.L.; Frederick, R.D.; Tooley, P.W.; Luster, D.G.; Campos, B.; Winegar, R.A.; Melcher, U.; Fletcher, J.; Blagden, T. Annotation and analysis of the mitochondrial genome of Coniothyrium glycines, causal agent of red leaf blotch of soybean, reveals an abundance of homing endonucleases. PLoS ONE 2018, 13, e0207062. [Google Scholar] [CrossRef] [PubMed]
- Kouvelis, V.N.; Ghikas, D.V.; Typas, M.A. The analysis of the complete mitochondrial genome of Lecanicillium muscarium (synonym Verticillium lecanii) suggests a minimum common gene organization in mtDNAs of Sordariomycetes: Phylogenetic implications. Fungal. Genet. Biol. 2004, 41, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Korovesi, A.G.; Ntertilis, M.; Kouvelis, V.N. Mt-rps3 is an ancient gene which provides insight into the evolution of fungal mitochondrial genomes. Mol. Phylogenet Evol. 2018, 127, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Wai, A.; Shen, C.; Carta, A.; Dansen, A.; Crous, P.W.; Hausner, G. Intron-encoded ribosomal proteins and N-acetyltransferases within the mitochondrial genomes of fungi: Here today, gone tomorrow? Mitochondr. DNA A 2019, 30, 573–584. [Google Scholar] [CrossRef]
- Megarioti, A.H.; Kouvelis, V.N. The coevolution of fungal mitochondrial introns and their homing endonucleases (GIY-YIG and LAGLIDADG). Genome Biol. Evol. 2020, 12, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Barker, F.K.; Lutzoni, F.M. The utility of the incongruence length difference test. Syst. Biol. 2002, 51, 625–637. [Google Scholar] [CrossRef]
- Crous, P.W.; Luangsa-Ard, J.J.; Wingfield, M.J.; Carnegie, A.J.; Hernandez-Restrepo, M.; Lombard, L.; Roux, J.; Barreto, R.W.; Baseia, I.G.; Cano-Lira, J.F.; et al. Fungal Planet description sheets: 785-867. Persoonia 2018, 41, 238–417. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Burgess, T.I.; Hardy, G.E.S.J.; Gené, J.; Guarro, J.; Baseia, I.G.; García, D.; Gusmão, L.F.P.; Souza-Motta, C.M.; et al. Fungal Planet description sheets: 716-784. Persoonia 2018, 40, 240–393. [Google Scholar] [CrossRef] [PubMed]
- Sandor, S.; Zhang, Y.-J.; Xu, J. Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 2018, 102, 9433–9448. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [PubMed] [Green Version]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Johansen, S.; Haugen, P. A new nomenclature of group I introns in ribosomal DNA. RNA 2001, 7, 935–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, Y.-J. Proposal of a new nomenclature for introns in protein-coding genes in fungal mitogenomes. IMA Fungus 2019, 10, 15. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Mata, A.F.; Sánchez-DelBarrio, J.C.; Rico, S.G.; Librado, P.; Ramos-Onsins, S.E.; Gracia, A.S. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Blair, C.; He, X. RASP 4: Ancestral state reconstruction tool for multiple genes and characters. Mol. Biol. Evol. 2020, 37, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | IBT 7711 | IBT 40288 | IBT 40293 | IBT 40285 | JCM 22618 | CBS 120646 |
|---|---|---|---|---|---|---|
| Species | Stachybotrys chartarum | Stachybotrys chartarum | Stachybotrys chartarum | Stachybotrys chlorohalonata | Memnoniella echinata | Myrothecium inundatum |
| Geography | Denmark | Oakland CA, USA | Oakland CA, USA | Oakland CA, USA | -- | -- |
| Isolation source | Building materials | Building materials | Building materials | Building materials | Unknown | Unknown |
| Run accession | SRR628249 | SRR628247 | SRR628248 | SRR628246 | DRR032509 | SRR3439795 |
| Mitogenome size (bp) | 30,745 | 30,718 | 30,745 | 31,375 | 28,743 | 30,041 |
| Accession no. | OK267386 | OK267387 | OK267388 | OK267389 | OK267390 | OK267391 |
| AT% | 73.7 | 73.7 | 73.7 | 73.8 | 74.5 | 73.2 |
| AT skew | −0.02 | −0.02 | −0.02 | −0.01 | −0.02 | 0.00 |
| GC skew | 0.12 | 0.12 | 0.12 | 0.12 | 0.15 | 0.13 |
| No. standard PCGs | 14 | 14 | 14 | 14 | 14 | 14 |
| No. free-standing ORFs | 2 | 2 | 2 | 2 | 1 | 3 |
| No. rRNAs | 2 | 2 | 2 | 2 | 2 | 2 |
| No. tRNAs | 28 | 28 | 28 | 28 | 28 | 27 |
| No. introns | 2 | 2 | 2 | 3 | 2 | 2 |
| No. intronic ORFs | 2 | 2 | 2 | 3 | 2 | 2 |
| Intronic region (nt) † | 3884 | 3884 | 3884 | 5123 | 3905 | 4029 |
| Intron-containing genes | rnl; cox1 | rnl; cox1 | rnl; cox1 | rnl; cox1 | rnl; cox1 | rnl; cox1 |
| Genic region (nt) ‡ | 25,239 | 25,230 | 25,239 | 26,410 | 24,526 | 25,700 |
| Genic region (%) | 82.1 | 82.1 | 82.1 | 84.2 | 85.3 | 85.6 |
| Intergenic regions ‡ (nt) | 5506 | 5488 | 5506 | 4965 | 4217 | 4341 |
| Intergenic regions (%) | 17.9 | 17.9 | 17.9 | 15.8 | 14.7 | 14.4 |
| Node | Age (95% HPD) | Note | |
|---|---|---|---|
| Normal Prior | Uniform Prior | ||
| 1 | 225 (160, 293) | 228 (172, 289) | |
| 2 | 206 (149, 269) | 208 (159, 264) | |
| 3 | 192 (136, 248) | 194 (146, 245) | |
| 4 | 189 (133, 246) | 191 (144, 243) | |
| 5 | 170 (118, 223) | 172 (127, 223) | |
| 6 | 147 (102, 194) | 148 (106, 194) | Clavicipitaceae/Ophiocordycipitaceae |
| 7 | 71 (44, 100) | 72 (45, 101) | Clavicipitaceae |
| 8 | 83 (46, 125) | 88 (49, 132) | Hypocreaceae |
| 9 | 119 (71, 171) | 119 (73, 169) | Cordycipitaceae |
| 10 | 149 (100, 200) | 149 (105, 199) | Sarocladiaceae/Bionectriaceae/incertaesedis |
| 11 | 141 (89, 193) | 142 (94, 193) | Stachybotryaceae |
| 12 | 112 (67, 159) | 120 (74, 169) | Nectriaceae |
| 13 | 64 (32, 100) | 65 (33, 99) | |
| 14 | 11 (4, 20) | 12 (4, 20) | Stachybotrys |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, L.-Y.; Zhang, S.; Zhang, Y.-J. Comparative Mitogenomics of Fungal Species in Stachybotryaceae Provides Evolutionary Insights into Hypocreales. Int. J. Mol. Sci. 2021, 22, 13341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413341
Ren L-Y, Zhang S, Zhang Y-J. Comparative Mitogenomics of Fungal Species in Stachybotryaceae Provides Evolutionary Insights into Hypocreales. International Journal of Molecular Sciences. 2021; 22(24):13341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413341
Chicago/Turabian StyleRen, Li-Yuan, Shu Zhang, and Yong-Jie Zhang. 2021. "Comparative Mitogenomics of Fungal Species in Stachybotryaceae Provides Evolutionary Insights into Hypocreales" International Journal of Molecular Sciences 22, no. 24: 13341. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413341